Novel CSF biomarkers for frontotemporal lobar degenerations[image] (original) (raw)
Abstract
Objective:
To identify antemortem CSF diagnostic biomarkers that can potentially distinguish between the 2 main causes of frontotemporal lobar degeneration (FTLD), i.e., FTLD with TDP-43 pathology (FTLD-TDP) and FTLD with tau pathology (FTLD-tau).
Methods:
CSF samples were collected antemortem from 23 patients with FTLD with known pathology to form a autopsy cohort as part of a comparative biomarker study that additionally included 33 living cognitively normal subjects and 66 patients with autopsy-confirmed Alzheimer disease (AD). CSF samples were also collected from 80 living patients clinically diagnosed with frontotemporal dementia (FTD). Levels of 151 novel analytes were measured via a targeted multiplex panel enriched in neuropeptides, cytokines, and growth factors, along with levels of CSF biomarkers for AD.
Results:
CSF levels of multiple analytes differed between FTLD-TDP and FTLD-tau, including Fas, neuropeptides (agouti-related peptide and adrenocorticotropic hormone), and chemokines (IL-23, IL-17). Classification by random forest analysis achieved high sensitivity for FTLD-TDP (86%) with modest specificity (78%) in the autopsy cohort. When the classification algorithm was applied to a living FTD cohort, semantic dementia was the phenotype with the highest predicted proportion of FTLD-TDP. When living patients with behavioral variant FTD were examined in detail, those predicted to have FTLD-TDP demonstrated neuropsychological differences vs those predicted to have FTLD-tau in a pattern consistent with previously reported trends in autopsy-confirmed cases.
Conclusions:
Clinical cases with FTLD-TDP and FTLD-tau pathology can be potentially identified antemortem by assaying levels of specific analytes that are well-known and readily measurable in CSF.
GLOSSARY
AD
= Alzheimer disease;
AgRP
= Aguti-related protein;
ANG-2
= angiopoietin-2;
ACTH
= adrenocorticotropic hormone;
ALS
= amyotrophic lateral sclerosis;
ApoB
= apolipoprotein B;
bv-FTD
= behavioral variant FTD;
CBS
= corticobasal syndrome;
FTD
= frontotemporal dementia;
FTLD
= frontotemporal lobar degeneration;
FTLD-tau
= frontotemporal lobar degeneration with tau pathology;
FTLD-TDP
= frontotemporal lobar degeneration with TDP-43 pathology;
IL
= interleukin;
MDC
= macrophage-derived chemokine;
PNFA
= progressive nonfluent aphasia;
PPA
= primary progressive aphasia;
PSP
= progressive supranuclear palsy;
S100b
= S100 calcium binding protein b;
SemD
= semantic dementia;
TRAIL-R3
= tumor necrosis factor-related apoptosis-inducing ligand receptor 3.
Frontotemporal lobar degeneration (FTLD) includes neurodegenerative disorders which lead to progressive behavioral or language abnormalities.1–3 There are 2 major forms of FTLD: FTLD-TDP with neuronal and glial inclusions immunoreactive to TAR DNA binding protein of ∼43 kD (TDP-43), and FTLD-tau containing fibrillar and hyperphosphorylated tau inclusions.2,3 The main FTLD lesions likely reflect distinct disease mechanisms,2 although antemortem diagnosis of the underlying pathology remains clinically challenging. Certain phenotypes and neuropsychological profiles have been proposed as predictors for FTLD subtypes,4–6 but differences at the group level are difficult to translate into pathologic prediction at the individual patient level. Patterns of atrophy may offer clues to the pathology, although structural changes often reflect phenotypes rather than pathology.7
Compared to clinical and imaging predictors, chemical biomarkers may better identify the underlying FTLD pathology. CSF levels of peptides related to Alzheimer disease (AD), including total tau, tau phosphorylated at threonine 181 (p-tau181), and Aβ1-42 (Aβ42), represent the most established biomarkers in neurodegenerative disease research.8 Altered levels of these peptides can help confirm AD in elderly patients with dementia,9 and identify patients with clinical FTLD features due to atypical presentations of pathologic AD.9,10 No definitive CSF biomarker yet exists to distinguish between FTLD-TDP or FTLD-tau, although some studies suggest that TDP-43 levels in plasma and CSF may predict FTLD-TDP.11,12 Recently, we performed a targeted proteomic search for novel CSF biomarkers for AD compared to other neurodegenerative disorders including autopsy-confirmed cases of FTLD-TDP and FTLD-tau.13 In this context, we hypothesize that certain CSF biomarkers exist to distinguish between FTLD-TDP and FTLD-tau.
METHODS
Standard protocol approvals, registrations, and patient consents.
All protocols were approved by the Institutional Review Board at the University of Pennsylvania. Written consent was obtained from all patients and their representatives.
Subjects.
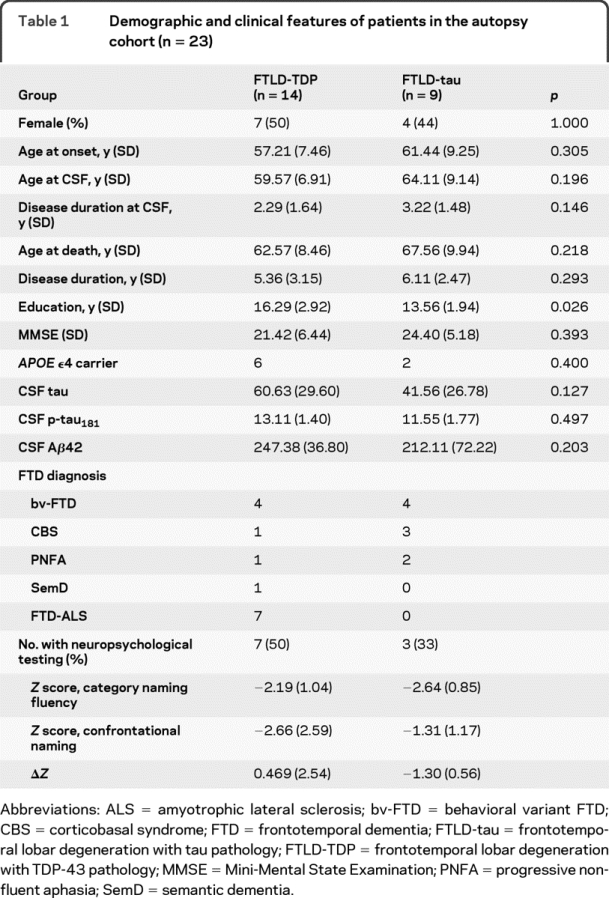
Patients and control subjects were recruited and longitudinally followed at the University of Pennsylvania in specialty services dedicated to the evaluation and management of neurodegenerative diseases. The autopsy cohort was previously described.14 Briefly, each patient in the autopsy cohort had undergone detailed cognitive, neurologic, neuroimaging, and laboratory examinations to ensure the accuracy of clinical diagnosis according to established criteria for AD,15 FTD,16 and amyotrophic lateral sclerosis (ALS).17 Autopsy-confirmed cases (n = 82) were characterized neuropathologically with detailed immunohistochemical analysis.18 Clinical history was reviewed in a blinded manner to confirm FTD syndromic diagnosis according to consensus criteria for behavioral variant FTD (bv-FTD),1,16 semantic dementia (SemD),16 progressive nonfluent aphasia (PNFA),16 and corticobasal syndrome (CBS).19 Seven patients with clinical FTD and ALS but no autopsy were added to the FTLD-TDP group, as these cases nearly always have TDP-43 pathology.20 APOE genotyping was performed for all subjects in the autopsy cohort. Compared to patients with FTLD-tau (n = 9), patients with FTLD-TDP (n = 14) had more years of education (p = 0.026). The 2 groups were otherwise similar in gender, disease duration to CSF, and cognitive performance measured by Mini-Mental State Examination (table 1).
Table 1 Demographic and clinical features of patients in the autopsy cohort (n = 23)
Patients clinically diagnosed with bv-FTD or primary progressive aphasia (PPA) without autopsy (n = 80) were also recruited to form the living cohort. A subset of these living patients (n = 65) had neuropsychological analysis for category naming fluency (n = 65) and confrontational naming (n = 56). These measures were selected because of their potential use in distinguishing patients with FTLD-tau from patients with FTLD-TDP, in that relatively better performance in category naming fluency than confrontational naming was suggestive of FTLD-TDP.6
Procedures.
Baseline CSF samples were obtained during routine diagnostic lumbar puncture as previously described.9 Briefly, lumbar puncture was performed with a 20- or 24-guage spinal needle, and CSF was transferred into polypropylene tubes. Aliquots (0.5 mL) were prepared, bar-coded, and then stored in polypropylene vials at −80°C. Samples were interrogated by Rules-Based Medicine (RBM), Inc. (Austin, TX) to assay levels of 151 analytes using the Human DiscoveryMAP™ panel and a Luminex 100 platform as described.13 Measures of CSF Aβ42, total tau, and p-tau181 were performed using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3, Ghent, Belgium) immunoassay kit-based reagents as described.9
Statistical analysis.
Statistical analysis was performed in SPSS 12.0 except for classification. Mann-Whitney U test was used to identify analytes that differed between autopsy-confirmed FTLD-TDP and FTLD-tau at the univariate level. Given the high dimensional data with a small sample size, random forests (http://www.stat.berkeley.edu/∼breiman/RandomForests/) was used for classification in the autopsy cohort. Analytes were entered with nodes optimized for best classification of FTLD-TDP vs FTLD-tau. Out-of-box error rate was used to derive diagnostic accuracy, with sensitivity and specificity derived from the confusion matrix. Receiver operating characteristics curves were used to derive cutoff values for each individual analyte. The established random forests structure was then used to classify each patient in the living cohort as likely to have FTLD-TDP or FTLD-tau.
For neuropsychological analysis, Z score was calculated for each neuropsychological subtest according to cognitively normal control subjects. According to previously observed patterns in subjects with autopsy-confirmed FTLD,6 a relative performance score was calculated between confrontational naming and category naming fluency (ΔZ = Z score of category naming fluency − Z score of confrontational naming). A positive relative performance score was taken as suggestive of predicted FTLD-TDP.
RESULTS
As in our previous study,13 not all analytes were sufficiently abundant to be reliably measured and here 106 of the 151 analytes in the MAP had measurable levels for analysis. To determine the best biomarkers of FTLD-TDP, we used 3 independent analytical strategies to identify MAP analytes associated with FTLD-TDP vs FTLD-tau.
Autopsy cohort.
Mann-Whitney U test identified 10 analytes that differ between FTLD-TDP and FTLD-tau (table 2, figure 1), including interleukin-17 (IL-17), interleukin-23 (IL-23), Eotaxin-3, adrenocorticotropic hormone (ACTH), Fas, angiopoietin-2 (ANG-2), apolipoprotein B (ApoB), macrophage-derived chemokine (MDC), S100 calcium binding protein b (S100b), and tumor necrosis factor-related apoptosis-inducing ligand receptor 3 (TRAIL-R3). As the number of analytes is significantly larger than the number of cases in the training set, we performed additional classification algorithms using random forest analysis to identify putative biomarkers for each FTLD subtype and to classify patients in the test set.
Table 2 Candidate biomarkers for FTLD-TDP according to Mann-Whitney U test and random forests analysis
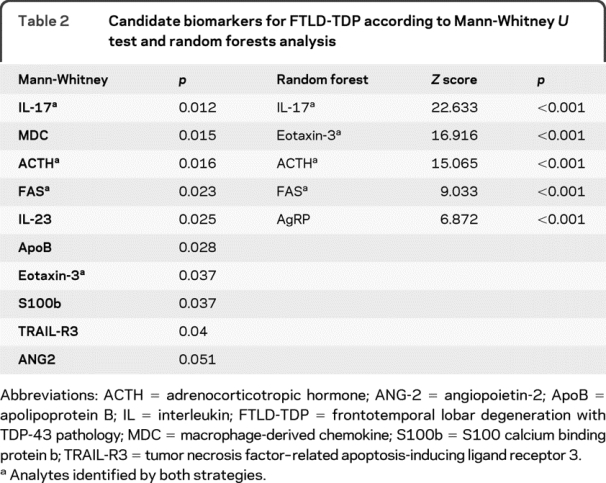

Figure 1 Box plots showing median values, quartiles, and outliers (circles) of novel CSF analytes useful in the distinction between frontotemporal lobar degeneration with TDP-43 pathology (gray) and frontotemporal lobar degeneration with tau pathology (white), along with total tau levels
Values shown are normalized to mean values of cognitively normal subjects. AgRP = Aguti-related protein; ANG-2 = angiopoietin-2; ACTH = adrenocorticotropic hormone; ApoB = apolipoprotein B; FTLD = frontotemporal lobar degeneration; IL = interleukin; MDC = macrophage-derived chemokine; S100b = S100 calcium binding protein b; TRAIL-R3 = tumor necrosis factor-related apoptosis-inducing ligand receptor 3.
Random forests analysis using demographic information (age at CSF collection, gender), CSF biomarkers for AD (levels of tau, p-tau181, and Aβ42), and 106 MAP analyte levels identified a short list of analytes that differentiated between FTLD-TDP and FTLD-tau through a tree-based classification algorithm. Optimal classification was achieved by using the top 5 analytes identified by random forests, including IL-17, Eotaxin-3, ACTH, Fas, and Aguti-related protein (AgRP) (table 2, figure 1). These biomarkers were associated with a diagnostic accuracy of 82.6%, with 85.7% sensitivity and 77.8% specificity for FTLD-TDP. To determine the relative performance of individual analyte alone, we derived cutoff values for each analyte using receiver operating characteristic curves: 0.1350 ng/mL for ACTH (sensitivity 71.4%, specificity 77.8%), 53.0 for AgRP pg/mL (sensitivity 57.1%, specificity 88.9%), 52.5 pg/mL for Eotaxin-3 (sensitivity 78.6%, specificity 88.9%), 0.455 for FAS ng/mL (sensitivity 64.3%, specificity 77.8%), and 9.25 pg/mL for IL-17 (14.3% sensitivity, 77.8% specificity).
While some biomarkers identified by Mann-Whitney U test contributed little to the distinction of FTLD-TDP from FTLD-tau by random forests, we made the observation that FTLD-TDP and FTLD-tau cases differed in both IL-23 and IL-17 levels. This was suggestive of an altered IL-23 pathway, as IL-23 induces the differentiation of naïve T-cells into IL-17-releasing helper T cells.21 We thus additionally analyzed levels of IL-23 and IL-17 in FTLD-TDP and FTLD-tau cases to determine the direction and magnitude of these changes (figure 2). When levels of IL-23 and IL-17 were normalized to average levels in cognitively healthy control subjects (n = 33) previously reported,14 patients with FTLD-TDP (p = 0.074) and FTLD-tau (p = 0.069) both had a relative increase in IL-23 levels compared to IL-17. With this increase in IL-23, FTLD-tau cases were found to have similar levels of IL-17 as control subjects, while FTLD-TDP had lower IL-17 levels than control subjects (p = 0.002).
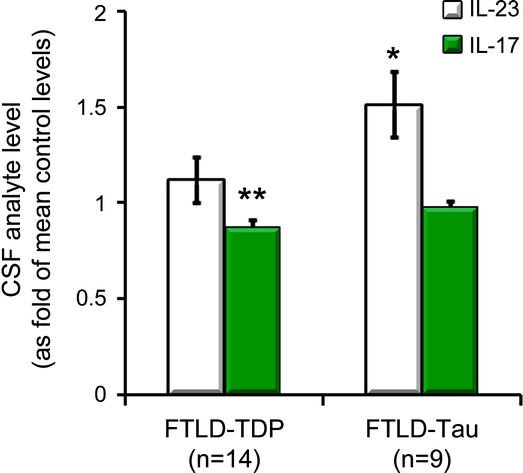
Figure 2 Relative levels of CSF chemokines involved in the IL-23/IL-17 axis in control subjects, and patients with FTLD-TDP and FTLD-tau (±SEM)
*p < 0.005 by Mann-Whitney U test compared to control subjects and FTLD-TDP (FTLD-tau); **p < 0.02 compared to control subjects and FTLD-tau. FTLD-tau = frontotemporal lobar degeneration with tau pathology; FTLD-TDP = frontotemporal lobar degeneration with TDP-43 pathology; IL = interleukin.
Living cohort.
Finally, we sought to determine the likelihood of FTLD-TDP vs FTLD-tau as the underlying pathology in an independent living cohort consisting of 80 patients with bv-FTD or PPA (table 3, table e-1 on the _Neurology_® Web site at www.neurology.org). The most common syndromic diagnosis was bv-FTD (n = 37, 46%), with PPA (n = 22) and CBS (n = 21) similar in number. Among these 80 patients, 53 were predicted by random forests to have FTLD-TDP, including 26 with bv-FTD (70.2%), 12 with CBS (57.1%), 5 with PNFA (50%), and 10 with SemD (83.3%). By comparison, among 23 patients with neuropathologic information, 12 of 14 patients predicted to have TDP-43 pathology had FTLD-TDP, and 7 of 9 patients predicted to have tau pathology had FTLD-tau.
Table 3 Clinical and neuropsychological features of patients predicted by random forests analysis to have FTLD-TDP or FTLD-tau in the living cohort (n = 80)
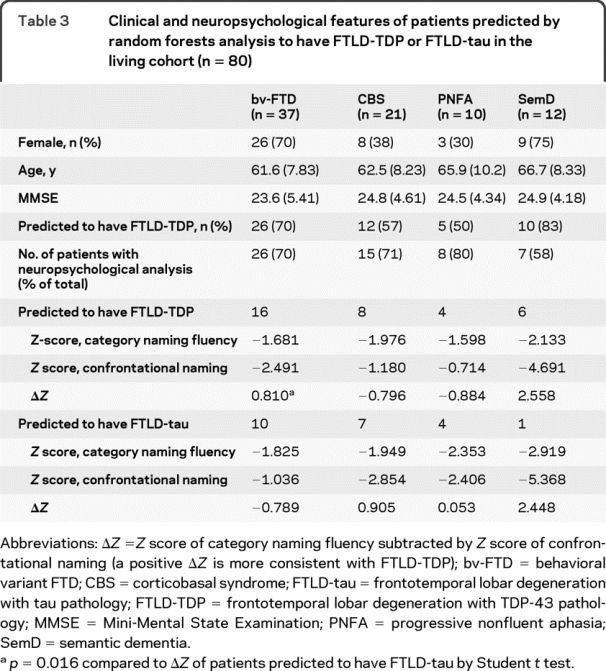
As relative performance in certain neuropsychological tests can reflect differential brain region involvement associated with underlying FTLD pathology,6 we analyzed the neuropsychological profiles of patients in the living cohort. A total of 56 patients underwent testing for both category naming fluency and confrontational naming, and a Z score difference (category naming fluency − confrontation naming) was calculated for each patient with a more positive score suggestive of FTLD-TDP. Compared to those predicted by random forests to have FTLD-tau, patients predicted to have FTLD-TDP performed relatively worse on confrontational naming than category naming fluency (Z score difference of 0.541 vs 0.05, p = 0.314). This difference became most pronounced when the largest phenotypic subgroup—bv-FTD (n = 26)—was analyzed, with patients predicted to have FTLD-TDP having higher _Z_-score differences than those predicted to have FTLD-tau (p = 0.016).
DISCUSSION
FTLD-TDP and FTLD-tau can each lead to clinical FTD syndromes, although the underlying pathologic substrate is difficult to predict on clinical grounds alone. Using autopsy-confirmed cases of FTLD-TDP and FTLD-tau as our training set, we identified novel CSF biomarkers that can improve the distinction between FTLD-TDP and FTLD-tau, including AgRP, ACTH, IL-17, Eotaxin-3, and Fas, with high sensitivity and modest specificity for FTLD-TDP. While the involvement of these candidate biomarkers in the development and progression in FTLD remains to be determined, these analytes offer promise in the antemortem differential diagnosis of FTLD-TDP vs FTLD-tau.
Prior to the current study, only TDP-43 itself has been examined as a potential biomarker for nonfamilial cases of FTLD-TDP.11,22 One study showed elevated plasma TDP-43 levels in 46% of clinical FTD cases and 22% of clinical AD cases, but the lack of pathologic confirmation in these groups limited confident interpretation of results.11 Levels of TDP-43 also appeared to be elevated in CSF samples from patients with FTD-ALS,12 but the significant overlap in TDP-43 levels between patients and healthy subjects emphasized the need for improved assays. Other studies have also sought to identify biomarkers in disorders associated with FTLD, including ALS and progressive supranuclear palsy (PSP). Potential biomarkers of ALS have included elevated levels of TDP-43,12 inflammatory proteins (GM-CSF,23 G-CSF,23 MCP-1,23,24 MIP-1a/b,23 and multiple interleukins including IL-2, IL-6, IL-8, IL-15, and IL-1723,24), axonal structural proteins (neurofilament light chain25), and growth factors (FGF basic protein and VEGF23), and decreased levels of cystatin C,26 insulin-like growth factor 1,27 IL-10,23 interferon-γ,23 erythropoietin,28 and angiotensin II.29 While alterations in some biomarkers are likely specific for FTLD-TDP spectrum disorders, changes in many likely can occur in either FTLD-TDP or FTLD-tau. For example, neurofilament light chains were proposed as a biomarker for ALS,25 but elevated levels were independently found in PSP and CBS.30 Hence, any discovery or validation biomarker work in FTLD-TDP or FTLD-tau needs to incorporate both disorders comparatively with neuropathologic confirmation.
Some of the proposed ALS biomarkers above were specifically evaluated in our multiplex panel.23 MCP-1 was found by 2 previous studies23,24 to be elevated in ALS, and MCP-1 was increased in our FTLD-TDP cases compared to control subjects (p = 0.01) and FTLD-tau (p = 0.089). However, MCP-1 did not emerge as a reliable biomarker for FTLD-TDP in the current study despite the difference in levels. Further analysis showed that MCP-1 levels were highly correlated with Fas levels (R = 0.698, p = 0.005), and Fas was identified by multiple methods as a key discriminator between FTLD-TDP and FTLD-tau. Our multiplex approach thus identified Fas as a more robust proxy biomarker for FTLD-TDP than MCP-1 for a similar underlying biological process. As apoptosis induced by Fas and Fas-associated death domain protein is associated with significant macrophage recruitment and MCP-1 upregulation,31 our finding of elevated Fas levels in FTLD-TDP (compared to FTLD-tau and control subjects) would suggest that Fas-induced apoptosis is more associated with FTLD-TDP than FTLD-tau. Among the remaining analytes that distinguished FTLD-TDP from FTLD-tau, AgRP and ACTH are both hypothalamic neuropeptides, and their elevation in the CSF may reflect hypothalamic dysfunction. No specific hypothalamic dysfunction has been previously described in FTLD-TDP, but disinhibited behaviors common in bv-FTD and hypothalamic dysfunction such as an eating disorder can both be linked to amygdala abnormalities.32 Clinically, elevated AgRP may contribute to the common hyperoral behavior in clinical FTD through its appetite-promoting effect. Analytes identified only by Mann-Whitney U test may also be biologically associated with FTLD. For example, CSF levels of ANG-2 were elevated in FTLD-TDP, and this elevation may be associated with respiratory status of patients with FTLD-TDP.33 IL-23 levels differed between FTLD subtypes by Mann-Whitney U test, and IL-23 promotes the development of helper T cells that release IL-17 (identified by both analytical strategies).21 These T-helper 17 cells have been implicated in multiple sclerosis,33 and microglia can themselves release IL-17 in the presence of IL-23.34 As IL-23 is relatively increased in both FTLD-TDP and FTLD-tau, there may be a common IL-17 dysfunction in FTLD. Whether higher IL-23 levels are protective or harmful in FTLD remains to be determined, along with the biological significance of decreased IL-17 levels in FTLD-TDP despite the upregulated IL-23 levels.
The true diagnostic accuracy of the novel analytes cannot be determined without a fully validated test set, but we predicted the underlying FTLD pathology in a group of living patients with clinical bv-FTD, PPA, and CBS. Consistent with previous reports,4,35,36 we found SemD to have the highest proportion of patients predicted to have FTLD-TDP among all FTD phenotypes. Patients with bv-FTD were next most likely to have FTLD-TDP, and patients with CBS and PNFA were least likely to have FTLD-TDP. However, the proportion of patients predicted to have FTLD-TDP in the bv-FTD, CBS, and PNFA groups were on the higher ends of previously reported ranges.4,35 This could be due to the higher sensitivity at the cost of specificity observed in the autopsy cohort, or bias associated with referral or research participation. At the same time, the pattern of relative performance in category naming fluency and confrontational naming in autopsy-confirmed cases of FTLD-TDP6 was also noted among patients with bv-FTD, which is the most prevalent phenotype. While this pattern of relative neuropsychological performance was not observed in other phenotypes, the limited power within each non-bv-FTD phenotype may mask any such trend in two tasks. The classification results are thus in keeping with reported clinicopathologic correlations, although continued follow-up of these patients to autopsy or validation in an independent autopsy cohort would be necessary to determine the diagnostic performance of novel biomarkers reported here.
The limited sample size in the autopsy cohort may bias the results of our classification, and replication in independent cohorts and platforms will be necessary. There also exists pathologic heterogeneity within FTLD-TDP and FTLD-tau. For example, FTLD-TDP is associated with multiple combinations of pathologic inclusions,2 and different types of FTLD-tau (corticobasal degeneration vs PSP) may uniquely associate with certain analytes. While we aimed to identify analytes common to members within the main pathologic groups, analytes associated with specific pathologic subgroups may also be of biological and clinical significance. Additional analytes may also help distinguish between FTLD-TDP and FTLD-tau, such as levels of tau (which did not significantly differ between FTLD-TDP and FTLD-tau cases in the current study) or phosphorylated TDP-43 peptides in the CSF using more sensitive assays. Improved assays may also determine the utility of previously reported candidate ALS biomarkers, as we were unable to detect levels of GM-CSF, G-CSF, IL-2, IL-6, or IL-15 using the standard RBM protocols. Nevertheless, based on this novel exploratory study of FTLD biomarkers, we propose a stepwise workup of patients clinically diagnosed with FTLD spectrum disorders using first the more validated biomarker assays of CSF AD biomarkers (p-tau181, tau, and Aβ42) to exclude cases of clinical FTD due to atypical AD. This can then be followed by measurements of novel FTLD biomarkers like those reported here to further distinguish between FTLD-TDP and FTLD-tau.
AUTHOR CONTRIBUTIONS
W.T.H. conducted all statistical analyses. W.T.H., M.G., and J.Q.T. have full access to all of the data, take full responsibility for the data, the analyses and interpretation, and the conduct of the research, and have the right to publish any and all data separate and apart from any sponsor.
DISCLOSURE
Dr. Hu has a patent pending re: Cerebrospinal fluid and plasma biomarkers for AD; has received royalties from a publication in CONTINUUM: Lifelong Learning in Neurology (Lippincott Williams & Wilkins, 2010); and receives research support from the Penn-Pfizer Alliance and the American Academy of Neurology Foundation (Clinical Research Training Fellowship). Dr. Chen-Plotkin has patents pending re: Use of the discovery of the gene TMEM106B as a risk factor for development of frontotemporal lobar degeneration and Use of the discovery of 11 plasma proteins as biomarkers for cognitive decline in Parkinson's disease; and receives research support from the Burroughs Wellcome Fund and the American Academy of Neurology (Clinician-Scientist Training Award). Dr. Grossman serves on a scientific advisory board for Allon Therapeutics Inc.; serves as Editor of Cognitive and Behavioral Neurology; has served as a consultant for Pfizer Inc; and receives research support from the NIH (AG17586 [project and core leader], AG15116 [PI], NS44266 [PI] and NS53488 [project leader]). Dr. Arnold serves on a scientific advisory board for Eli Lilly and Company; serves on the editorial board of the Translational Neuroscience Schizophrenia Bulletin; serves as a consultant for the Beasley Firm; and receives research support from Eli Lilly and Company, Pfizer Inc, Elan Corporation, Wyeth, Janssen, the NIH (AG-10124 [Clinical Core Leader and Center Associate Director]) and from the Marian S. Ware Family Foundation. Dr. Clark serves as Medical Director of and holds stock options in Avid Radiopharmaceuticals, Inc.; has served on scientific advisory boards for Elan Corporation and Wyeth; serves as a consultant for Bristol-Myers Squibb, Elan Corporation, Myriad Genetics, Inc., and Ono Pharmaceutical Co. Ltd.; and receives research support from the Department of Health, Commonwealth of Pennsylvania. Dr. Shaw has served on a scientific advisory board for Bristol-Myers Squibb; has received funding for travel and speaker honoraria from Pfizer Inc; serves on the editorial board of Therapeutic Drug Monitoring; may potentially receive revenue for patent pending re: O-methylated rapamycin derivatives for alleviation and inhibition of lymphoproliferative disorders, licensed by the University of Pennsylvania to Novartis; receives royalties from publication of Applied Pharmacokinetics and Pharmacodynamics: Principles of Therapeutic Drug Monitoring (Wolters Kluwer/Lippincott Williams & Wilkins, 2005); receives research support from the NIH (AG024904 [Co-PI Biomarker Core Laboratory] and 1RC 2AG-036535 [ARRA] [PI Biomarker Core Laboratory]); and receives board of directors' compensation and holds stock options in Saladax Biomedical. Dr. McCluskey reports no disclosures. Dr. Elman has received royalties from publications in UpToDate; and receives salary support from the ALS Association and the Muscular Dystrophy Association. Dr. Hurtig served on a grant review panel for the Michael J. Fox Foundation for Parkinson's Research; serves on the editorial board of Parkinsonism and Related Disorders; serves as Movement Disorders Section Editor for UpToDate; has received speaker honoraria from Teva Pharmaceutical Industries Ltd.; receives research support from Teva Pharmaceutical Industries Ltd., Boehringer Ingelheim, Bayer Schering Pharma, Kyowa Hakko Kirin Pharma, Inc., PRA International, Novartis, GlaxoSmithKline, Avid Radiopharmaceuticals, Inc., St Jude Medical, Amarin Corporation, Prestwick Pharmaceutical, Inc., HP Therapeutics Foundation, Inc., Cephalon, Inc., NIH (NINDS P50 NS053488-01 [Core Leader and PI] and NINDS U10 NS044451-023 [Site PI]); and holds stock in Teva Pharmaceutical Industries Ltd. Dr. Siderowf serves on a scientific advisory board for and has received speaker honoraria from Teva Pharmaceutical Industries Ltd.; serves/has served as a consultant to Supernus Pharmaceuticals, Inc., Teva Pharmaceutical Industries Ltd., Merck Serono, and Schering-Plough Corp.; receives research support from the NIH (NINDS U10 NS0444451 [Site PI], NINDS P50 NS053488 [Co-I], NINDS R43NS0636071 [Co-I], and NINDS R01NS065087 [Co-I]), the US Department of Defense, and from the Department of Health, Commonwealth of Pennsylvania; and has served as a consultant on manganese litigation. Dr. Lee has received funding for travel or speaker honoraria from Takeda Pharmaceutical Company Limited, Genentech, Inc. and Pfizer Inc; serves on the editorial boards of Laboratory Investigation, Neurorehabilitation and Neural Repair, NeuroSignals, Neuron, and Experimental Neurology and on the Board of Reviewing Editors for Science Magazine; holds and has patents pending re: Modified Avidin-Biotin Technique, Method of Stabilizing Microtubules to Treat Alzheimer's Disease, Method of Screening for Alzheimer's Disease or Disease Associated with the Accumulation of Parid Helical Filaments, Compositions and Methods for Producing and Using Homogeneous Neuronal Cell Transplants, Animal Model for Alzheimer's Disease, Method of Identifying, Diagnosing and Treating α-Synuclein Positive Neurodegenerative Disorders, Identification and Characterization of AB-Negative Plaques and Methods of Diagnosing Alzheimer's Disease, and Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary Frontotemporal Dementia and Parkinsonism Linked to Chromosome-17; and receives research support from the NIH (NIA P30-AG 009215-19 [project leader], and from Ware Benaroya). Dr. Soares has a patent pending re: Describing plasma based biomarkers for Alzheimer's disease; and is a full-time employee of and owns stock in Pfizer Inc. Dr. Trojanowski has received funding for travel and honoraria from Takeda Pharmaceutical Company Ltd.; has received speaker honoraria from Pfizer Inc; serves as an Associate Editor of Alzheimer's & Dementia; may accrue revenue on patents re: Modified Avidin-Biotin Technique, Method of Stabilizing Microtubules to Treat Alzheimer's Disease, Method of Detecting Abnormally Phosphorylated Tau, Method of Screening for Alzheimer's Disease or Disease Associated with the Accumulation of Paired Helical Filaments, Compositions and Methods for Producing and Using Homogeneous Neuronal Cell Transplants, Rat Comprising Straight Filaments in Its Brain, Compositions and Methods for Producing and Using Homogeneous Neuronal Cell Transplants to Treat Neurodegenerative Disorders and Brain and Spinal Cord Injuries, Diagnostic Methods for Alzheimer's Disease by Detection of Multiple MRNAs, Methods and Compositions for Determining Lipid Peroxidation Levels in Oxidant Stress Syndromes and Diseases, Compositions and Methods for Producing and Using Homogenous Neuronal Cell Transplants, Method of Identifying, Diagnosing and Treating Alpha-synuclein Positive Neurodegenerative Disorders, Mutation-specific Functional Impairments in Distinct Tau Isoforms of Hereditary Frontotemporal Dementia and Parkinsonism Linked to Chromosome-17: Genotype Predicts Phenotype, Microtubule Stabilizing Therapies for Neurodegenerative Disorders; and Treatment of Alzheimer's and Related Diseases with an Antibody; and receives research support from the NIH (NIA P01 AG 09215-20 [PI], NIA P30 AG 10124-18 [PI], NIA PO1 AG 17586-10 [Project 4 Leader], NIA 1PO1 AG-19724-07 [Core C Leader], NIA 1 U01 AG 024904-05 [Co-PI Biomarker Core Laboratory], NINDS P50 NS053488-02 [PI], NIA UO1 AG029213-01 [Co-I]; RC2NS069368 [PI], RC1AG035427 [PI], and NIA P30AG036468 [PI]), and from the Marian S. Ware Alzheimer Program.
Address correspondence and reprint requests to Dr. John Q. Trojanowski, Center for Neurodegenerative Disease Research, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, HUP, Maloney 3rd Floor, 36th and Spruce Streets, Philadelphia, PA 19104-4283 trojanow@mail.med.upenn.edu
Supplemental data at www.neurology.org
e-Pub ahead of print on November 3, 2010, at www.neurology.org.
Study funding: Supported by the Penn-Pfizer Alliance and the NIH (AG-10124, AG-17586, and NS-44266).
Disclosure: Author disclosures are provided at the end of the article.
Received April 2, 2010. Accepted in final form August 3, 2010.
REFERENCES
- 1.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol 2001;58:1803–1809. [DOI] [PubMed] [Google Scholar]
- 2.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009;117:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48. [DOI] [PubMed] [Google Scholar]
- 5.Hu WT, Mandrekar JN, Parisi JE, et al. Clinical features of pathologic subtypes of behavioral-variant frontotemporal dementia. Arch Neurol 2007;64:1611–1616. [DOI] [PubMed] [Google Scholar]
- 6.Grossman M, Xie SX, Libon DJ, et al. Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology 2008;70:2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitwell JL, Jack CR Jr, Senjem ML, et al. MRI correlates of protein deposition and disease severity in postmortem frontotemporal lobar degeneration. Neurodegener Dis 2009;6:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw LM, Korecka M, Clark CM, Lee VM, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov 2007;6:295–303. [DOI] [PubMed] [Google Scholar]
- 9.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tapiola T, Alafuzoff I, Herukka SK, et al. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol 2009;66:382–389. [DOI] [PubMed] [Google Scholar]
- 11.Foulds P, McAuley E, Gibbons L, et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer's disease and frontotemporal lobar degeneration. Acta Neuropathol 2008;116:141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinacker P, Hendrich C, Sperfeld AD, et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 2008;65:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu WT, Chen-Plotkin A, Arnold SE, et al. Novel CSF biomarkers for Alzheimer's disease and mild cognitive impairment. Acta Neuropathol 2010;119:669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu WT, Chen-Plotkin A, Arnold SE, et al. Biomarker discovery for Alzheimer's disease, frontotemporal lobar degeneration, and Parkinson's disease. Acta Neuropathol 2010;120:385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–746. [DOI] [PubMed] [Google Scholar]
- 16.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 17.Ross MA, Miller RG, Berchert L, et al. Toward earlier diagnosis of amyotrophic lateral sclerosis: revised criteria: rhCNTF ALS Study Group. Neurology 1998;50:768–772. [DOI] [PubMed] [Google Scholar]
- 18.Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009;117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Litvan I, Bhatia KP, Burn DJ, et al. Mov Disord Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003;18:467–486. [DOI] [PubMed] [Google Scholar]
- 20.Hu WT, Seelaar H, Josephs KA, et al. Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol 2009;66:1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol 2009;5:325–331. [DOI] [PubMed] [Google Scholar]
- 22.Kasai T, Tokuda T, Ishigami N, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol 2009;117:55–62. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell RM, Freeman WM, Randazzo WT, et al. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 2009;72:14–19. [DOI] [PubMed] [Google Scholar]
- 24.Kuhle J, Lindberg RL, Regeniter A, et al. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol 2009;16:771–774. [DOI] [PubMed] [Google Scholar]
- 25.Zetterberg H, Jacobsson J, Rosengren L, Blennow K, Andersen PM. Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: impact of SOD1 genotype. Eur J Neurol 2007;14:1329–1333. [DOI] [PubMed] [Google Scholar]
- 26.Pasinetti GM, Ungar LH, Lange DJ, et al. Identification of potential CSF biomarkers in ALS. Neurology 2006;66:1218–1222. [DOI] [PubMed] [Google Scholar]
- 27.Bilic E, Bilic E, Rudan I, et al. Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls Eur J Neurol 2006;13:1340–1345. [DOI] [PubMed] [Google Scholar]
- 28.Brettschneider J, Widl K, Ehrenreich H, Riepe M, Tumani H. Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci Lett 2006;404:347–351. [DOI] [PubMed] [Google Scholar]
- 29.Kawajiri M, Mogi M, Higaki N, et al. Reduced angiotensin II levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol Scand 2009;119:341–344. [DOI] [PubMed] [Google Scholar]
- 30.Constantinescu R, Rosengren L, Johnels B, Zetterberg H, Holmberg B. Consecutive analyses of cerebrospinal fluid axonal and glial markers in Parkinson's disease and atypical parkinsonian disorders. Parkinsonism Relat Disord 2010;16:142–145. [DOI] [PubMed] [Google Scholar]
- 31.Schaub FJ, Han DK, Liles WC, et al. Fas/FADD-mediated activation of a specific program of inflammatory gene expression in vascular smooth muscle cells. Nat Med 2000;6:790–796. [DOI] [PubMed] [Google Scholar]
- 32.Kling AS, Tachiki K, Lloyd R. Neurochemical correlates of the Kluver-Bucy syndrome by in vivo microdialysis in monkey. Behav Brain Res 1993;56:161–170. [DOI] [PubMed] [Google Scholar]
- 33.Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing T H 17 cells in multiple sclerosis. Ann Neurol 2009;66:390–402. [DOI] [PubMed] [Google Scholar]
- 34.Kawanokuchi J, Shimizu K, Nitta A, et al. Production and functions of IL-17 in microglia. J Neuroimmunol 2008;194:54–61. [DOI] [PubMed] [Google Scholar]
- 35.Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406. [DOI] [PubMed] [Google Scholar]