Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA (original) (raw)
Abstract
Telomerase is a unique ribonucleoprotein complex that catalyzes the addition of telomeric DNA repeats onto the 3′ ends of linear chromosomes. All vertebrate telomerase RNAs contain a catalytically essential core domain that includes the template and a pseudoknot with extended helical subdomains. Within these helical regions is an asymmetric 5-nt internal bulge loop (J2a/b) flanked by helices (P2a and P2b) that is highly conserved in its location but not sequence. NMR structure determination reveals that J2a/b forms a defined S-shape and creates an ∼90 ° bend with a surprisingly low twist (∼10 °) between the flanking helices. A search of RNA structures revealed only one other example of a 5-nt bulge, from hepatitis C virus internal ribosome entry site, with a different sequence but the same structure. J2a/b is intrinsically flexible but the interhelical motions across the loop are remarkably restricted. Nucleotide substitutions in J2a/b that affect the bend angle, direction, and interhelical dynamics are correlated with telomerase activity. Based on the structures of P2ab (J2a/b and flanking helices), the conserved region of the pseudoknot (P2b/P3, previously determined) and the remaining helical segment (P2a.1–J2a.1 refined using residual dipolar couplings and the modeling program MC-Sym) we have calculated an NMR-based model of the full-length pseudoknot. The model and dynamics analysis show that J2a/b serves as a dominant structural and dynamical element in defining the overall topology of the core domain, and suggest that interhelical motions in P2ab facilitate nucleotide addition along the template and template translocation.
Telomeres are DNA-protein complexes that cap the ends of linear chromosomes. During each round of cell replication, telomeres shorten due to incomplete replication of telomere DNA repeats, and shortening of telomeres below a critical length leads to telomere fusions and cell senescence (1). Telomerase, a unique reverse transcriptase first discovered in Tetrahymena about two decades ago, is essential for maintaining the telomere length and the stability of chromosomes in most eukaryote species (2–4). A high level of telomerase activity is associated with cell proliferation in most (∼90%) cancers (5). The telomerase holoenzyme is a large complex comprising a unique reverse transcriptase protein (telomerase reverse transcriptase, TERT) which contains the active site for nucleotide addition, an essential RNA component [telomerase RNA (TR)] which contains the template for telomere DNA synthesis (6), and several species-specific proteins required for function in vivo such as assembly and localization (7).
In addition to providing the template, the TR contains subdomains (8, 9) required for catalytic activity, localization, TR 3′ end processing, and accumulation (10). Telomerase deficiency due to mutations in TR has been linked to several inherited human diseases, such as dyskeratosis congenita, aplastic anemia, myelodysplasia, and idiopathic pulmonary fibrosis (11). The minimal components of vertebrate TR required for interaction with TERT and catalytic activity in vitro are the core domain (12) (also called the pseudoknot or pseudoknot/core domain) and the CR4/CR5 domain (Fig. 1) (13, 14). Although the size of TR varies from ∼150 nt in ciliates to ∼450 nt in vertebrates to more than 1,000 nt in the budding and fission yeasts (9, 15), the catalytically essential core domain has a secondary structure that appears to be conserved across all species. The conserved secondary structure elements of the core domain are a large loop containing the template and 5′ template boundary elements, a pseudoknot (P2/P3 in vertebrates), and a short loop-closing helix (P1 in vertebrates) (9, 16, 17). Despite the importance of these RNA elements for telomerase function, little is known about the overall TR tertiary structure and how it interacts with TERT and contributes to activity. TERT contains, in addition to the usual reverse transcriptase motifs, additional RNA binding domains that interact with both the CR4/CR5 and core domain (14, 18–20). TERT binds to the P6.1 helix of CR4/CR5 but its specific binding site on the core domain other than the template has not been identified. A structure of the putative TERT from the Tribolium castaneum has revealed that it forms a ring-shaped structure around the potential template-primer binding site similar to other reverse transcriptases (21, 22), but other interactions with TR remain to be elucidated.
Fig. 1.

The secondary structure of human telomerase RNA (hTR) (Upper) and the sequence and secondary structure of the hTR core domain with helical and loop regions labeled (Lower). The hTERT (gray ellipse) interacts with both the hTR core and hTR CR4/CR5 domains. The P2/P3 pseudoknot contains three subdomains: P2a.1-J2a.1-P2a (P2a1a) [colored blue-black-gold], P2a-J2a/b-P2b (P2ab) [colored gold-green-red], and the P2b-P3 pseudoknot [colored red-pink].
In human telomerase RNA (hTR), the full-length P2/P3 pseudoknot can be divided into three subdomains, P2a.1-J2a.1-P2a (P2a1a), P2a-J2a/b-P2b (P2ab), and P2b-P3 and associated loops (P2b-P3 pseudoknot), which comprise all the helical elements of the core domain except for P1 helix (Fig. 1). Within P2/P3, the three-dimensional structure of only the P2b-P3 pseudoknot has been determined, and its dynamics have been extensively investigated (23, 24). This region of the TR contains almost all of the highly conserved nucleotides in the full-length P2/P3. P2b-P3 forms a compact H-type pseudoknot with extensive tertiary interactions between the loop nucleotides and the stems. Folding of the pseudoknot, and in particular the pseudoknot tertiary interactions, have been shown to be essential for function (23, 25, 26). Detailed NMR dynamics analysis showed that P2b-P3 has little global motion. P2a.1 and the adjacent internal loop J2a.1 are a mammalian specific extension to P2a (8, 27). P2ab is located at the center of the P2/P3 pseudoknot, where the J2a/b bulge loop is flanked by helices P2a and P2b. The location of J2a/b is highly conserved in all vertebrate TRs and the asymmetric loop in mammalian TRs is usually a 5-nt bulge loop. Its sequence is pyrimidine-rich except for a relatively conserved G at the 5′ end (61% in vertebrates, 83% in mammalian TRs) (8, 27). Previous biochemical studies of J2a/b and adjacent nucleotides have shown that swapping the sequence or replacing it with its counterpart from the mouse TR had relatively little effect on telomerase activity (12, 28). Nevertheless, its conserved central location in the P2/P3 pseudoknot suggests that it might play a role in defining the overall topology of the core domain.
The secondary structure of the core domain constrains the position of the template relative to the P2/P3 pseudoknot. Single molecule FRET-assisted low-resolution modeling (29) and biochemical cross-linking experiments (26) have provided evidence that, in the free hTR, the template is positioned near to the P3 stem of the P2b-P3 pseudoknot. During each cycle of processive telomere repeat synthesis, the template needs to move relative to the TERT active site by 6 nt (for a complete TTAGGG vertebrate telomere repeat) and subsequently translocate back to the original position. Thus, the large pseudoknot might also encode intrinsic conformational plasticity that would facilitate these movements during each telomere DNA repeat addition cycle. It has previously been proposed that a conformational switch between a completely folded and partially unfolded pseudoknot (hairpin) might play a role in translocation (30, 31). However, current mutational data are consistent with a requirement for a folded pseudoknot for optimal telomerase activity (23, 32).
To provide insight into the conformational constraints imposed by the P2/P3 pseudoknot in positioning the template into the active site of telomerase, we have investigated the role of the secondary structure elements of the P2/P3 pseudoknot in defining the topology of the core domain. We determined the solution structure of P2ab and quantified the local and global dynamics within and across the J2a/b loop by NMR. J2a/b induces a large bend between the two helices that has a surprisingly limited interhelical flexibility. Based on the structure and dynamics, we designed a series of nucleotide substitutions in J2a/b to investigate its functional roles in telomerase enzymatic activity. Activity assays of telomerase with nucleotide substitutions in J2a/b indicate that the bend angle, direction, and flexibility induced by J2a/b are important for function. In contrast to J2a/b, NMR analysis and structure modeling of P2a1a using a combined RDC-MC-Sym approach indicate that the internal J2a.1 loop does not induce a significant bend or flexibility between P2a.1 and P2a. The subdomain structures of the P2/P3 pseudoknot were assembled to determine an NMR based structure model of the entire helical region of the hTR core domain. Our data show that J2a/b serves as a dominant structural and dynamical element in defining the overall topology of the core domain and suggest that interhelical motions in P2ab facilitate nucleotide addition along the template and template translocation.
Results
Determination and Overview of the Solution Structure of P2ab.
To assess the structural role of the J2a/b region in the overall core domain, we designed an RNA construct of the P2a-J2a/b-P2b subdomain (P2ab). The P2a stem was extended with one additional GC base pair to facilitate in vitro transcription, the C92-G122 base pair in P2b was replaced with a U-A base pair to facilitate resonance assignments in the otherwise GC-containing stem, and P2b was capped with a closing UUCG tetraloop (Fig. 2A). Resonance assignments and structure calculations are described in Materials and Methods and the SI Appendix. A total of 785 NOE restraints (an average of 22 NOEs per nucleotide), 230 dihedral angle restraints, and 76 residual dipolar couplings (RDCs) were used in the structure calculation. It is important to note that during the refinement with RDCs, we first performed an order tensor analysis to determine the degree of alignment of the individual P2a and P2b helices (33). The analysis showed that there was a significant difference between their alignments, and therefore it was necessary to refine the structures using two independent axial (D a) and rhombic (R) alignment values for each helical domain, while one common pseudo molecule representing the orientation of the alignment was shared between the two helices. The fact that P2a and P2b had different degrees of alignment indicates that the conformational freedom in the J2a/b bulge results in interhelical motions. The ensemble structures of P2ab should therefore be considered as dynamically averaged rather than static solution structures (discussed below).
Fig. 2.

Solution structure of hTR P2ab. (A) Sequence and secondary structure of the P2ab construct used in the solution NMR study. (B) Superposition of the 20 lowest-energy structures over all heavy atoms. (C) Stereoview of the lowest-energy structure, which shows the S-shaped conformation of the J2a/b loop. (D) Space-filling rendering of the lowest-energy structure of P2ab. In all panels, P2a is gold, J2a/b is green, P2b is red, and the UUCG tetraloop is gray.
The ensemble of the 20 lowest-energy structures is well determined with an rmsd to the mean of 1.14 ± 0.26 Å for all heavy atoms (Fig. 2B and Table 1). P2a and P2b are well-defined A-form helices with an rmsd to the mean of 0.59 ± 0.19 Å and 0.54 ± 0.11 Å, respectively. J2a/b introduces a large bend between P2a and P2b, with an average interhelical angle of 89 ± 3° across the major groove (Fig. 2C). The backbone of J2a/b displays an overall S-shape conformation, with the change in helical direction occurring at U86. The large interhelical bend together with the local conformation of J2a/b creates an open interface between P2a and P2b (Fig. 2D). The bend disrupts the stacking interactions between the helices, weakening the stabilities of the neighboring base pairs of J2a/b, U83-A126, and C89-G125, as evidenced by the disappearance of imino resonances of U83 and G125 at 20 °C. The nucleotides at the ends of the bulge are well defined, with G84 stacking above P2a, and C88 stacking below P2b C89. Although the positions of the three other J2a/b residues are less well defined due to the large local dynamics (Fig. 2B, see below), C85 and U86 tend to form continuous stacking interactions with G84, and C87 tends to stack with C88. Remarkably, the S-shape conformation of the 5-nt bulge results in almost no twist between P2a and P2b (10 ± 10°) (Fig. 2B).
Table 1.
Structure statistics for P2ab
| NOE distance and dihedral constraints | |
|---|---|
| Distance restraints | |
| Total NOE | 785 |
| Intraresidue | 335 |
| Interresidue | 450 |
| Hydrogen bond | 73 |
| Total dihedral angle restraints | 230 |
Total RDCs (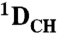 and and 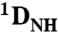 ) ) |
76 |
| Violations | |
| Distance constraints, Å | 0.018 ± 0.003 |
| Dihedral angle constraints, ° | 0.121 ± 0.016 |
| Dipolar couplings, Hz | 0.222 ± 0.026 |
| Deviation from idealized geometry | |
| Bond lengths, Å | 0.005 ± 0.0003 |
| Bond angles, ° | 1.064 ± 0.011 |
| Impropers, ° | 0.484 ± 0.018 |
| Average rmsd (Å) from the mean | |
| all heavy atoms | 1.14 ± 0.26 |
| all heavy atoms without bulge (G8–C12) | 0.86 ± 0.20 |
| No. of NOE violations > 0.2, Å | 0.75 ± 0.85 |
| No. of NOE violations > 0.5, Å | 0 |
| No. of dihedral violations > 5, ° | 0 |
| No. of RDC violations > 2, Hz | 0 |
J2a/b Bulge Serves as a Highly Flexible Hinge for Interhelical Motions.
To investigate the range of motion between P2a and P2b induced by the J2a/b bulge, we have analyzed NMR spin relaxation rates in addition to RDCs. For these studies, we applied a domain elongation strategy to P2ab, where P2a was elongated using a stretch of unlabeled A-U or G-C base pairs with otherwise 13C, 15N-labeled G/C or A/U nucleotides, respectively (E-P2ab) (Fig. 3A) (34). NMR spectra of the E-P2ab RNAs and nonelongated P2ab show excellent agreement between chemical shifts in common, indicating that the elongation does not affect the structure of P2ab (SI Appendix, Fig. S1). In contrast, the nonexchangeable resonance intensities from nonconstant time 1H-13C heteronuclear single quantum coherence (HSQC) spectra of the E-P2ab constructs differ from P2ab and reveal the existence of complex dynamics within this RNA. The elongated P2a helix within E-P2ab was designed to dominate the overall molecular tumbling, and as expected displays uniformly low peak intensities, providing an internal dynamics reference (34). Peak intensities from P2b within E-P2ab are in general higher than the reference P2a, consistent with the order tensor analysis of the RDCs that indicated that P2b is moving collectively relative to P2a (see SI Appendix). In particular, J2a/b nucleotides have significantly higher peak intensities, indicating a highly flexible interface between P2a and P2b. Among the J2a/b residues, U86 exhibits the most unrestricted motions and G84 is the least dynamic, consistent with its stacking on top of P2a.
Fig. 3.

Dynamical characterization of P2ab. (A) Normalized resonance intensities of the elongated P2ab (E-P2ab) from nonconstant-time 2D 1H-13C HSQC experiments. Sequence and secondary structure of E-P2ab is shown in the inset. The horizontal dashed line represents the value of 0.1. (B) Quantitative characterization of P2ab dynamics by Modelfree analysis of 15N spin relaxation data. Motional amplitudes (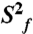 ) at the picosecond to subnanosecond timescale in P2a and P2b are shown in gold and dark red, respectively. Motional amplitudes (
) at the picosecond to subnanosecond timescale in P2a and P2b are shown in gold and dark red, respectively. Motional amplitudes (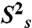 ) at the nanosecond timescale in P2b are shown in red. Time constants (t) of motions in P2a and P2b are shown in gold and red. Numerical values are given in SI Appendix, Table S3. The horizontal dashed line represents the amplitude (
) at the nanosecond timescale in P2b are shown in red. Time constants (t) of motions in P2a and P2b are shown in gold and red. Numerical values are given in SI Appendix, Table S3. The horizontal dashed line represents the amplitude (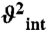 ) of interhelical motion from order tensor analysis of RDCs.
) of interhelical motion from order tensor analysis of RDCs.
Quantitative characterization of the internal motions in P2ab on the pico- to nanosecond timescale was obtained by model-free analysis of 15N spin relaxation rates, _R_1, _R_2, and {1H}-15N heteronuclear NOEs, measured for all observable imino resonances in E-P2ab (SI Appendix, Table S2) (34–37). Two layers of motions were observed that are faster than the overall molecular tumbling rate (21.7 ± 1.6 ns): First, N-H bonds fluctuate locally across the entire E-P2ab on the picosecond timescale (τ e = 0.13 ± 0.05 ns) with amplitudes 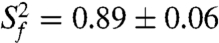 (
(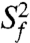 varies between zero and one for maximum and minimum motions); and second, P2b moves collectively relative to P2a at a slower nanosecond timescale (τ s = 1.83 ± 0.19 ns) with larger amplitude motions (
varies between zero and one for maximum and minimum motions); and second, P2b moves collectively relative to P2a at a slower nanosecond timescale (τ s = 1.83 ± 0.19 ns) with larger amplitude motions ( ) (Fig. 3B and SI Appendix, Table S3). Although the spin relaxation provides dynamical information on the timescale of pico- to nanoseconds (38), RDCs have a wider sensitivity for dynamics up to micro- to millisecond timescales (33, 39, 40). The order tensor analysis of RDCs measured from the individual domains showed an even larger amplitude of interhelical motions, as characterized by _ϑ_int = 0.69 ± 0.04, where _ϑ_int = _ϑ_P2a/_ϑ_P2b varies between zero and one for maximum and minimum motions (33). Assuming a cone motional model, these values correspond to P2a moving relative to P2b with an amplitude of ∼39°. Given that J2a/b is a 5-nt bulge, it is surprising that the motional freedom between P2a and P2b is so limited. As a comparison, the 3-nt bulge transactivation response element (TAR) RNA from HIV-1, whose dynamics have been systematically characterized, displays a much larger motional amplitude as characterized by _ϑ_int = 0.45 ± 0.05 (41). Because the interhelical freedom encoded in a 5-nt bulge would in general be expected to be significantly larger than that in a 3-nt bulge (42), this inherently restricted motional amplitude is surprising and suggests that both the bend and the allowed range of motions of P2a relative to P2b across the J2a/b bulge might be important for telomerase activity.
) (Fig. 3B and SI Appendix, Table S3). Although the spin relaxation provides dynamical information on the timescale of pico- to nanoseconds (38), RDCs have a wider sensitivity for dynamics up to micro- to millisecond timescales (33, 39, 40). The order tensor analysis of RDCs measured from the individual domains showed an even larger amplitude of interhelical motions, as characterized by _ϑ_int = 0.69 ± 0.04, where _ϑ_int = _ϑ_P2a/_ϑ_P2b varies between zero and one for maximum and minimum motions (33). Assuming a cone motional model, these values correspond to P2a moving relative to P2b with an amplitude of ∼39°. Given that J2a/b is a 5-nt bulge, it is surprising that the motional freedom between P2a and P2b is so limited. As a comparison, the 3-nt bulge transactivation response element (TAR) RNA from HIV-1, whose dynamics have been systematically characterized, displays a much larger motional amplitude as characterized by _ϑ_int = 0.45 ± 0.05 (41). Because the interhelical freedom encoded in a 5-nt bulge would in general be expected to be significantly larger than that in a 3-nt bulge (42), this inherently restricted motional amplitude is surprising and suggests that both the bend and the allowed range of motions of P2a relative to P2b across the J2a/b bulge might be important for telomerase activity.
Directional Bend and Flexibility of J2a/b Is Important for Telomerase Activity.
In order to test the importance of the J2a/b bulge structure and dynamics in telomerase catalytic activation, we systematically changed the length, strand location, and sequence of J2a/b (Fig. 4A). The effects of J2a/b on telomerase activity (nucleotide addition) and processivity (telomere DNA repeat addition) were analyzed by direct telomerase activity assays (28, 43), in which active telomerase was reconstituted in vitro in rabbit reticulocyte lysate (RRL) using human TERT (hTERT) and in vitro transcribed full-length hTR with various substitutions in the J2a/b region. We successively shortened the length of the J2a/b by deleting nucleotides symmetrically from the center of the bulge. Deletion of the central nucleotide in J2a/b, residue U86, reduced the telomerase activity by ∼40% (Fig. 4). Further deletion of bulge nucleotides decreased the activity accordingly, to less than 20% (greater than a fivefold decrease) when the bulge was deleted entirely. The processivity of the in vitro reconstituted telomerase also gradually decreased as the length of the bulge became shorter (Fig. 4). To test if the orientation as well as the size of the bulge loop is important for telomerase activity, the bulge nucleotides were moved to the opposite strand. This substitution had the most dramatic effect on telomerase activity, reducing it to ∼10% of WT with a decrease in processivity comparable to deleting the bulge completely (Fig. 4). Taken together with the structure of P2ab, these results support the hypothesis that the directional bend induced by J2a/b is important for properly orienting the TR for optimal catalytic activation.
Fig. 4.

The J2a/b loop is important for human telomerase activity. (A) The sequence and position of J2a/b loop substitutions. (B) Direct telomerase activity assay of in vitro assembled human telomerase from hTERT and full-length hTR with J2a/b loop substitutions. Numbers of telomeric repeats synthesized during the enzymatic reaction are shown on the right. LC is a 15 nt loading control. Lane C is the control where no hTR was added during in vitro reconstitution of human telomerase. (C) Relative activity of each mutant to that of WT hTR. The error bars are the standard deviation calculated from 2–3 independent reactions. (D) The effects of each J2a/b substitution on the processivity of in vitro reconstituted human telomerase. The processivity was determined as R_1/2 = 2 ln 2/(2.303_k), where k is the slope.
To investigate whether the inherent dynamics encoded in J2a/b are important for telomerase activity, we made additional nucleotide substitutions that would primarily affect the internal dynamics while minimizing effects on the structure, and we tested their effect on telomerase activity (Fig. 4). The entire J2a/b bulge was substituted with five cytidines (J2ab-C5) or an additional two cytidines were inserted on either side of U86 (J2ab-C + 2). J2ab-C5 was expected to have similar dynamics to J2ab-WT, whereas the additional two residues in J2ab-C + 2 were expected to increase the interhelical freedom between P2a and P2b. J2ab-C5 has only a small decrease in activity and small increase in processivity, whereas telomerase with the J2ab-C + 2 substitution is both more active and more processive than the wild type. These results support the hypothesis that the overall bend and inherent flexibility encoded in J2a/b are important for telomerase activity and processivity.
Although the J2a/b sequence is not highly conserved among 42 vertebrate TRs, the consensus sequence for the 5-nt bulge is G83Y78Y87Y96Y87, where Y is pyrimidine and the subscripts are percent out of 23 mammalian TRs (15). To investigate the effect of the 5-nt bulge sequence on activity, we made an additional substitution in which all five bulge nucleotides were G. A previous study had shown that flipping the sequence of J2a/b 5′ to 3′ decreased activity slightly (12), similar to our J2ab-C5 substitution. J2ab-G5 decreased the activity even more, to ∼1/3 of WT, but surprisingly the processivity was about the same as WT and J2ab-C5. We therefore acquired 1H-13C HSQC and RDC data on a P2ab-G5 construct, where J2a/b in P2ab was replaced with five Gs in order to investigate its dynamics. 1H-13C HSQC spectra of P2ab-G5 were almost identical to P2ab (for P2a and P2b), indicating that the bulge substitution did not affect the helical structure, as expected. Consistent with this result, analysis of measured RDCs (30 on P2a, 28 on P2b) gave perfect fits to the individual stems. The RDC analysis showed that P2ab-G5 has an interhelical angle of 103 ± 4°, about 14° larger than P2ab, and approximately the same twist (4 ± 15°). Contrary to our expectation that the G5 substitution would decrease the flexibility of J2a/b due to the tendency of purines to stack on one another, P2ab-G5 has the same global dynamics as P2ab (SI Appendix, Table S5). Therefore the results on P2ab-G5 are consistent with flexibility induced by the J2a/b bulge being important for facilitating template translocation, and therefore processivity. The G5 substitution significantly changed the purine content of J2a/b. The decrease in telomerase activity for P2ab-G5 may be due to a requirement for pyrimidines in the loop for proper folding in the context of TERT association. Overall, the results of the activity assays show that interhelical flexibility in P2ab induced by J2a/b is correlated with processivity, and that J2a/b bending in a defined direction is required for activity. The consensus sequence of J2a/b may therefore have evolved for both these functions. We hypothesize that the bend is important for correctly positioning the template in the active site of TERT, whereas the flexibility of P2a relative to P2b is important for releasing steric strain generated by processive addition of nucleotides on the RNA template for each telomere repeat.
Mammalian-Specific P2a.1-J2a.1 Does Not Induce an Additional Bend in the Pseudoknot.
P2a.1 is connected to P2a by an asymmetric J2a.1 internal loop that can base pair to form an irregular helix with some bulge bases (Fig. 1). NMR spectra of a P2a.1-J2a.1-P2a construct (P2a1a) show resonance line broadening and missing peaks throughout J2a.1, indicative of conformational exchange among alternative base pairs (SI Appendix, Fig. S3). We therefore developed a method to determine the structure of P2a1a, called RDC-MC-Sym, which uses a combination of NMR RDCs and computational modeling with MC-Sym (44). An initial set of P2a1a structures were generated using MC-Sym, and were used to extract initial sets of distance restraints for J2a.1 and flanking base pairs. These were refined, using RDCs measured for P2a.1 and P2a and ideal A-form distances for these helices, following standard NIH-XPLOR protocols (see details in the SI Appendix). The RDC-MC-Sym structure calculations produce different clusters of similar structures, which all showed that P2a.1 is essentially a linear extension of P2a. In the lowest-energy structure cluster, there is a negligible (6 ± 3°) bend and a twist of 135 ± 14° between the two helices across J2a.1. This amount of twisting would correspond to about four base pairs, consistent with J2a.1 forming an irregular helix. Other structure clusters display similar bends (4 ± 1°–10 ± 5°) and twists (110 ± 17°–132 ± 15°) (SI Appendix, Fig. S4). The J2a.1 region has observable structural variations between clusters, consistent with our NMR observation of conformational exchange in this region (SI Appendix, Fig. S4). Despite the conformational flexibility within J2a.1, there are no significant motions between P2a.1 and P2a, based on the high value of _ϑ_int = _ϑ_P2a.1/_ϑ_P2a = 0.85 ± 0.03 obtained from order tensor analysis of the RDCs. Thus, in contrast to the bulge loop J2a/b, which induces a large bend between P2ab and significant dynamics between P2a and P2b, the asymmetric internal loop J2a.1 does not induce a bend or significant dynamics between P2a.1 and P2a.
The structure and dynamics results on P2a1a are consistent with phylogeny, biochemical results, and disease mutations in this region of the pseudoknot. Nucleotide substitutions (12, 28) and the C72G mutation associated with aplastic anemia (45) that disrupt base pairs flanking J2a.1 decrease telomerase activity, consistent with the expectation that they would increase the size of J2a.1 and therefore increase flexibility and possible bending between P2a.1 and P2a. P2a.1-J2a.1 is not present in nonmammalian vertebrates, which instead have a single long P2a helix. Consistent with the absence of an internal loop in nonmammalian vertebrates, J2a.1 does not induce additional flexibility or bending in this region of the hTR pseudoknot.
Structure of the Full-length Pseudoknot in the Core Domain.
P2ab is located in the center of the hTR core domain, flanked by the catalytically essential P2b-P3 pseudoknot and P2a.1-J2a.1. Based on the results on P2ab structure, dynamics, and importance of J2a/b for activity reported here, we hypothesized that P2ab may act as the dominant structural element in defining the overall topology of the hTR core by directing the relative orientation between the other two helical regions. To address this hypothesis in more detail, we modeled the structure of the full-length P2/P3 pseudoknot containing all nucleotides except for the nonconserved single strand region of J2a/3. With the structures of P2a1a, P2ab, and P2b-P3 pseudoknot (24) in hand, we applied a domain approach to obtain a model structure of the entire helical region of the core domain by linking the overlapping ends of P2a.1a, P2ab, and P2b-P3 and refining them as a single entity using the AMBER force field together with short-range NOE restraints from the individual constructs (see details in SI Appendix). The ensemble of the 20 lowest-energy structures is well determined with an rmsd to the mean of 1.13 ± 0.22 Å for all heavy atoms (Fig. 5A). We note that the AMBER refinement maintained the major structural features from the individual structural elements, such as bending, twisting, and the triple helix in P2b-P3.
Fig. 5.

Model structure of the hTR core domain. (A) Superposition of the 20 lowest-energy structures over all heavy atoms of the AMBER refined hTR P2/P3 pseudoknot (model includes all residues except J2a/3 nucleotides 148–168). (B) Model representation of the hTR core domain in complex with 10-nt telomeric DNA. The lowest-energy structure of P2/P3 pseudoknot is shown together with a 10 base-pair hybrid A-form hTR template-primer DNA helix. Putative positions of the template-adjacent single strand regions and J2a/3 nucleotides 148–168 are shown as gray lines, and the P1 helix as dark green lines. The template RNA is shown in purple and the complementary telomeric DNA is shown in cyan. All other regions are colored as in Fig. 1. (C) Comparison of the structure of the 5-nt bulge found in P2ab (Left) and the HCV IRES domain IIa (Right; PDB ID code 1P5P). (D) Schematic representation of the core domain showing the proposed change in the J2a/b bend during the telomere addition cycle. Helical regions of the core domain are shown as cylinders, the template and adjacent single strand are shown as purple and gray lines, respectively, the telomere DNA is shown as cyan line, and TERT is shown as a gray ellipse with the active site depicted as a yellow star.
The model structure of full-length (P2/P3) pseudoknot revealed that the overall topology of the core domain is defined by the large bend across J2a/b (88 ± 3° in the P2/P3 structures). Stacking interactions of the terminal bulge residues play a critical role in maintaining the interhelical bend (Fig. 5). The 3′ end of the J2a/3 loop that is present in the P2b/P3 pseudoknot lies in the minor groove, and the remainder of J2a/3 would be expected to continue across the J2a/b bulge loop and along the minor groove to connect with U147 at the end of P2a.1 and would be on the outer surface of the core domain (Fig. 5B). The other strand of P2a.1 connects to the single strand region containing the 10 nt template and adjacent nucleotides (6 + 8). These 24 nt need to span the distance between the 5′ end of P2a.1 and 3′ end of the pseudoknot. As both the P2b/P3 pseudoknot and P2a1a are ∼50 Å long, the J2a/b bend creates a ∼70 Å end-to-end distance between P2b/P3 and P2a.1 (Fig. 5A). Assuming full base pairing between the template and telomere DNA at the end of one cycle of nucleotide addition, the 10 bp template would span ∼26 Å (in both length and diameter) and the template adjacent nucleotides could span a maximum of ∼83 Å. Thus, the 24-nt single strand containing the template region can span the distance between the P2b/P3 pseudoknot and P2a.1 domains without significant stretching. Without the J2a/b-induced bend in the full-length pseudoknot, the end-to-end distance would be too long to satisfy the steric requirements for template placement on TERT during telomere addition and translocation.
Discussion
J2a/b Conformation Determines the Overall Helical Structure of the hTR Core Domain.
The core domain of human telomerase is essential for telomerase catalytic activation. We have shown that J2a/b, a centrally located asymmetric bulge in the extended stem P2 of the pseudoknot, directs the overall shape of the core domain by introducing a large bend which brings the ends of P2 and P3 within 70 Å of each other, despite the low level of sequence conservation in vertebrate J2a/b. The other helical elements of the full-length pseudoknot, P2a.1 and the P2b/P3 pseudoknot, have no significant effect on either static or dynamic bending.
P2b/P3 forms an H-type pseudoknot in which J2b/3 interacts with the major groove of P3 to form a conserved U-A-U triple-helix (Fig. 5 A and B), which has been shown to be important for catalysis (23, 24). Based on the effect of 2′H and 2′O methyl ribose substitutions on activity, it has been proposed that A176 2′OH, in the last U-A-U triplet, contributes directly to catalysis (26). In our model structure of the full-length pseudoknot, A176 2′OH is on the inside surface of the bent RNA, facing toward where the template would be, and would thus be positioned to interact with TERT (Fig. 5B). The conserved single nucleotide bulge, U177, which also contributes to activity, is also facing in this direction (24). This region of the pseudoknot is more protected from chemical probing in vivo than the free TR is in vitro (46). An engineered _cis_-telomerase, in which a 6 nt DNA primer is linked to the 3′ end of the pseudoknot, has its maximal activity with a linker length of 5 nt or longer, which corresponds to a maximal distance of 33 Å between the end of the pseudoknot and the 5′ end of the TR template (47). This result is consistent with the close proximity of the end of the pseudoknot to the template seen in our model structure. The relative orientation of the helical ends of the P2/P3 pseudoknot in our model structure, as well as structural details of the internal loops, differ significantly from a low-resolution FRET model (6.5–8.0 Å) of the hTR core domain [Protein Data Bank (PDB) ID code 2INA] incorporating distances between fluorescently labeled peptide nucleic acids hybridized to single stranded regions of the hTR core domain (SI Appendix, Fig. S5) (29). Finally, we note that the 70 Å distance between the two ends of the P2/P3 pseudoknot corresponds approximately to the diameter of the crystal structure of the putative TERT from Tribolium castaneum (21, 22), so that the arc of the pseudoknot would fit nicely either parallel or perpendicular to the primer-template binding site (SI Appendix, Fig. S6).
Structurally Encoded Dynamics of J2a/b May Be Important for Activity.
The restricted interhelical motions fit very well with the template displacement during nucleotide addition and template translocation during telomere repeat synthesis. As both P2b/P3 pseudoknot and P2a1a have been shown to exhibit little interdomain motion, the overall helical motion within the entire helical region of the core domain must be dominated by the flexing of P2a and P2b across J2a/b. Our dynamical characterization showed that P2a is moving relative to P2b with an amplitude of ∼39° assuming a cone motional model. Applying this motional amplitude onto our model structure, the end-to-end distance between the two ends of the full-length pseudoknot can change by ∼28 Å. This displacement agrees well with the ∼17 Å translocation of the template during the synthesis of the telomeric DNA repeats. Although these motions are for the free TR, the inherent and limited flexibility of J2a/b lead us to hypothesize that these motions may be functionally relevant. Thus, during every cycle of telomeric DNA repeat synthesis, the interdomain motions within the pseudoknot/template core domain may be correlated with the movement of the template from the 3′ to 5′ end. Our data support the hypothesis that the J2a/b-induced bend is an important element in positioning the template into the active site of hTERT, and that the limited flexing of the bend facilitates movement of the template through the active site during processive nucleotide addition (Fig. 5D).
J2a/b Bulge Belongs to a Rare 5-nt Bulge Family.
A surprising result of the investigation into J2a/b dynamics was that the interhelical dynamics were much smaller than those of the previously well-studied 3-nt bulge TAR RNA from HIV-1 and the predicted range of dynamics of 4-nt loops (41, 42). We therefore searched for 5-nt bulges flanked by helices using RNA FRABASE (48). To our surprise, in the entire PDB there is only one other set of structures of a 5-nt bulge, which is found in domain II of hepatitis C virus (HCV) internal ribosome entry site (IRES). The 5-nt bulge in HCV IRES has a completely different sequence than J2a/b, but the structure is remarkably similar (Fig. 5C) (49). Five nucleotide bulges may thus represent a rare RNA structure motif. It has been proposed that the bend in domain II is a conserved structural feature of HCV-like IRESs that is structurally but not sequence encoded (50). Mutations within domain II that affect its overall shape also impair function. In telomerase, J2a/b is also not conserved in sequence but is conserved in location. We propose that J2a/b is a structurally encoded functional domain. Because telomerase is highly active in most cancer cell lines but not in somatic cells, it has attracted wide interest as a potential drug target. The unusual features of J2a/b make it an attractive target for small molecules that could bind the bulge and affect the bend angle and dynamics. Indeed, small molecule inhibitors of HCV that target the 5-nt bulge in HCV IRES have been developed (51, 52).
Materials and Methods
NMR Sample Preparation and Spectroscopy.
Unlabeled, uniformly 13C, 15N-labeled, and base-specifically (G, C, A, and U) 13C, 15N-labeled RNA samples (P2ab, P2ab-G5, E-AU-P2ab, E-GC-P2ab, and P2a1a) were prepared by in vitro transcription using T7 polymerase (53). NMR experiments were carried out on Bruker DRX 500, 600, and Avance 800 MHz spectrometers equipped with 5 mm triple-resonance cryogenic probes or QXI (HCP) probe (500 MHz) at 283 K for exchangeable and at 293 K for nonexchangeable proton spectra. NMR data for assignments and structure determination were acquired and analyzed as described (53, 54). One-bond C-H and N-H RDCs were measured on uniformly 13C, 15N-labeled P2ab, P2ab-G5, and P2a.1 samples in ∼15 mg/mL Pf1 phage (ASLA Biotech, Ltd.) on 800 MHz spectrometer using 2D 1H-13C S3CT-HSQC and standard 1H-15N HSQC experiments (41) (SI Appendix, Tables S1 and S4). NMR dynamic characterization of P2ab were measured on E-AU-P2ab and E-GC-P2ab sample at 293 K on 600 MHz spectrometer (34). Spin relaxation data were analyzed using the extended model-free formalism (35, 55) as implemented in Modelfree (Version 4.16 for Linux) from Palmer and co-workers (37) (SI Appendix, Table S3). The lowest-energy P2ab structure where the P2a helix was extended with 22 A-form base pairs was used as the input structure for the analysis. Detailed descriptions of sample preparation, NMR experiments and assignment methodology, and analysis of spin relaxation data are given in the SI Appendix.
Structure Calculations and Modeling.
The structure of P2ab was calculated using standard NIH-XPLOR protocols (56) as described previously (24, 57). For refinement with RDCs, all 76 C-H and N-H RDCs were normalized to C-H bond with bond length of 1.0 Å. The optimal values for the magnitude and asymmetry of the alignment tensor are D a = -20.7 Hz and -29.8 Hz and R = 0.19 and 0.25, for P2a and P2b domain, respectively. The force constants for RDCs were gradually increased from 0.001 to 1.0 and 0.5 kcal·mol-1·Hz-2 for P2a and P2b to account for the difference in the D a values. For modeling of P2a1a, we developed a method using a combination of NMR RDCs and computational modeling with MC-Sym (44) (RDC-MC-Sym approach). The MC-Sym program was used to generate structure models to provide short-range distance restraints for the J2a.1 and neighboring residues, and NMR RDCs were used to provide long-range angular restraints for the interhelical orientation between P2a.1 and P2a. These complimentary angular and distance restraints, together with ideal A-form restraints for P2a.1 and P2a helices, were combined for structure calculations of P2a1a using NIH-XPLOR following standard protocols. Detailed procedures of the RDC-MC-Sym calculations of P2a1a are described in the SI Appendix. A model of the P2/P3 pseudoknot was generated by computationally linking the overlapping ends of P2a1a, P2ab, and P2b-P3 pseudoknot (24) structures (SI Appendix, Fig. S7) and refining with AMBER software (58) as described (59) and detailed in the SI Appendix, using the ff99 force field (60). The 20 lowest-energy structures (ranked by total Amber energy) are reported (Fig. 5A). The restraints and structure statistics for these 20 structures are summarized in SI Appendix, Table S6. The coordinates of the P2a1a and P2/P3 model structures will be available upon request to the authors. For all structural analysis, the interhelical bend and twist angles and errors were computed by using EularRNA (61) as described previously (41).
In Vitro Reconstitution of Telomerase and Direct Telomerase Activity Assays.
Full-length WT and mutant hTRs were prepared as previously described (23). Telomerase was reconstituted in RRL using the TnT quick coupled transcription/translation system (Promega) as described (28, 43, 62) (see details in the SI Appendix). Telomerase activity was characterized as the total intensity of the extended DNA products after correcting the background and normalizing against the loading control. The activities of telomerases with mutant hTRs are presented as relative activity to the WT. In order to quantify the telomerase processivity, the intensities of each telomere repeat were measured, corrected for the number of dGTP incorporated per repeat, and normalized to the second telomere repeat whose intensity was set to be 100. The normalized intensities were then plotted in logarithmic scale against the telomere repeat number. The processivity was determined as R_1/2 = 2 ln 2/(2.303_k), where k is the slope (28, 63).
Supplementary Material
Supporting Information
Acknowledgments.
The authors thank Professor Julian J-L Chen and his laboratory for training on how to perform direct telomerase activity assays. This work was supported by National Institutes of Health Grant GM048123 and National Science Foundation MCB51770 (to J.F.). Q.Z. is a Baltimore Family Fellow of the Life Sciences Research Foundation.
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2009.
The authors declare no conflict of interest.
Data deposition: The chemical shifts, coordinates, and restraints for the 20 lowest-energy structures of P2ab have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2L3E).
References
- 1.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 2.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 3.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–279. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12:1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- 5.Blasco MA. Telomeres and human disease: Ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 6.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 7.Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006;7:484–494. doi: 10.1038/nrm1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–514. doi: 10.1016/s0092-8674(00)80687-x. [DOI] [PubMed] [Google Scholar]
- 9.Theimer CA, Feigon J. Structure and function of telomerase RNA. Curr Opin Struct Biol. 2006;16:307–318. doi: 10.1016/j.sbi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Collins K. Physiological assembly and activity of human telomerase complexes. Mech Ageing Dev. 2008;129:91–98. doi: 10.1016/j.mad.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armanios M. Syndromes of telomere shortening. Annu Rev Genom Hum G. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ly H, Blackburn EH, Parslow TG. Comprehensive structure-function analysis of the core domain of human telomerase RNA. Mol Cell Biol. 2003;23:6849–6856. doi: 10.1128/MCB.23.19.6849-6856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Autexier C, Pruzan R, Funk WD, Greider CW. Reconstitution of human telomerase activity and identification of a minimal functional region of the human telomerase RNA. EMBO J. 1996;15:5928–5935. [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell JR, Collins K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol Cell. 2000;6:361–371. doi: 10.1016/s1097-2765(00)00036-8. [DOI] [PubMed] [Google Scholar]
- 15.Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ. The telomerase database. Nucleic Acids Res. 2008;36:D339–343. doi: 10.1093/nar/gkm700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, et al. A universal telomerase RNA core structure includes structured motifs required for binding the telomerase reverse transcriptase protein. Proc Natl Acad Sci USA. 2004;101:14713–14718. doi: 10.1073/pnas.0405879101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JL, Greider CW. An emerging consensus for telomerase RNA structure. Proc Natl Acad Sci USA. 2004;101:14683–14684. doi: 10.1073/pnas.0406204101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JL, Opperman KK, Greider CW. A critical stem-loop structure in the CR4-CR5 domain of mammalian telomerase RNA. Nucleic Acids Res. 2002;30:592–597. doi: 10.1093/nar/30.2.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu Rev Biochem. 2006;75:493–517. doi: 10.1146/annurev.biochem.75.103004.142412. [DOI] [PubMed] [Google Scholar]
- 20.Robart AR, Collins K. Investigation of human telomerase holoenzyme assembly, activity, and processivity using disease-linked subunit variants. J Biol Chem. 2010;285:4375–4386. doi: 10.1074/jbc.M109.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008;455:633–637. doi: 10.1038/nature07283. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010;17:513–518. doi: 10.1038/nsmb.1777. [DOI] [PubMed] [Google Scholar]
- 23.Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17:671–682. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Kim NK, et al. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J Mol Biol. 2008;384:1249–1261. doi: 10.1016/j.jmb.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shefer K, et al. A triple helix within a pseudoknot is a conserved and essential element of telomerase RNA. Mol Cell Biol. 2007;27:2130–2143. doi: 10.1128/MCB.01826-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiao F, Cech TR. Triple-helix structure in telomerase RNA contributes to catalysis. Nat Struct Mol Biol. 2008;15:634–640. doi: 10.1038/nsmb.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie M, et al. Structure and function of the smallest vertebrate telomerase RNA from teleost fish. J Biol Chem. 2008;283:2049–2059. doi: 10.1074/jbc.M708032200. [DOI] [PubMed] [Google Scholar]
- 28.Chen JL, Greider CW. Determinants in mammalian telomerase RNA that mediate enzyme processivity and cross-species incompatibility. EMBO J. 2003;22:304–314. doi: 10.1093/emboj/cdg024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gavory G, Symmons MF, Krishnan Ghosh Y, Klenerman D, Balasubramanian S. Structural analysis of the catalytic core of human telomerase RNA by FRET and molecular modeling. Biochemistry. 2006;45:13304–13311. doi: 10.1021/bi061150a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comolli LR, Smirnov I, Xu L, Blackburn EH, James TL. A molecular switch underlies a human telomerase disease. Proc Natl Acad Sci USA. 2002;99:16998–17003. doi: 10.1073/pnas.262663599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theimer CA, Finger LD, Trantirek L, Feigon J. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc Natl Acad Sci USA. 2003;100:449–454. doi: 10.1073/pnas.242720799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JL, Greider CW. Functional analysis of the pseudoknot structure in human telomerase RNA. Proc Natl Acad Sci USA. 2005;102:8080–8085. doi: 10.1073/pnas.0502259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prestegard JH, Al-Hashimi HM, Tolman JR. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 35.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 36.Clore GM, et al. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 37.Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 38.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 39.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 40.Getz M, Sun X, Casiano-Negroni A, Zhang Q, Al-Hashimi HM. NMR studies of RNA dynamics and structural plasticity using NMR residual dipolar couplings. Biopolymers. 2007;86:384–402. doi: 10.1002/bip.20765. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–1267. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 42.Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science. 2010;327:202–206. doi: 10.1126/science.1181085. [DOI] [PubMed] [Google Scholar]
- 43.Huard S, Moriarty TJ, Autexier C. The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic Acids Res. 2003;31:4059–4070. doi: 10.1093/nar/gkg437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 45.Chen JL, Greider CW. Telomerase RNA structure and function: Implications for dyskeratosis congenita. Trends Biochem Sci. 2004;29:183–192. doi: 10.1016/j.tibs.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Antal M, Boros E, Solymosy F, Kiss T. Analysis of the structure of human telomerase RNA in vivo. Nucleic Acids Res. 2002;30:912–920. doi: 10.1093/nar/30.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiao F, Goodrich KJ, Cech TR. Engineering cis-telomerase RNAs that add telomeric repeats to themselves. Proc Natl Acad Sci USA. 2010;107:4914–4918. doi: 10.1073/pnas.0909366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popenda M, et al. RNA FRABASE 2.0: An advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinformatics. 2010;11:231. doi: 10.1186/1471-2105-11-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Structure of HCV IRES domain II determined by NMR. Nat Struct Biol. 2003;10:1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 50.Locker N, Easton LE, Lukavsky PJ. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007;26:795–805. doi: 10.1038/sj.emboj.7601549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parsons J, et al. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat Chem Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paulsen RB, et al. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc Natl Acad Sci USA. 2010;107:7263–7268. doi: 10.1073/pnas.0911896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dieckmann T, Feigon J. Assignment methodology for larger RNA oligonucleotides: Application to an ATP-binding RNA aptamer. J Biomol NMR. 1997;9:259–272. doi: 10.1023/a:1018622708674. [DOI] [PubMed] [Google Scholar]
- 54.Peterson RD, Theimer CA, Wu H, Feigon J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J Biomol NMR. 2004;28:59–67. doi: 10.1023/B:JNMR.0000012861.95939.05. [DOI] [PubMed] [Google Scholar]
- 55.Clore GM, et al. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 56.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 57.Wu H, et al. A novel family of RNA tetraloop structure forms the recognition site for Saccharomyces cerevisiae RNase III. EMBO J. 2001;20:7240–7249. doi: 10.1093/emboj/20.24.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Case DA, et al. AMBER 7. San Francisco: University of California; 2002. [Google Scholar]
- 59.Stefl R, Wu H, Ravindranathan S, Sklenar V, Feigon J. DNA A-tract bending in three dimensions: Solving the dA4T4 vs. dT4A4 conundrum. Proc Natl Acad Sci USA. 2004;101:1177–1182. doi: 10.1073/pnas.0308143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 61.Bailor MH, et al. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat Protoc. 2007;2:1536–1546. doi: 10.1038/nprot.2007.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim NK, Theimer CA, Mitchell JR, Collins K, Feigon J. Effect of pseudouridylation on the structure and activity of the catalytically essential P6.1 hairpin in human telomerase RNA. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq525. 10.1093/nar/gkq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang F, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information