The Genome Sequence of Taurine Cattle: A window to ruminant biology and evolution (original) (raw)
. Author manuscript; available in PMC: 2010 Sep 21.
Published in final edited form as: Science. 2009 Apr 24;324(5926):522–528. doi: 10.1126/science.1169588
Abstract
To understand the biology and evolution of ruminants, the cattle genome was sequenced to ∼7× coverage. The cattle genome contains a minimum of 22,000 genes, with a core set of 14,345 orthologs shared among seven mammalian species of which 1,217 are absent or undetected in non-eutherian (marsupial or monotreme) genomes. Cattle-specific evolutionary breakpoint regions in chromosomes have a higher density of segmental duplications, enrichment of repetitive elements, and species-specific variations in genes associated with lactation and immune responsiveness. Genes involved in metabolism are generally highly conserved, although five metabolic genes are deleted or extensively diverged from their human orthologs. The cattle genome sequence thus provides an enabling resource for understanding mammalian evolution and accelerating livestock genetic improvement for milk and meat production.
Domesticated cattle (Bos taurus and Bos taurus indicus) provide a significant source of nutrition and livelihood to nearly 6.6 billion humans. Cattle belong to a phylogenetically distant clade compared to humans and rodents, the Cetartiodactyl order of eutherian mammals, which first appeared ∼60 million years ago (1). Cattle represent the Ruminantia, which occupy diverse terrestrial environments with their ability to efficiently convert low quality forage into energy-dense fat, muscle and milk. These biological processes have been exploited by humans since domestication, which began in the Near East some 8,000-10,000 years ago (2). Since then, over 800 cattle breeds have been established representing an important world heritage and an enabling scientific resource for understanding the genetics of complex traits.
The cattle genome was assembled with methods similar to the rat and sea urchin genomes (3, 4). The most recent assemblies, Btau3.1 and Btau4.0, combined bacterial artificial chromosome (BAC) and whole genome shotgun (WGS) sequences. Btau3.1 was used for gene-specific analyses. Btau4.0, which includes finished sequence data and used different mapping methods to place the sequence on chromosomes, was used for all global analyses other than gene prediction. The contig N50 (50% of the genome is in contigs of this size or greater) is 48.7kb for both assemblies; the scaffold N50 for Btau4.0 is 1.9 Mb. In the Btau4.0 assembly, 90% of the total genome sequence was placed on the 29 autosomes and X chromosome and validated (3). Of 1.04 million expressed sequence tag (EST) sequences, 95.0% were contained in the assembled contigs. With an equivalent gene distribution in the remaining 5% of the genome, the estimated genome size is 2.87 Gbp. Comparison to 73 finished BACs and single nucleotide polymorphism (SNP) linkage data (5, 6) confirmed this assembly quality with greater than 92% genomic coverage and fewer than 0.8% of SNP were incorrectly positioned at the resolution of these maps (3, 4).
We used the cattle genome to catalog protein-coding genes, microRNA genes and ruminant-specific interspersed repeats and manually annotated over 4000 genes. The consensus protein-coding gene set for Btau3.1 (OGSv1), from six predicted gene sets (4), consists of 26,835 genes with a validation rate of 82% (4). On this basis we estimate that the cattle genome contains at least 22,000 protein-coding genes. We identified 496 microRNA genes of which 135 were putative novel microRNAs (4). About half of the cattle miRNA occur in 60 genomic microRNA clusters, containing 2 to 7 microRNA genes separated by less than 10 kbp (Fig. S2). The overall GC content of the cattle genome is 41.7%, with an observed-to-expected CpG ratio of 0.234, similar to other mammals.
The cattle genome has transposable element classes similar to other mammals as well as large numbers of ruminant-specific repeats (Table S4) that comprise 27% of its genome. The consensus sequence of BovB, a non-LTR LINE retrotransposon, lacked a functional open reading frame (ORF), suggesting it was inactive (7). However, BovB repeats with intact ORF were identified in the genome and their phylogeny (Fig. S4) indicates that some are still actively expanding and evolving. Mapping chromosomal segments of high and low density ancient repeats, L2/MIR (a LINE/SINE pair) and BovB, and more recent repeats, BovB/Art2A (BovB derived SINE pair), revealed that the genome consists of ancient regions enriched for L2/MIR and recent regions enriched for BovB/Art2A (Fig. S7). Exclusion of BovB/Art2A from contiguous blocks of ancient repeats suggests that evolution of the ruminant/cattle genome experienced invasions of new repeats into regions lacking ancient repeats. Alternatively, older repeats may have been destroyed by insertion of ruminant/cattle-specific repeats. AGC trinucleotide repeats, the most common simple sequence repeat (SSR) in artiodactyls (which include cattle, pigs and sheep), are 90 and 142 fold over-represented in cattle compared to human and dog, respectively (Fig. S10). 39% of the AGC repeats in the cattle genome were associated with Bov-A2 SINE elements.
A comparative analysis examined the rate of protein evolution and the conservation of gene repertoires among orthologs in the genomes of dog, human, mouse and rat (representing placental mammals), opossum (marsupial), and platypus (monotreme). Orthology was resolved for >75% of cattle and >80% of human genes (Fig. 1A). There were 14,345 orthologous groups with representatives in human, cattle or dog, mouse or rat, and opossum or platypus, which represent 16,749 cattle and 16,177 human genes, respectively of which 12,592 are single copy orthologs. We also identified 1,217 placental mammal-specific orthologous groups with genes present in human, cattle or dog, mouse or rat, but not in opossum or platypus. About 1,000 orthologs shared between rodents and laurasiatherians (cattle and dog), many of which encode G-protein coupled receptors, appear to have been lost or may be mis-annotated in the human genome (Fig. 1B). Gene repertoire conservation among these mammals correlates with conservation at the amino-acid sequence level (Fig. 1C). The elevated rate of evolution in rodents relative to other mammals (10) was supported by the higher amino acid sequence identity between human and dog or cattle proteins relative to that between human and rodent proteins. However, maximum-likelihood analysis of amino acid substitutions in single-copy orthologs supports the accepted sister lineage relationship of primates and rodents (1) (Fig. 1D).
Fig. 1.

Protein orthology comparison among genomes of cattle, dog, human, mouse and rat (Bos taurus, Canis familiaris, Homo sapiens, Mus musculus, Rattus norvegicus, representing placental mammals), opossum (Monodelphis domestica; marsupial), and platypus (Ornithorhynchus anatinus; monotreme). (A) The majority of mammalian genes are orthologous, with over half preserved as single-copies (dark blue); a few thousand have species-specific duplications (blue); another few thousand have been lost in specific lineages (orange). We also show those lacking confident orthology assignment (green), and those that are apparently lineage specific [unique (white)]. Placental-specific orthologs are shown in pink. Single- or multiple-copy genes were defined on the basis of representatives in human, bovine or dog, mouse or rat, and opossum or platypus. (B) Venn diagram showing shared orthologous groups (duplicated genes were counted as one) between laurasiatherians (cattle and dog), human, rodents (mouse and rat), and non-placental mammals (opossum and platypus) on the basis of the presence of a representative gene in at least one of the grouped species (as in A). (C) Distribution of ortholog protein identities between human and the other species for a subset of strictly conserved single-copy orthologs. (D) A maximum likelihood phylogenetic tree using all single-copy orthologs supports the accepted phylogeny and quantifies the relative rates of molecular evolution expressed as the branch lengths.
Alternative splicing is a major mechanism for transcript diversification (8), yet the extent of its evolutionary conservation and functional impact remain unclear. We used the cattle genome to analyze the conservation of the most common form of alternative splicing, exon skipping, defined as a triplet of exons in which the middle exon is absent in some transcripts, in a set of 1,930 exon-skipping events across human, mouse, dog and cattle (4). We examined 277 cases, with different conservation patterns between human and mouse, in 16 different cattle tissues with RT-PCR (4). These splicing events were divided into a shared set (163 in both human and mouse) and a non-shared set (114 in human but not in mouse). Of the 277, we detected exon skipping for 188 cases in cattle (Table S5) suggesting that the majority of genes with exon-skipping in human were present and regulated in cattle, and that if an event is shared between human and mouse, it was more likely to be found in cattle. It was estimated that at most 40% of exon skipping is conserved among mammals and our data agrees with the upper bound from previous analyses with human and rodents [e.g. (9)].
We constructed a cattle-human Oxford Grid (Fig. S12) (4) to conduct synteny-based chromosomal comparisons which reinforced that human genome organization is more similar to cattle than to rodents because most cattle chromosomes primarily correspond to part of one human chromosome; albeit with multiple rearrangements [e.g. (10)]. In contrast, the cattle-mouse Oxford Grid shows poorer chromosomal correspondence. Lineage-specific evolutionary breakpoints were identified for cattle, artiodactyls, and ferungulates (a group encompassing artiodactyls and carnivores, represented by cattle, pig and dog), and are shown with cattle (Fig. S11) and human sequence coordinates (Fig. 2) (4). Primate, dog, rodent, mouse, and rat lineage-specific breakpoint positions were similarly identified. A total of 124 evolutionary breakpoint regions (EBRs) were identified in the cattle lineage, of which 100 were cattle/ruminant specific and 24 were artiodactyl-specific (e.g. Fig. 2). Nine additional EBRs represent presumptive ferungulate-specific rearrangements. Bos taurus chromosome 16 (BTA16) is populated with four ferungulate specific EBRs, suggesting that this region was rearranged before the Artiodactyla and Carnivora divergence (Fig. 2). Such conserved regions demonstrate many inversions that occurred prior to the divergence of the carnivores and artiodactyls have probably been retained in the ancestral form within the human genome. In contrast to the cattle genome, a pig physical map identified only 77 lineage-specific EBRs. Interchromosomal rearrangements and inversions characterize most of the lineage-specific rearrangements observed in the cattle, dog, and pig genomes.
Fig. 2.
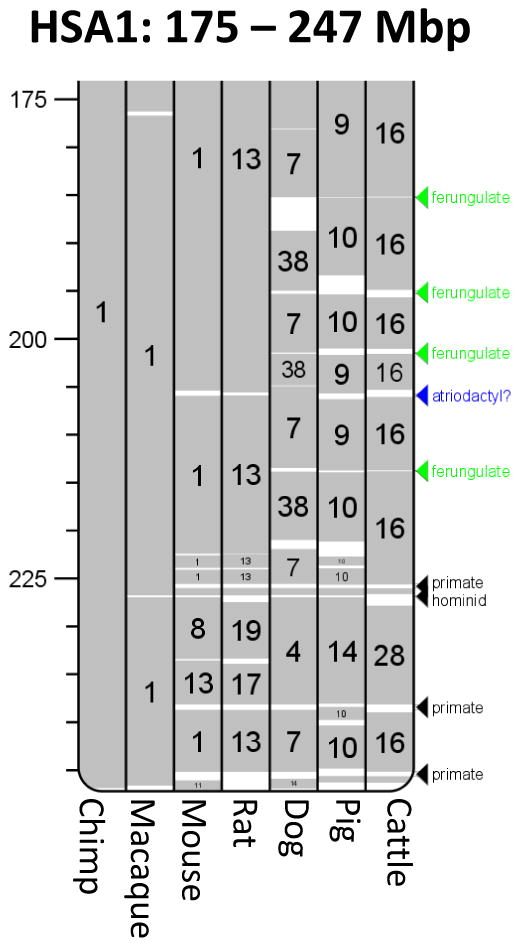
Examples of evolutionary breakpoint regions (EBRs). Ferungulate- artiodactyl- and primate-specific EBRs on HSA1 at 175-247 Mbp (other lineage-specific EBRs not shown). Homologous synteny blocks constructed for the macaque, chimp, cattle, dog, mouse, rat and pig genomes were used for pair-wise comparisons (4). White areas correspond to EBRs. Arrows to the right of the chromosome ideogram indicate positions of representative cattle-specific, artiodactyl-specific (specific to the chromosomes of pigs and cattle), ferungulate-specific (cattle, dog and pig), primate-specific (human, macaque, chimp), and hominoid-specific (human and chimp) rearrangements. Opossum is shown as an outgroup to the eutherian clade, which allows classification of ferungulate-specific EBRs.
An examination of repeat families and individual transposable elements within cattle-, artiodactyl- and ferungulate-specific EBRs showed a significantly higher density of LINE-L1 elements and the ruminant-specific LINE-RTE repeat family (11) in cattle-specific EBRs relative to the remainder of the cattle genome (Table S6). In contrast, the SINE-BovA repeat family and the more ancient tRNAGlu–derived SINE repeats (12) were present in lower density in cattle-specific EBRs, similar to other LINEs and SINEs (Table S7). The differences in repeat densities were generally consistent in cattle-, artiodactyl- and ferungulate-specific EBRs, with the exception of the tRNAGlu–derived and LTR-ERVL repeats, which are at higher densities in artiodactyl EBRs compared to the rest of the genome.
The tRNAGlu (CHRS) repeats originated in the common ancestor of Suina (pigs and peccaries), Ruminantia and Cetacea (whales) (12), suggesting that tRNAGlu –derived SINEs were involved in ancestral artiodactyl chromosome rearrangements. Furthermore, the lower density of the more ancient repeat families in cattle-specific EBRs suggests that either more recently arising repeat elements were inserted into regions lacking ancient repeats or that older repeats were destroyed by this insertion (Table S7). The differing density of repeat elements in EBRs were also found in regions of homologous synteny suggesting that repeats may promote evolutionary rearrangements (see below). Differences in repeat density in cattle-specific EBRs are thus unlikely to be caused by the accumulation of repeats in EBRs after such rearrangements occur. We identified a cattle-specific EBR associated with a bidirectional promoter (Figs. S14 and S15), that may affect control of the expression of the CYB5R4 gene which has been implicated in human diabetes and therefore may be important in the regulation of energy flow in cattle (4).
1,020 segmental duplications (SDs) corresponding to 3.1% (94.4 Mbp) of the cattle genome were identified (4). Duplications assigned to a chromosome showed a bipartite distribution with respect to length and percent identity (Fig. S16) and interchromosomal duplications were shorter (median length 2.5 kbp) and more divergent (<94% identity), relative to intrachromosomal duplications (median length 20 kbp, ∼97% identity), and tended to be locally clustered (Fig. S17). Twenty-one of these duplications were >300 kbp and located in regions enriched for tandem duplications (e.g. BTA18, Fig. S18). This pattern is reminiscent of the duplication pattern of the dog, rat and mouse but different from that of primate/great-ape genomes (13, 14). On average cattle SDs >10 kbp represent 11.7% of base pairs in 10 kbp intervals located within cattle-specific EBRs and 23.0% of base pairs located within the artiodactyl-specific EBRs. By contrast, in the remainder of the genome sequence assigned to chromosomes the fraction of SDs was 1.7% (p< 1 × 10-12). These data indicate that SDs play a role in promoting chromosome rearrangements by non-allelic homologous recombination [e.g. (15)] and suggest that either a significant fraction of the SDs observed in cattle occurred before the Ruminant-Suina split, and/or that the sites for accumulation of SDs are non-randomly distributed in artiodactyl genomes.
SDs involving genic regions may give rise to new functional paralogs. Seventy six percent (778/1,020) of the cattle SDs correspond to complete or partial gene duplications with high sequence identity (median 98.7%). This suggests that many of these gene duplications are specific to either the artiodactyla or the Bos lineage and tend to encode proteins that often interface with the external environment, particularly immune proteins and sensory/olfactory receptors. Several of these gene duplications are also duplicated in other mammalian lineages (e.g. cytochrome P450, sulfotransferase, ribonuclease A, defensins and pregnancy-associated glycoproteins). Paralogs located in segmental duplications present exclusively in cattle may have functional implications for the unique physiology, environment and diet of cattle.
An over-representation of genes involved in reproduction in cattle SDs (Tables S8 and S9) is associated with several gene families expressed in the ruminant placenta. These families encode the intercellular signaling proteins pregnancy associated glycoproteins (on BTA29), trophoblast Kunitz domain proteins (on BTA13) and interferon tau (IFNT) (on BTA8). A gene family encoding prolactin-related proteins (on BTA23) was only identified in the assembly-dependent analysis of SDs. These genes regulate ruminant-specific aspects of fetal growth, maternal adaptations to pregnancy and the coordination of parturition (16, 17). While Type I IFN genes are primarily involved in host defense (18), IFNT prevents regression of the corpus luteum during early pregnancy resulting in a uterine environment receptive to early conceptus development (19).
Signatures of positive selection (obtained by measurement of their rates of synonymous and nonsynonymous substitutions) identified 71 genes (4) including ten immune related genes (i.e. IFNAR2, IFNG, CD34, TREM1, TREML1, FCER1A, IL23R, IL24, IL15 and LEAP2). As previously mentioned, immune genes are over-represented in SDs (see Table 1 and Fig S20). Examples of genes varying in cattle relative to mouse include a cluster of β-defensin genes, which encode antimicrobial peptides, the anti-microbial cathelicidin genes [which show increased sequence diversity of the mature cathelicidin peptides (20)], changes in the numbers of interferon genes (21) and the number and organization of genes involved in adaptive immune responses in cattle compared to human and mouse (4). This extensive duplication and divergence of genes involved innate immunity may be because of the substantial load of microorganisms present in the rumen of cattle which increases the risk of opportunistic infections at mucosal surfaces and positive selection for the traits that enabled stronger and more diversified innate immune responses at these locations. Another possibility is that immunity may have been under selection due to the herd structure which can promote rapid disease transmission. Also, immune function-related duplicated genes have gained non-immune functions e.g. IFNT (see above), and the C-class lysozyme genes, which are involved in microbial degradation in the rumen, a fermentative foregut (see below).
Table 1.
Changes in the number of genes in innate immune gene families
| Gene Family | Bovine | Human | Murine |
|---|---|---|---|
| Cathelicidin | 10 | 1 | 1 |
| RNase | 21 | 13 | 25 |
| BPI-like | 13 | 9 | 11 |
| BPI/LBP | 3 | 2 | 2 |
| β-Defensin1 | ∼106 | 39 | 52 |
| Interferon subfamilies2 | |||
| IFNK | 1 | 1 | 1 |
| IFNE | 1 | 1 | 1 |
| IFNB | 6 | 1 | 1 |
| IFNA | 13 | 13 | 14 |
| IFNW | 24 | 1 | 0 |
| IFNT | 3 | 0 | 0 |
| IFNX4 | 3 | 0 | 0 |
| IFNL | 0 | 3 | 2 |
| IFNZ | 0 | 0 | 2 |
| C-type Lysozyme | 10 | 1 | 3 |
| ULBP3 | 30 | 3 | 1 |
There has been substantial reorganization of gene families encoding proteins present in milk. One such rearrangement affecting milk composition involves the histatherin (HSTN) gene within the casein gene cluster on BTA6 (Fig. S21). In the cattle genome HSTN is juxtaposed to a regulatory element (BCE) important (22) for β-casein (CSN2) expression, and as a probable consequence HSTN is regulated like the casein genes during the lactation cycle. This rearrangement that led to the juxtaposition of HSTN next to the BCE is also the probable cause of deletion of one of the two copies of α-S2-like casein genes (CSN1S2A) present in other mammalian genomes (23). The biological implications of this change in casein gene copy number are not yet clear.
Additionally, the cattle serum amyloid A (SAA) gene cluster arose from both a laurasiatherian SD and a cattle-specific EBR, resulting in two mammary gland-expressed _SAA3_- like genes, SAA3.1 and SAA3.2 on BTA29, and an _SAA3_-like gene on BTA15 (Fig. S21). SAA3.2 has been shown to inhibit microbial growth (24) Two additional milk protein genes were associated with SDs: cathelicidin (CATHL1) and beta-2 microglobulin (B2M) - part of the neonatal Fc receptor (FcRn) that transfers IgG immunoglobulin across epithelial cells of many tissues including the gut and mammary gland (25, 26). IgG is the predominant immunoglobulin in cow's milk compared to IgA in human milk (27), and unlike humans, which acquire passive immunity from the mother via placental transfer of immunoglobulins during pregnancy, calves acquire passive immunity via ingestion of IgG in milk (27). B2M is also redistributed in epithelial cells upon calving and it protects IgG from degradation (25). A genetic variant of B2M has negative effects on passive immune transfer (28), The additional copy of the gene encoding B2M may potentially be associated with the abundance of IgG in cows' milk and an increased capacity for uptake in the neonatal gut. Considering that the passive transfer of immunity to the calf is one of the important functions of milk, it is striking that lactation-related genes affected by genomic rearrangements encode immune-related proteins in milk.
Cattle metabolic pathways demonstrated a strong degree of conservation among the comprehensive set of genes involved in core mammalian metabolism (4) and permitted an examination of unique genetic events that may be related to ruminant-specific metabolic adaptations. However, among 1,032 genes examined from the human metabolic pathways, five were deleted or extensively diverged in cattle: PLA2G4C (phospholipase A2, group IVC), FAAH2 (fatty acid amide hydrolase 2), IDI2 (isopentenyl-diphosphate delta isomerase 2), GSTT2 (glutathione S-transferase theta 2) and TYMP (thymidine phosphorylase), which may be adaptations that impact on fatty acid metabolism, the mevalonate pathway (synthesis of dolichols, vitamins, steroid hormones and cholesterol), detoxification, pyrimidine metabolism, respectively. Phylogenetic analysis shows that PLA2G4C was deleted ∼87-97 Mya in the Laurasiatherian lineages (Fig. S22). Strikingly, ∼20% of the sequences from two abomasum (last chamber of the cattle stomach) EST libraries (a total of 2,392 sequences) correspond to three C-type lysozyme genes. Lysozyme primarily functions in animals as an antibacterial protein suggesting they probably function in the abomasum (similar to the monogastric stomach) to degrade the cell walls of bacteria entering from the foregut (29). The cattle genome contains 10 C-type lysozyme genes (Table S14, Fig. S23) and EST evidence (Fig. S23) shows that six of the seven remaining C-type lysozyme genes are expressed primarily in the rumen and/or intestine suggesting additional roles for the encoded proteins in ruminant digestion.
In summary, the biological systems most impacted by changes in the number and organization of genes in the cattle lineage include reproduction, immunity, lactation, and digestion. We highlighted the evolutionary activity associated with chromosomal breakpoint regions and their propensity for promoting gene birth and rearrangement. These changes in the cattle lineage probably reflect metabolic and immune adaptations due to microbial fermentation in the rumen, the herd environment and its influence on disease transmission, and the reproductive strategy of cattle. The cattle genome and associated resources will facilitate the identification of novel functions and regulatory systems of general importance in mammals and may provide an enabling tool for genetic improvement within the beef and dairy industries.
Supplementary Material
SOM
Acknowledgments
The master accession for this WGS sequencing project is AAFC03000000. The individual WGS sequences are AAFC03000001-AAFC03131728, and the scaffold records are CM000177-CM000206 (chromosomes) and DS490632-DS495890 (unplaced scaffolds).
Funded by: the National Human Genome Research Institute (NHGRI U54 HG003273); the U.S. Department of Agriculture's Agricultural Research Service (USDA ARS Agreement No. 59-0790-3-196) and Cooperative State Research, Education, and Extension Service National Research Initiative (Grant No. 2004-35216-14163); the state of Texas; Genome Canada through Genome British Columbia; The Alberta Science and Research Authority; The Commonwealth Scientific and Industrial Research Organization of Australia (CSIRO); Agritech Investments Ltd., Dairy Insight, Inc. and AgResearch Ltd., all of New Zealand; the Research Council of Norway; the Kleberg Foundation; and the National, Texas and South Dakota Beef Check-off Funds.
Principal Investigator: Richard A. Gibbs1
Analysis Project Leadership: Christine G. Elsik2,3, Ross L. Tellam4
Sequencing Project Leadership: Richard A. Gibbs1, Donna M. Muzny1, George M. Weinstock5,1
Analysis Group Organization: David L. Adelson6, Evan E. Eichler7,8, Laura Elnitski9, Christine G. Elsik2,3, Roderic Guigó10, Debora L. Hamernik11, Steve M. Kappes12, Harris A. Lewin13,14, David J. Lynn15, Frank W. Nicholas16, Alexandre Reymond17, Monique Rijnkels18, Loren C. Skow19, Ross L. Tellam4, Kim C. Worley1, Evgeny M. Zdobnov20,21,22
Sequencing Project White Paper: Richard A. Gibbs1, Steve M. Kappes12, Lawrence Schook13, Loren C. Skow19, George M. Weinstock5,1, James Womack23
Gene Prediction and Consensus Gene Set: Tyler Alioto10, Stylianos E. Antonarakis20, Alex Astashyn24, Charles E. Chapple10, Hsiu-Chuan Chen24, Jacqueline Chrast17, Francisco Câmara10, Christine G. Elsik2,3 (leader), Olga Ermolaeva24, Roderic Guigó10, Charlotte N. Henrichsen17, Wratko Hlavina24, Yuri Kapustin24, Boris Kiryutin24, Paul Kitts24, Felix Kokocinski25, Melissa Landrum24, Donna Maglott24, Kim Pruitt24, Alexandre Reymond17, Victor Sapojnikov24, Stephen M. Searle25, Victor Solovyev26, Alexandre Souvorov24, Catherine Ucla20, George M. Weinstock5,1, Carine Wyss20
Experimental Validation of Gene Set: Tyler Alioto10, Stylianos E. Antonarakis20, Charles E. Chapple10, Jacqueline Chrast17, Francisco Câmara10, Roderic Guigó10 (leader), Charlotte N. Henrichsen17, Alexandre Reymond17, Catherine Ucla20, Carine Wyss20
MicroRNA Analysis: Juan M. Anzola3, Daniel Gerlach20,21, Evgeny M. Zdobnov20,21,22 (leader)
GC Composition Analysis: Eran Elhaik27,28, Christine G. Elsik2,3 (leader), Dan Graur27, Justin T. Reese2
Repeat Analysis: David L. Adelson6 (leader), Robert C. Edgar29, John C. McEwan30, Gemma M. Payne30, Joy M. Raison31
Protein Ortholog Analysis: Thomas Junier19,20, Evgenia V. Kriventseva32, Evgeny M. Zdobnov20,21,22 (leader)
Exon Skipping Analysis: Jacqueline Chrast17, Eduardo Eyras33,34, Charlotte N. Henrichsen17, Mireya Plass34, Alexandre Reymond17 (leader)
Evolutionary Breakpoint Analysis and Oxford Grid: Ravikiran Donthu13, Denis M. Larkin13,14, Harris A. Lewin13,14 (leader), Frank W. Nicholas16
Bidirectional Promoter Analysis: Laura Elnitski9 (leader), Denis M. Larkin13,14, Harris A. Lewin13,14, James Reecy35, Mary Q. Yang9
Segmental Duplication Analysis: David L. Adelson6, Lin Chen7, Ze Cheng7, Carol G. Chitko-McKown36, Evan E. Eichler7,8 (leader), Laura Elnitski9, Christine G. Elsik2,3, George E. Liu37, Lakshmi K. Matukumalli38,37, Jiuzhou Song39, Bin Zhu39
Analysis of Gene Ontology in Segmental Duplications: Christine G. Elsik2,3, David J. Lynn15 (leader), Justin T. Reese2
Adaptive Evolution: Daniel G. Bradley40, Fiona S.L. Brinkman15, Lilian P.L. Lau40, David J. Lynn15 (leader), Matthew D. Whiteside15
Innate Immunity: Ross L. Tellam4 (leader), Angela Walker41, Thomas T. Wheeler42
Lactation: Theresa Casey43, J. Bruce German44,45, Danielle G. Lemay45, David J. Lynn15, Nauman J. Maqbool46, Adrian J. Molenaar42, Monique Rijnkels18 (leader)
Metabolism: Harris A. Lewin13,14 (leader), Seongwon Seo47, Paul Stothard48
Adaptive Immunity: Cynthia L. Baldwin49, Rebecca Baxter50, Candice L. Brinkmeyer-Langford19, Wendy C. Brown51 Christopher P. Childers2, Timothy Connelley52, Shirley A. Ellis53, Krista Fritz19, Elizabeth J. Glass50, Carolyn T.A. Herzig49, Antti Iivanainen54, Kevin K. Lahmers51, Loren C. Skow19 (leader)
Annotation Data Management: Anna K. Bennett2, Christopher P. Childers2, C. Michael Dickens3, Christine G. Elsik2,3 (leader), James G.R. Gilbert25, Darren E. Hagen2, Justin T. Reese2, Hanni Salih3
Manual Annotation Organization: Jan Aerts55, Alexandre R. Caetano56, Brian Dalrymple4, Christine G. Elsik2,3, Jose Fernando Garcia57, Richard A. Gibbs1, Clare A. Gill3,58, Debora L. Hamernik11, Stefan G. Hiendleder59, Erdogan Memili60, Frank W. Nicholas16, James Reecy35, Monique Rijnkels18, Loren C. Skow19, Diane Spurlock35, Paul Stothard48, Ross L. Tellam4, George M. Weinstock5,1, John L. Williams61, Kim C. Worley1
cDNA Tissues, Libraries and Sequencing: Lee Alexander62, Michael J. Brownstein63, Leluo Guan48, Robert A. Holt64 (leader), Steven J.M. Jones64 (leader), Marco A. Marra64 (leader), Richard Moore64, Stephen S. Moore48 (leader), Andy Roberts62, Masaaki Taniguchi65,48, Richard C. Waterman62
Genome Sequence Production: Joseph Chacko1, Mimi M. Chandrabose1, Andy Cree1 (leader), Marvin Diep Dao1, Huyen H. Dinh1 (leader), Ramatu Ayiesha Gabisi1, Sandra Hines1, Jennifer Hume1 (leader), Shalini N. Jhangiani1, Vandita Joshi1, Christie L. Kovar1 (leader), Lora R. Lewis1, Yih-shin Liu1, John Lopez1, Margaret B. Morgan1, Donna M. Muzny1 (leader), Ngoc Bich Nguyen1, Geoffrey O. Okwuonu1, San Juana Ruiz1, Jireh Santibanez1, Rita A. Wright1
Sequence Finishing: Christian Buhay1 (leader), Yan Ding1, Shannon Dugan-Rocha1 (leader), Judith Herdandez1, Michael Holder1, Aniko Sabo1
Automated BAC Assembly: Amy Egan1, Jason Goodell1, Katarzyna Wilczek-Boney1
Sequence Production Informatics: Gerald R. Fowler1 (leader), Matthew Edward Hitchens1, Ryan J. Lozado1, Charles Moen1, David Steffen66,1, James T. Warren1, Jingkun Zhang1
BAC Mapping: Readman Chiu64, Steven J.M. Jones64, Marco A. Marra64 (leader), Jacqueline E. Schein64
Genome Assembly: K. James Durbin67,1, Paul Havlak68,1, Huaiyang Jiang1, Yue Liu1, Xiang Qin1, Yanru Ren1, Yufeng Shen1,69, Henry Song1, George M. Weinstock5,1, Kim C. Worley1 (leader)
Sequence Library Production: Stephanie Nicole Bell1, Clay Davis1, Angela Jolivet Johnson1, Sandra Lee1, Lynne V. Nazareth1 (leader), Bella Mayurkumar Patel1, Ling-Ling Pu1, Selina Vattathil1, Rex Lee Williams, Jr.1
BAC Production: Stacey Curry1, Cerissa Hamilton1, Erica Sodergren5,1 (leader)
Sequence Variation Detection: Lynne V. Nazareth1, David A. Wheeler1
Markers and Mapping: David L. Adelson6, Jan Aerts55, Wes Barris4, Gary L. Bennett36, Brian Dalrymple4, André Eggen70, Clare A. Gill3,58, Ronnie D. Green71, Gregory P. Harhay36, Matthew Hobbs72, Oliver Jann50, Steve M. Kappes12 (leader), John W. Keele36, Matthew P. Kent73, Denis M. Larkin13,14, Harris A. Lewin13,14, Sigbjørn Lien73, John C. McEwan30, Stephanie D. McKay74, Sean McWilliam4, Stephen S. Moore48, Frank W. Nicholas16, Gemma M. Payne30, Abhirami Ratnakumar75,4, Hanni Salih3, Robert D. Schnabel74, Timothy Smith36, Warren M. Snelling36, Tad S. Sonstegard37, Roger T. Stone36, Yoshikazu Sugimoto76, Akiko Takasuga76, Jeremy F. Taylor74, Ross L. Tellam4, Curtis P. Van Tassell37, John L. Williams61
Genomic DNA: Michael D. MacNeil62
Manual Annotation: Antonio R.R. Abatepaulo77, Colette A. Abbey3, Jan Aerts55, Virpi Ahola78, Iassudara G. Almeida57, Ariel F. Amadio79, Elen Anatriello77, Suria M. Bahadue2, Cynthia L. Baldwin49, Rebecca Baxter50, Anna K. Bennett2, Fernando H. Biase13, Clayton R. Boldt3, Candice L. Brinkmeyer-Langford19, Wendy C. Brown51, Alexandre R. Caetano56, Jeffery A. Carroll80, Wanessa A. Carvalho77, Theresa Casey43, Eliane P. Cervelatti57, Elsa Chacko81, Jennifer E. Chapin3, Ye Cheng35, Christopher P. Childers2, Jungwoo Choi3, Adam J. Colley82, Timothy Connelley52, Tatiana A. de Campos56, Marcos De Donato83, Isabel K.F. de Miranda Santos56,77, Carlo J.F. de Oliveira77, Heather Deobald84, Eve Devinoy85, C. Michael Dickens3, Kaitlin E. Donohue2, Peter Dovc86, Annett Eberlein87, Shirley A. Ellis53, Carolyn J. Fitzsimmons59, Alessandra M. Franzin77, Krista Fritz19, Gustavo R. Garcia77, Jose Fernando Garcia57, Sem Genini61, J. Bruce German44,45, James G.R. Gilbert25, Clare A. Gill3,58, Cody J. Gladney3, Elizabeth J. Glass50, Jason R. Grant48, Marion L. Greaser88, Jonathan A. Green74, Darryl L. Hadsell18, Darren E. Hagen2, Hatam A. Hakimov89, Rob Halgren43, Jennifer L. Harrow25, Elizabeth A. Hart25, Nicola Hastings90,50, Marta Hernandez91, Carolyn T.A. Herzig49, Stefan G. Hiendleder59, Matthew Hobbs72, Zhi-Liang Hu35, Antti Iivanainen54, Aaron Ingham4, Terhi Iso-Touru78, Catherine Jamis2, Oliver Jann50, Kirsty Jensen50, Dimos Kapetis61, Tovah Kerr51, Sari S. Khalil2, Hasan Khatib92, Davood Kolbehdari48,93, Charu G. Kumar13, Dinesh Kumar94,35, Richard Leach50, Justin C-M Lee2, Danielle G. Lemay45, Changxi Li95,48, George E. Liu37, Krystin M. Logan96, Roberto Malinverni61, Nauman J. Maqbool46, Elisa Marques48, William F. Martin45, Natalia F. Martins56, Sandra R. Maruyama77, Raffaele Mazza97, Kim L. McLean84, Juan F. Medrano98, Erdogan Memili60, Adrian J. Molenaar42, Barbara T. Moreno57, Daniela D. Moré77, Carl T. Muntean3, Hari P. Nandakumar19, Marcelo F.G. Nogueira99, Ingrid Olsaker100, Sameer D. Pant82, Francesca Panzitta61, Rosemeire C.P. Pastor57, Mario A. Poli101, Nathan Poslusny2, Satyanarayana Rachagani35, Shoba Ranganathan81,102, Andrej Razpet86, James Reecy35, Penny K. Riggs3,58, Monique Rijnkels18, Gonzalo Rincon98, Nelida Rodriguez-Osorio60,103, Sandra L. Rodriguez-Zas13, Natasha E. Romero3, Anne Rosenwald2, Lillian Sando4, Sheila M. Schmutz84, Seongwon Seo47, Libing Shen2, Laura Sherman48, Loren C. Skow19, Bruce R. Southey104, Diane Spurlock35, Ylva Strandberg Lutzow4, Jonathan V. Sweedler104, Imke Tammen72, Masaaki Taniguchi65,48, Ross L. Tellam4, Bhanu Prakash V.L. Telugu74, Jennifer M. Urbanski2, Yuri T. Utsunomiya57, Chris P. Verschoor82, Ashley J. Waardenberg4,105, Angela Walker41, Zhiquan Wang48, Robert Ward106, Rosemarie Weikard87, Thomas H. Welsh, Jr.3,58, Thomas T. Wheeler42, Stephen N. White51,107, John L. Williams61, Laurens G. Wilming25, Kris R. Wunderlich3, Jianqi Yang108, Feng-Qi Zhao109
1Human Genome Sequencing Center, Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA
2Department of Biology, 406 Reiss, Georgetown University, 37th & O Streets NW, Washington, DC, 20057, USA
3Department of Animal Science, Texas A&M University, 2471 TAMU, College Station, TX, 77843-2471, USA
4Livestock Industries, Commonwealth Scientific and Industrial Research Organization (CSIRO), 306 Carmody Road, St. Lucia, Queensland, 4067, Australia
5The Genome Center at Washington University, Washington University School of Medicine, 4444 Forest Park Ave, St. Louis, MO, 63108, USA
6School of Molecular and Biomedical Science, School of Agriculture, Food and Wine, The University of Adelaide, Adelaide, SA, 5005, Australia
7Department of Genome Sciences, University of Washington, 1705 NE Pacific St, Seattle, WA, 98195-5065, USA
8Howard Hughes Medical Institute, Seattle, WA, 98195, USA
9National Human Genome Research Institute, National Institutes of Health, 5625 Fishers Lane, Rockville, MD, 20878, USA
10Center for Genomic Regulation and Grup de Recerca en Informática Biomédica, Institut Municipal d'Investigació Mèdica, Universitat Pompeu Fabra, 08003 Barcelona, Catalonia, Spain
11USDA- Cooperative State Research, Education, & Extension Service, 1400 Independence Ave SW, Stop 2220, Washington, DC, 20250-2220, USA
12National Program Staff, USDA - Agricultural Research Service, 5601 Sunnyside Avenue, Beltsville, MD, USA
13Department of Animal Sciences, University of Illinois at Urbana-Champaign, 1201 West Gregory Drive, Urbana, IL, 61801, USA
14Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1201 West Gregory Drive, Urbana, IL, 61801, USA
15Department of Molecular Biology and Biochemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC, V5A 1S6, Canada
16Faculty of Veterinary Science, University of Sydney, Sydney, NSW, 2006, Australia
17Center for Integrative Genomics, University of Lausanne, Lausanne, 1015, Switzerland
18USDA/ARS Children's Nutrition Research Center, Department of Pediatrics-Nutrition, Baylor College of Medicine, 1100 Bates Street, Houston, TX, 77030-2600, USA
19Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, TX, 77843, USA
20Department of Genetic Medicine and Development, University of Geneva Medical School, 1 rue Michel-Servet, Geneva, 1211, Switzerland
21Swiss Institute of Bioinformatics, 1 rue Michel-Servet, Geneva, 1211, Switzerland
22Division of Molecular Biosciences, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
23Department of Veterinary Pathobiology, Texas A&M University, College Station, TX, 77843, USA
24National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, 20892, USA
25Informatics Department, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1HH, UK
26Department of Computer Science, University of London, Royal Holloway, Egham, Surrey, TW20 0EX, UK
27Department of Biology and Biochemistry, University of Houston, Houston, TX, 77204, USA
28McKusick - Nathans Institute of Genetic Medicine, BRB 579, Johns Hopkins University School of Medicine, 733 N. Broadway, Baltimore, MD, 21205, USA
2945 Monterey Drive, Tiburon, CA, 94920, USA
30Animal Genomics, AgResearch, Invermay, PB 50034, Mosgiel, 9053, New Zealand
31eResearch SA, University of Adelaide, North Terrace, Adelaide, South Australia, 5005, Australia
32Department of Structural Biology and Bioinformatics, University of Geneva Medical School, 1 rue Michel-Servet, Geneva, 1211, Switzerland
33Catalan Institution for Research and Advanced Studies, 08010 Barcelona, Catalonia, Spain
34Computational Genomics, Universitat Pompeu Fabra, 08003 Barcelona, Catalonia, Spain
35Department of Animal Science, Iowa State University, 2255 Kildee Hall, Ames, IA, 50011-3150, USA
36Meat Animal Research Center, USDA - Agricultural Research Service, Clay Center, NE, 68933, USA
37Bovine Functional Genomics Laboratory, USDA - Agricultural Research Service, BARC-East, Beltsville, MD, 20705, USA
38Department of Bioinformatics and Computational Biology, George Mason University, 10900 University Blvd, Manassas, VA, 20110, USA
39Department of Bioengineering, University of Maryland, College Park, MD, 20742, USA
40Smurfit Institute of Genetics, Trinity College Dublin, Dublin 2, Ireland
41Department of Veterinary Pathobiology, 245 Bond Life Sciences Center, University of Missouri, Columbia, MO, 65211, USA
42Dairy Science and Technology Section, AgResearch, Ruakura Research Centre, East Street, Private Bag 3123, Hamilton, 3240, New Zealand
43Department of Animal Science, Michigan State University, East Lansing, MI, 48824-1225, USA
44Nestlé Research Centre, Vers chez les Blanc CH, Lausanne 26, 1000, Switzerland
45Department of Food Science and Technology, University of California-Davis, Davis, CA, 95616, USA
46Bioinformatics, Mathematics and Statistics, AgResearch, Ruakura Research Centre, East Street, Private Bag 3123, Hamilton, 3240, New Zealand
47Division of Animal Science and Resource, Chungnam National University, Daejeon, 305-764, Korea
48Department of Agricultural, Food and Nutritional Science, University of Alberta, 410 AgFor Centre, Edmonton, Alberta, T6G 2P5, Canada
49Department of Veterinary and Animal Sciences, University of Massachusetts, Amherst, MA, 01003, USA
50The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK
51Department of Veterinary Microbiology and Pathology, Washington State University, Pullman, WA, 99164, USA
52Division of Infection and Immunity, The Roslin Institute, Royal (Dick) School of Veterinary Science, University of Edinburgh, Roslin, Midlothian, EH25 9RG, UK
53Immunology Division, Institute for Animal Health, Compton, RG20 7NN, UK
54Department of Basic Veterinary Sciences, University of Helsinki, POB 66, Helsinki, FIN-00014, Finland
55Genome Dynamics and Evolution, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK
56Embrapa Recursos Genéticos e Biotecnologia, Final Av. W/5 Norte, Brasilia, DF, 70770-900, Brazil
57Animal Production and Health Department, UNESP - Sao Paulo State University, Aracatuba, SP, 16050-680, Brazil
58Texas AgriLife Research, College Station, TX, 77843, USA
59JS Davies Epigenetics and Genetics Group, School of Agriculture, Food & Wine and Research Centre for Reproductive Health, The University of Adelaide, Roseworthy Campus, Roseworthy, SA, 5371, Australia
60Department of Animal and Dairy Sciences, Mississippi Agricultural and Forestry Experiment Station, Mississippi State University, Mississippi State, MS, 39762, USA
61Parco Tecnologico Padano, Via Einstein, Polo Universitario, Lodi, 26900, Italy
62Fort Keogh Livestock and Range Research Laboratory, USDA - Agricultural Research Service, Miles City, MT, 59301, USA
63Building 49, B1EE16, 49 Convent Dr, Bethesda, MD, 20892, USA
64Genome Sciences Centre, British Columbia Cancer Agency, 675 West 10th Avenue, Vancouver, British Columbia, V5Z 1L3, Canada
65Division of Animal Sciences, National Institute of Agrobiological Sciences, Tsukuba, Ibaraki, 305-8602, Japan
66Bioinformatics Research Center, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA
67Department of Biomolecular Engineering, University of California at Santa Cruz, Santa Cruz, CA, 95064, USA
68Department of Computer Science, University of Houston, Houston, TX, 77204-3010, USA
69Department of Computer Science and Center for Computational Biology and Bioinformatics, Columbia University, New York, NY, 10032, USA
70INRA, Animal Genetics and Integrative Biology, Bovine Genetics and Genomics, 78350 Jouy-en-Josas, France
71Pfizer Animal Genetics, Pfizer Animal Health, New York, NY, 10017, USA
72Faculty of Veterinary Science, University of Sydney, Camden, NSW, 2570, Australia
73Centre for Integrative Genetics and Department of Animal and Aquacultural Sciences, Norwegian University of Life Sciences, Arboretveien 6, Ås, 1432, Norway
74Division of Animal Sciences, University of Missouri, 920 East Campus Drive, Columbia, MO, 65211, USA
75Department of Medical Biochemistry and Microbiology, Uppsala University, Uppsala Biomedical Centre Husargatan 3, Uppsala, 75 123, Sweden
76Shirakawa Institute of Animal Genetics, Nishigo, Fukushima 961-8061, Japan
77Department of Biochemistry and Immunology, Ribeirão Preto Medical School, University of São Paulo, Av Bandeirantes 3900, Ribeirão Preto, SP, 14049-900, Brazil
78Biotechnology and Food Research, MTT Agrifood Research Finland, Jokioinen, FI-31600, Finland
79EEA Rafaela, Instituto Nacional de Tecnología Agropecuaria (INTA), Ruta 34 Km 227, Rafaela, Santa Fe, 2300, Argentina
80Livestock Issues Research Unit, USDA - Agricultural Research Service, Lubbock, TX, 79403, USA
81Department of Chemistry and Biomolecular Sciences & ARC Centre of Excellence in Bioinformatics, Macquarie University, Sydney, 2109, NSW, Australia
82Department of Animal and Poultry Science, University of Guelph, Guelph, ON, N1G2W1, Canada
83Instituto de Investigaciones en Biomedicina y Ciencias Aplicadas, Universidad de Oriente, Av. Universidad, Cumana, Sucre, 6101, Venezuela
84Department of Animal and Poultry Science, University of Saskatchewan, Saskatoon, SK, S7N 5A8, Canada
85INRA – UR1196 Génomique et Physiologie de la Lactation, F78352 Jouy-en-Josas, France
86Department of Animal Science, University of Ljubljana, Groblje 3, Domzale, SI-1230, Slovenia
87Research Unit Molecular Biology, Research Institute for the Biology of Farm Animals (FBN), Dummerstorf, 18196, Germany
88Department of Animal Sciences, University of Wisconsin-Madison, 1805 Linden Drive, Madison, WI, 53706, USA
89Department of Molecular and Cellular Biology, University of Guelph, Guelph, ON, N1G 2W1, Canada
90Cell Biology and Biophysics, EMBL-Heidelberg, Meyerhofstraβe 1, Heidelberg, Germany
91Laboratory of Molecular Biology, Instituto Tecnologico Agrario de Castilla y Leon (ITACyL), Ctra. Burgos km 119, Valladolid, 47071, Spain
92Department of Dairy Science, University of Wisconsin, Madison, WI, 53706, USA
93Monsanto Company, 3302 SE Convenience Blvd, Ankeny, Iowa, 50021, USA
94Genes & Genetic Resources Molecular Analysis Lab, National Bureau of Animal Genetic Resources, Baldi Bye Pass, Karnal, Haryana, 132001, India
95Lacombe Research Centre, Agriculture and Agri-Food Canada, Lacombe, Alberta, T4L 1W1, Canada
96Biomedical Sciences, University of Guelph, Guelph, ON, N1G2W6, Canada
97Zootechnics Institute, Università Cattolica del Sacro Cuore, via Emilia Parmense 84, Piacenza, 29100, Italy
98Department of Animal Science, University of California, Davis, Davis, CA, 95616, USA
99Departamento de Ciências Biológicas, Faculdade de Ciências e Letras, UNESP – São Paulo State University, Av Dom Antônio 2100, Vila Tênis Clube, Assis, SP, 19806-900, Brazil
100Department of Basic Sciences and Aquatic Medicine, Norwegian School of Veterinary Science, P.O. Box 8146 Dep, Oslo, NO-0033, Norway
101Instituto de Genética Ewald Favret, Instituto Nacional de Tecnología Agropecuaria (INTA), Las Cabañas y de Los Reseros s/n CC25, Castelar, Buenos Aires, B1712WAA, Argentina
102Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, Singapore, 117597, Singapore
103Grupo CENTAURO, Universidad de Antioquia, Medellín, Colombia
104Department of Chemistry, University of Illinois, Urbana, IL, 61801, USA
105Eskitis Institute for Cell and Molecular Therapies, Griffith University, Nathan, Queensland, 4111, Australia
106Nutrition and Food Sciences, Utah State University, Logan, UT, 84322, USA
107Animal Disease Research Unit, USDA - Agricultural Research Service, Pullman, WA, 99164, USA
108Department of Pharmacology, 2-344 BSB, University of Iowa, 51 Newton Road, Iowa City, IA, 52242, USA
109Department of Animal Science, 211 Terrill, University of Vermont, 570 Main Street, Burlington, VT, 05405, USA
References
- 1.Murphy WJ, Pevzner PA, O'Brien SJ. Trends Genet. 2004 Dec;20:631–639. doi: 10.1016/j.tig.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Willham RL. Journal of Animal Science. 1986;62:1742–1758. [Google Scholar]
- 3.Liu Y, et al. BMC Genomics. In Press. [Google Scholar]
- 4.Materials, methods, and additional discussion are available on Science online.
- 5.Nilsen H, et al. Anim Genet. 2008 Apr;39:97–104. doi: 10.1111/j.1365-2052.2007.01686.x. [DOI] [PubMed] [Google Scholar]
- 6.Prasad A, et al. BMC Genomics. 2007;8:310. doi: 10.1186/1471-2164-8-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malik HS, Eickbush TH. Mol Biol Evol. 1998 Sep;15:1123–1134. doi: 10.1093/oxfordjournals.molbev.a026020. [DOI] [PubMed] [Google Scholar]
- 8.Modrek B, Lee CJ. Nat Genet. 2003 Jun;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- 9.Sorek R, Shamir R, Ast G. Trends Genet. 2004 Feb;20:68–71. doi: 10.1016/j.tig.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Everts-van der Wind A, et al. Proc Natl Acad Sci U S A. 2005 Dec 20;102:18526–18531. doi: 10.1073/pnas.0509285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kordis D, Gubensek F. Gene. 1999 Sep 30;238:171–178. doi: 10.1016/s0378-1119(99)00260-7. [DOI] [PubMed] [Google Scholar]
- 12.Shimamura M, Abe H, Nikaido M, Ohshima K, Okada N. Mol Biol Evol. 1999 Aug;16:1046–1060. doi: 10.1093/oxfordjournals.molbev.a026194. [DOI] [PubMed] [Google Scholar]
- 13.Bailey JA, Eichler EE. Nat Rev Genet. 2006 Jul;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 14.Bailey JA, et al. Science. 2002 Aug 9;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 15.Murphy WJ, et al. Science. 2005 Jul 22;309:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- 16.Hashizume K, et al. Reprod Fertil Dev. 2007;19:79–90. doi: 10.1071/rd06118. [DOI] [PubMed] [Google Scholar]
- 17.Larson JH, et al. Physiol Genomics. 2006 May 16;25:405–413. doi: 10.1152/physiolgenomics.00307.2005. [DOI] [PubMed] [Google Scholar]
- 18.Zhang SY, et al. Immunol Rev. 2008 Dec;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 19.Roberts RM, Chen Y, Ezashi T, Walker AM. Semin Cell Dev Biol. 2008 Apr;19:170–177. doi: 10.1016/j.semcdb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scocchi M, Wang S, Zanetti M. FEBS Lett. 1997 Nov 17;417:311–315. doi: 10.1016/s0014-5793(97)01310-0. [DOI] [PubMed] [Google Scholar]
- 21.Katze MG, He Y, Gale M., Jr Nat Rev Immunol. 2002 Sep;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 22.Schmidhauser C, et al. Mol Biol Cell. 1992 Jun;3:699–709. doi: 10.1091/mbc.3.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rijnkels M, Elnitski L, Miller W, Rosen JM. Genomics. 2003 Oct;82:417–432. doi: 10.1016/s0888-7543(03)00114-9. [DOI] [PubMed] [Google Scholar]
- 24.Molenaar AJ, et al. Biomarkers. 2009;14:26–37. doi: 10.1080/13547500902730714. [DOI] [PubMed] [Google Scholar]
- 25.Mayer B, et al. J Dairy Res. 2005;72:107–112. doi: 10.1017/s0022029905001135. Spec No. [DOI] [PubMed] [Google Scholar]
- 26.Roopenian DC, Akilesh S. Nat Rev Immunol. 2007 Sep;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 27.Newby TJ, Stokes CR, Bourne FJ. Vet Immunol Immunopathol. 1982 Jan;3:67–94. doi: 10.1016/0165-2427(82)90032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clawson ML, et al. Mamm Genome. 2004 Mar;15:227–236. doi: 10.1007/s00335-003-2320-x. [DOI] [PubMed] [Google Scholar]
- 29.Irwin DM. J Mol Evol. 1995 Sep;41:299–312. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOM