MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences (original) (raw)
Abstract
Miniature inverted-repeat transposable elements (MITEs) are a special type of Class 2 non-autonomous transposable element (TE) that are abundant in the non-coding regions of the genes of many plant and animal species. The accurate identification of MITEs has been a challenge for existing programs because they lack coding sequences and, as such, evolve very rapidly. Because of their importance to gene and genome evolution, we developed MITE-Hunter, a program pipeline that can identify MITEs as well as other small Class 2 non-autonomous TEs from genomic DNA data sets. The output of MITE-Hunter is composed of consensus TE sequences grouped into families that can be used as a library file for homology-based TE detection programs such as RepeatMasker. MITE-Hunter was evaluated by searching the rice genomic database and comparing the output with known rice TEs. It discovered most of the previously reported rice MITEs (97.6%), and found sixteen new elements. MITE-Hunter was also compared with two other MITE discovery programs, FINDMITE and MUST. Unlike MITE-Hunter, neither of these programs can search large genomic data sets including whole genome sequences. More importantly, MITE-Hunter is significantly more accurate than either FINDMITE or MUST as the vast majority of their outputs are false-positives.
INTRODUCTION
Transposable elements (TEs) reside in all characterized eukaryotic genomes where they are often the largest component. For example, sequences derived from TEs make up at least 31% of the genome of dog (Canis familiaris), 38% of mouse (Mus musculus), 46% of human (Homo sapiens) and 85% of maize (Zea mays ssp. mays L.) (1–4). TEs have structural features and classification systems that serve to distinguish them from simpler repetitive sequences like microsatellite repeats. TEs are divided into two classes based on the molecule involved in transposition: retrotransposons (Class 1) move via a RNA intermediate while DNA is the intermediate of DNA transposons (Class 2). In each class, TEs are further divided into superfamilies and families (5). In plants, six Class 2 superfamilies have been identified thus far: Tc1/Mariner, PIF/Harbinger, hAT, MULE, CACTA and Helitron (5,6). With the exception of Helitrons, TEs in the other five superfamilies have terminal inverted repeats (TIRs) and transpose through a cut-and-paste mechanism. TEs are also classified as autonomous or non-autonomous elements based on whether they can produce functional transposase.
Miniature inverted-repeat TEs (MITEs) are a special type of Class 2 non-autonomous element that is present in high copy numbers in many eukaryotic genomes. For example, ∼56 000 MITEs were identified in sorghum (Sorghum bicolor) (7), 73 500 in rice (Oryza sativa) (8) and 150 000 in human (9). Ever since their discovery almost 20 years ago (10,11), MITEs have been the subject of increasing interest in both plants and animals (12–15). Unlike the ‘traditional’ low copy non-autonomous TEs (such as the Ds element of maize), MITEs are uniformly short (most <500 bp) and amplify rapidly from one or a few elements to very high copy numbers (16). The two largest MITEs families, Stowaway and Tourist, were found to be members of the Tc1/Mariner and the PIF/Harbinger superfamilies, respectively (12,17–19). MITEs have also been reported from the hAT and MULE superfamilies (13,20).
While the rapidly expanding databases of genomic sequence present an opportunity to expand the study of MITEs, it also poses a significant challenge to their correct and efficient annotation. Many TE annotation programs have been developed that use one or more of the following computational approaches: (i) homology-based, (ii) de novo, (iii) polymorphism based and (iv) structure based (21–23). Homology-based TE annotation is powerful at detecting TEs that share sequence similarity with known elements, but it is inadequate at identifying full length or novel TEs. Methods using de novo approaches can discover all TEs as long as they have multiple copies. However, the drawback of this approach is that its output is a mixture of TEs from all superfamilies and non-TE repeats. As such, the manual identification and classification of TEs from the output of de novo methods is often very tedious and time consuming. Polymorphism-based approaches can discover new TEs but the output is also a mixture of different types of sequences. More importantly, its application is limited to the comparison of data sets from very closely related species. When compared to the other algorithms, structure-based approaches are very effective at discovering certain TE types like LTR retrotransposons. However, currently available programs are less successful at identifying other TE types like non-autonomous Class 2 transposons (including MITEs) because they possess few distinguishing structural features.
To date three programs have been developed exclusively to find MITEs: TRANSPO (24), FINDMITE (15) and MUST (25). TRANSPO is a homology-based program that requires known MITE sequences. As such it is not effective at finding new MITEs (21). FINDMITE and MUST are structure-based TE discovery programs that can be used to discover new MITEs because they search for common MITE structural features rather than similar sequences. However, because MITEs have only two common structural features, TIRs and target site duplications (TSDs), many sequences that are not MITEs are in the outputs of FINDMITE and MUST. Thus, the false-positive rates of these programs are very high and extensive manual curation is required to filter false-positives from their output files.
Here, we present MITE-Hunter, a program that accurately discovers MITEs as well as other short non-autonomous ‘cut-and-paste’ Class 2 TEs in genomic data sets including those of whole genomes. To evaluate MITE-Hunter, we compared it with FINDMITE and MUST. We chose the rice genome to evaluate the performance of MITE-Hunter because rice harbors abundant and well-annotated Class 2 TEs and MITEs (8,26,27). In the examples reported in this study, MITE-Hunter missed only two known rice MITEs and discovered 16 previously unknown elements. Compared to FINDMITE and MUST, MITE-Hunter has a much lower false-positive rate and the output is easier to be checked and classified. MITE-Hunter and related programs can be freely downloaded at http://target.iplantcollaborative.org/.
MATERIALS AND METHODS
The MITE-Hunter pipeline
MITE-Hunter is a UNIX program pipeline composed mainly of Perl scripts. Given genomic sequences as the input data, MITE-Hunter identifies Class 2 non-autonomous TEs and produces outputs of consensus sequences classified into families. MITE-Hunter can use multiple processers (default 5 CPUs). The MITE-Hunter pipeline has five main steps that are summarized in Figure 1: (i) identify TE candidates through a structure-based approach, (ii) identify and filter false-positives using an approach based on the pairwise sequence alignment (PSA), (iii) generate exemplars, (iv) identify and filter false-positives using an approach based on the multiple sequence alignment (MSA), generate consensus sequences and predict TSDs and (v) group consensus sequences into families. Details of each step are presented in the results section.
Figure 1.

The five main steps of the MITE-Hunter pipeline. Gray bars are genomic sequences, black and red triangles are TSDs and TIRs, respectively, blue bars are predicted TEs, white bars are homolog sequences, dashed lines are gaps and yellow bars are sequences that are similar to each other but not to those represented by green bars (and vice versa). (A) Identification of candidate TEs. Three predicted candidate TEs are shown. (B) Filtering of false-positives based on the PSA. Four types of alignments are shown (a–d). Except for the candidates in (d), all the others are filtered as false-positives. (C) Selection of TE exemplars. (D) Filtering of false-positives based on the MSA, predicting TSDs and generating consensus sequences. (e) and (f) are two special types of MSA (see text for detail). (E) Selecting new exemplars and grouping TEs into families.
Data set and Programs
The build five rice IRGSP/RAP genome sequence was used (28) as was Repbase version 14.02 (29) and RepeatMasker 3.26 (Smit, A.F.A., Hubley,R. and Green,P., unpublished data; http://www.repeatmasker.org). TE copy number was calculated using a previously described method (4). Pair-wise sequences alignment (PSA) used BLAST (30) and multiple sequences alignment (MSA) used Muscle (31). All computation was done on a Linux cluster.
RESULTS
MITE discovery in rice
We applied MITE-Hunter to the rice genome with default parameters. MITE-Hunter completed the analysis in ∼44 h. Details of the algorithms and results of each step of MITE-Hunter are presented below.
- Identifying all candidates (Figure 1A). MITE-Hunter uses genomic sequences as the input data. Long input sequences are first cut into small fragments (default 2 kb) with overlaps (default 500 bp). TE candidates are identified from each fragment sequence as those that have TIR-like structures (default 10 bp with at most 1 bp mismatch) flanked by putative TSDs (2–10 bp; default is TA if TSD length = 2). Because low complexity sequences (LCS) are rare in MITEs but make up many TIR-like and TSD-like structures, TE candidates that have LCS in TIRs or have too many LCS within internal sequences are filtered as follows. First, TIRs that have stretches of tandem 1–2 nt units (default ≥8 bp) or have low G+C content (default <20%) are filtered. Second, a candidate will be filtered if it has too many LCS (default ≥20%) identified by DUST (Tatusov,R. and Lipman,D.J., unpublished data). Using rice genomic DNA sequences as the input data (∼380 Mb), 629 698 candidate TEs were identified and retrieved together with their flanking sequences (default 60 bp).
- Filtering false-positives based on the pairwise sequence alignment (PSA) (Figure 1B). Candidate TEs and their flanking sequences are submitted to an all-by-all BLASTN comparison (default _E_-value = 1_e_−10). To reduce the computational load, candidates are divided into groups based on their length (default interval = 100 bp) and BLASTN is performed separately for each group. From the BLASTN results, single copy candidates are identified and filtered. Of the remaining candidates, only those that share sequence similarity within but not in their flanking regions are retained. Four types of PSAs are shown in Figure 1B-a–d. In type (a), the similar region extends to both sides. In type (b) and (c), similar regions extend to the left and right of the flanking regions, respectively. Only in type (d) is the similar region within the TIRs and the candidate not filtered as a false-positive. Of the 629 698 rice candidates from Step I, 38 617 passed this filter. These candidates were trimmed of their flanking sequences before being sent to the next step.
- Identifying TE exemplars (Figure 1C). To reduce computational load in the following steps, MITE-Hunter clusters TE candidates based on their sequence similarity and picks one as the exemplar that best characterizes the features of each group. First, the candidates from Step II are subjected to an all-by-all BLASTN comparison. Based on the BLASTN results, candidates are clustered as follows: (a) the candidate that matches most of the others (default matched length percentage >90% and identity ≥80%) is selected as the exemplar, (b) the exemplar and the candidates that it matches are put into one group and will not be sampled again and (c) repeat 1 and 2 until no candidates remain. In this step, of 38 617 TE candidates from Step II, 3887 exemplars were selected and sent to the next step.
- Filtering false-positives using the multiple sequence alignment (MSA), generating consensus sequences and predicting TSDs (Figure 1D). Each exemplar identified in Step III is used as a query to perform BLASTN searches of the genomic database. Homologs are identified and retrieved together with their flanking sequences (default 60 bp) by a command line version of TARGeT (32). Candidates that have too few homologs (default ≤3) are filtered because many ultimately prove to be false-positives. A MSA is generated using homologs of each exemplar. To reduce the computational load, if there are too many homologs for an exemplar, only the top 35 with the highest BLASTN alignment scores are used. From each MSA, three average identity scores are calculated from the left flanking region (L), homologous region (H) and the right flanking region (R).
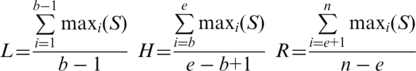
In these equations, b and e are the beginning and ending positions of homologs in the MSA, n is the total length of the MSA and S is the proportion of different nucleotides in each column of the MSA. Candidates whose H score is significantly higher than both L and R scores (default >0.25) are retained.
Two special situations that can potentially confound the results are addressed in MITE-Hunter. One concerns TE homologs with DNA inserts that cause large gaps in the MSA and significantly lower the H score. In this case, homologs with additional sequences (default >25 bp) are identified and filtered before calculating the H, L and R scores. An example is shown in Figure 1D-e, where the MSA has two long gaps caused by the additional sequences in homologs 1 and 4 (represented by red bars). After filtering homologs 1 and 4, a new MSA is generated using the remaining homolog sequences (2, 3 and 5). The other special situation is that for some MSAs, although the L and R scores are low, a subgroup of flanking sequences is very similar. Based on our experience, most candidates with this type of MSA are false-positives. An example is shown in Figure 1D-f, where the L and R scores are much lower than the H score of the MSA. However, in the left flanking region of this MSA, sequences in 1 and 2 are very similar but are different from the sequences in 3, 4 and 5, which are also similar to each other. To filter this type of false-positive, MITE-Hunter calculates the identity between flanking sequences from the MSA. In such cases, the candidate will be filtered if >50% of the homologs (default value) share >60% identity (default value) in their flanking regions.
For candidates that pass these filters, MITE-Hunter predicts TSDs. TSDs are predicted again in this step because it is more accurate to predict TSDs based on MSA than from a single sequence in Step I. Identical sequences flanking each homolog (default 2–10 bp) are identified as candidate TSDs. From these, actual (predicted) TSDs are considered those with the most common sequence length. For example, the actual TSD will be 3 bp if 8 of 10 candidate TSDs are 3 bp in length. Predicted TSDs are useful in the manual classification of MITE-Hunter output into superfamilies.
In general, consensus sequences better represent homologous TEs than exemplars. While exemplars are selected from real TEs that may have mutations that are different from other homologs, consensus TEs are generated from MSAs and are composed of residues that are most abundant in all of the homologs. MITE-Hunter generates consensus sequences by choosing the most frequent nucleotide from each column (default ≥70%) in the homologous region of the MSA. Of the 3887 TE exemplars from Step III, 2253 were verified and consensus sequences were generated and sent to the final step. - Identifying new exemplars and grouping into families (Figure 1E). To further condense the output, new exemplars are selected from the consensus sequences in Step IV using the same approach as in Step III. This step is necessary because after replacing the exemplar sequences with consensus sequences in Step IV, the similarity between many TE consensus sequences satisfies the grouping criteria. Of the 2253 TE consensus sequences from Step IV, 700 new exemplars were selected and used to execute the all-by-all BLASTN comparison. From the BLASTN results the exemplars were grouped into 446 families based on the 80-80-80 rule (5).
Accuracy evaluation of MITE-Hunter
To test the authenticity of the MITE-Hunter output we curated the 700 rice TEs (Figure 2). Each MSA file was manually analyzed for TIR and TSD structures that are characteristic of Class 2 TE superfamilies found in plant genomes. A TE is validated if it has at least three full-length copies and its ends, characterized by TIRs and TSDs, can be recognized from the MSA file. TEs that do not meet these criteria are considered to be false-positives. Using these strict parameters, we identified 46 false-positives. In addition, eight solo LTRs and four short Helitrons were identified and classified as false-positives. These 12 elements were in the MITE-Hunter output because they coincidentally have TIR-like and TSD-like structures near their ends. After removing these elements there were 642 TEs remaining from the original 700, resulting in a false-positive rate of 8.3% [(46 + 8 + 4)/700].
Figure 2.

Flowchart of the manual curation of rice Class 2 non-autonomous TEs from MITE-Hunter output. The authentication process began with 700 consensus TEs and was reduced by the number shown for each step. The numbers on the right are the remaining consensus TEs after each step (see text for detail). Three different types of compound TEs are shown (a, b and c). Open and solid bars represent different TEs from different families. (a) One TE inserted into another. (b) Two different adjacent TEs. (c) Two adjacent copies from the same TE family.
Classification of TEs discovered by MITE-Hunter
In addition to 58 false-positives, we were unable to classify 15 TEs into superfamilies. Although these sequences appeared to be TEs (based on their MSA files), their TSDs and TIRs were ambiguous because they contained too many mismatches. As such, they were classified as unknowns.
The remaining 627 TEs were confirmed to be ‘cut-and-paste’ Class 2 TEs and were classified into previously described superfamilies. However, during the classification process we found that several families contain TEs belonging to more than one superfamily. By comparing their sequences, we discovered that this problem was caused by 14 compound TEs that were formed by the insertion of one superfamily member into another (Figure2-a). Because TEs were grouped into families based on their similarity, these 14 compound TEs drag TEs from different superfamilies together. In addition, we identified another 12 compound TEs that were formed by the fusion of two TEs from the same superfamily (Figure 2-b and -c). These 26 compound TEs have low full-length copy number in the genome and were excluded from the following analysis. Thus 601 TE consensus sequences remained.
Manual curation reveals that some TE consensus sequences in the MITE-Hunter output miss or have additional sequences at their ends. This problem is caused by the existence of false-TIR and TSD structures near the authentic ones. The missing or additional sequences are mostly short and can be manually identified after locating the real TIRs and TSDs in the MSA files. After correcting the consensus sequences of the remaining 601 Class 2 TEs (by adding or trimming the missing or additional sequence), the similarity between some TE sequences satisfies the grouping criteria in Step III (Figure 1C). As such we ran the programs in Step III and V of MITE-Hunter and got the final data set composed of 551 TE consensus sequences grouped into 401 families. Of these, 97 Tc1/Mariner TEs are grouped into 86 families, 146 PIF/Harbingers into 104 families, 123 hATs into 95 families, 173 Mutators into 110 families and 12 CACTAs into 6 families.
Identification of MITEs from MITE-Hunter output
To identify and characterize MITEs from MITE-Hunter output, we performed a RepeatMasker search of the rice genomic database using the curated 551 TE sequences as the query. From the RepeatMasker output, we counted the copy number of each TE (data not shown). To distinguish MITEs from lower copy Class 2 non-autonomous TEs, we defined a MITE as a Class 2 non-autonomous TE of <800 bp and with at least 100 full-length copies in the genome. Potential MITEs that have not experienced significant amplification were defined as having fewer copies (10–99) but high sequence identity (identity ≥99%). Based on these criteria, we identified 132 rice MITEs from the MITE-Hunter output, including 15 hAT-MITEs, 22 Mutator-MITEs, 50 Stowaways and 45 Tourists. No additional CACTA MITEs were found.
Comparison of MITE-Hunter output to Repbase data
To estimate the false-negative rate of MITE-Hunter we used the rice Class 2 non-autonomous elements in the Repbase as the reference data set. Repbase was selected for this analysis because it is a collective TE database containing most, if not all, previously reported rice Class 2 TEs (29). However, because Repbase contains both Class 1 and 2 autonomous and non-autonomous TEs, the first step was to retrieve only rice Class 2 non-autonomous elements. From these we then selected 230 elements that were <1.7 kb because the longest rice TE found by MITE-Hunter has 1676 bp. The 230 elements were manually checked using the same approach that was applied to the MITE-Hunter output. Thirty-two of the 230 elements were excluded because they lack multiple full-length copies. In addition, 13 were excluded because their TIR and TSD structures could not be identified from MSA files. The remaining 185 Repbase TEs were classified into Class 2 TE superfamilies. By using the same approach as was used for identifying MITEs from the MITE-Hunter output, we identified 101 MITE-like elements from the 185 Repbase TEs, including 4 hAT-MITEs, 19 Mutator-MITEs, 40 Stowaways and 38 Tourists.
The false-negative rates of MITE-Hunter were calculated separately for Class 2 non-autonomous TEs and MITEs as follows. First, we used the curated 551 Class 2 non-autonomous TEs discovered by MITE-Hunter as the query to mask the Repbase data set using RepeatMasker. On average, 84.9% of the sequences in the Repbase data set were masked (Table 1, second column). Using a similar approach, 97.6% of MITE sequences in the Repbase were masked by the TEs in the MITE-Hunter output (Table 1, third column). Thus the false-negative rate of MITE-Hunter is 15.1% for Class 2 non-autonomous TEs and 2.4% for MITEs. MITE-Hunter failed to identify only two Tourist MITEs (OSTE23 and ID-4) that were in Repbase. In contrast, using the data of the Repbase as the libraries, 47.9% of Class 2 non-autonomous TEs and 83.4% of MITEs in the MITE-Hunter output were masked (Table 1, the last two columns). Sixteen MITEs discovered by MITE-Hunter were not found in Repbase including 1 Tourist, 11 hAT-MITEs and 4 Mutator-MITEs.
Table 1.
Comparison between MITE-Hunter output and rice TEs in Repbase
| Superfamily | Repbase data masked by MITE-Hunter output (%) | MITE-Hunter output masked by Repbase data (%) | ||
|---|---|---|---|---|
| Alla | MITEs onlyb | Allc | MITEs onlyd | |
| Tc1/Mariner | 93.3 | 100.0 | 72.5 | 99.9 |
| PIF/Harbinger | 83.8 | 94.6 | 53.1 | 93.0 |
| hAT | 85.8 | 100.0 | 25.6 | 28.4 |
| Mutator | 81.0 | 99.3 | 49.5 | 80.0 |
| CACTA | 88.2 | – | 81.7 | – |
| Together | 84.9 | 97.6 | 47.9 | 83.4 |
Evaluation of FINDMITE and MUST
We tested the ability of two previously published MITE finding programs, FINDMITE and MUST, to discover MITEs in the rice genomic data set using default parameters. Importantly, when we attempted to use the entire genomic sequence (∼372.8 Mb) as the input data, both FINDMITE and MUST reported errors and quit. As such we applied FINDMITE and MUST to a much smaller data set, rice chromosome 12 (∼28.2 Mb) (Table 2). MUST completed the task in ∼5 h and 30 min and generated 5485 putative TE sequences. Because FINDMITE requires users to define the TSD sequence and length, we chose ‘TA’, which is the TSD sequence of Stowaway MITEs. FINDMITE finished in <1 min and generated 10 864 putative Stowaways. To calculate the false-positive rate, we randomly sampled 100 TE sequences from the outputs of FINDMITE and MUST, respectively, and checked them using the same approach as was used for evaluating MITE-Hunter. With only 15 and 14 validated TEs for FINDMITE and MUST, respectively, both programs have a false-positive rate of over 80%. To perform an impartial comparison, we also applied MITE-Hunter to the rice chromosome 12 data set. Using default parameters, MITE-Hunter finished in 1 h and 40 min and generated 114 TE consensus sequences that were grouped into 88 families. Through manual curation, five TEs were identified as false-positives resulting in a false-positive rate of 4.4%. Because the input data is a small subset of the rice genome, we did not compare the results of FINDMITE and MUST to the Repbase data to calculate the false-negative rate.
Table 2.
Comparisons of MITE-Hunter with FINDMITE and MUST
| Program | Running timea | Predicted TEs | False-positives (%) |
|---|---|---|---|
| MITE-Hunter | 1.7 h | 114 | 4.4 |
| FINDMITEb | <1 min | 10 864 | 85.0 |
| MUST | 5.5 h | 5485 | 86.0 |
DISCUSSION
A necessary prerequisite for the comprehensive analysis of MITEs is their identification in newly sequenced genomes. Two programs were previously developed for this purpose, FINDMITE and MUST. However, as demonstrated in this study, both FINDMITE and MUST have very high false-positive rates (∼85%) and cannot efficiently utilize whole genomic data sets like that from rice. To remedy this situation, we developed MITE-Hunter, which is a structure-based program pipeline that can efficiently identify TEs that have TIR and TSD structures from whole genome data sets. Important features of MITE-Hunter are discussed below.
MITE-Hunter has an efficient approach to reduce the high false-positive rate, which is the main limitation of currently available MITE discovery programs. The vast majority of rice genomic sequences with TIR-like and TSD-like structures are not Class 2 TEs. MITE-Hunter has two modules to filter false-positives, that both exploit the principle that homologs of a true TE only share sequence similarity within the terminal structures. The main difference between the two modules is that one detects sequence similarity through the PSA approach while the other uses the MSA approach. The MSA-based module is more powerful at identifying false-positives but it is slower than the PSA-based module. To achieve both high speed and high sensitivity, the PSA-based module is first performed in Step II to filter most of the false-positives while the MSA-based module is performed in Step IV to filter the remaining false-positives. Because MITE-Hunter has such a system to identify and filter artificial TE candidates, the false-positive rate of MITE-Hunter (4.4–8.3%) is ten times lower than either FINDMITE (85%) or MUST (86%).
MITE-Hunter is competent at discovering Class 2 non-autonomous TEs especially MITEs. In our test, MITE-Hunter rediscovered most of the known rice Class 2 non-autonomous TEs (85%) and almost all MITEs (97.6%) in Repbase [Table 1, second and third columns]. Only two MITEs (OSTE23 and ID-4) in Repbase were missed by MITE-Hunter. OSTE23 is a very old MITE family and its TIR and TSD structures are difficult to detect even by manual examination of the MSA file. ID-4 has two mismatches in the TIRs that were not identified in Step I of MITE-Hunter.
Compared to other MITE discovery programs, the MITE-Hunter output is much easier to curate manually. First, the number of TEs in the MITE-Hunter output is very small because MITE-Hunter generates consensus sequences that best represent the whole TE data set of the genome being analyzed. As shown in the results section, MITE Hunter generated 700 consensus TEs from the entire rice genomic data set. In contrast, FINDMITE generated ∼10 000 putative Stowaway MITEs using only the smallest rice chromosome (#12) as the input data set. Using the same data set MUST generated about 5000 elements. Second, for each TE sequence in its output, MITE-Hunter generates a MSA file and predicts TSDs, which are useful for both TE validation and classification. The validity of each TE discovered by MITE-Hunter can be determined by identifying TIRs and TSDs from the MSA file by manual inspection. Finally, in the output of MITE-Hunter, identified TEs are automatically grouped into families based on the sequence similarity, which further helps manual curation by users. These features are of value to all users, especially those who need a TE data set that is 100% accurate and is classified into superfamilies
In summary, MITE-Hunter is the first program to efficiently and accurately identify MITEs from whole genome sequence. Whereas the rice Class 2 non-autonomous TEs in Repbase were the products of many studies, MITE-Hunter was able to find virtually all the MITEs in a relatively short time frame and to do so accurately. Finally, the MITE-Hunter output is easy to curate as it contains highly condensed TE consensus sequences that are grouped into families. The validity of a TE discovered by MITE-Hunter can be quickly judged from the automatically generated MSA file, which is, to our knowledge, a unique feature of MITE-Hunter.
FUNDING
The National Science Foundation (NSF) plant genome (0607123 to S.R.W.). Funding for open access charge: The NSF plant genome grant 0607123.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Yaowu Yuan for valuable discussions of both of the programs and the article. We thank Hao Wang for installing and running MUST.
REFERENCES
- 1.Kirkness EF, Bafna V, Halpern AL, Levy S, Remington K, Rusch DB, Delcher AL, Pop M, Wang W, Fraser CM, et al. The dog genome: survey sequencing and comparative analysis. Science. 2003;301:1898–1903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 2.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 5.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 6.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 8.Oki N, Yano K, Okumoto Y, Tsukiyama T, Teraishi M, Tanisaka T. A genome-wide view of miniature inverted-repeat transposable elements (MITEs) in rice, Oryza sativa ssp. japonica. Genes Genet. Syst. 2008;83:321–329. doi: 10.1266/ggs.83.321. [DOI] [PubMed] [Google Scholar]
- 9.Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc. Natl Acad. Sci. USA. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bureau TE, Wessler SR. Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell. 1994;6:907–916. doi: 10.1105/tpc.6.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bureau TE, Wessler SR. Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992;4:1283–1294. doi: 10.1105/tpc.4.10.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR. Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science. 2009;325:1391–1394. doi: 10.1126/science.1175688. [DOI] [PubMed] [Google Scholar]
- 13.Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, Jiang J, Buell CR, Baker B. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 2009;19:42–56. doi: 10.1101/gr.078196.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osborne PW, Luke GN, Holland PW, Ferrier DE. Identification and characterisation of five novel miniature inverted-repeat transposable elements (MITEs) in amphioxus (Branchiostoma floridae) Int. J. Biol. Sci. 2006;2:54–60. doi: 10.7150/ijbs.2.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu Z. Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito, Anopheles gambiae. Proc. Natl Acad. Sci. USA. 2001;98:1699–1704. doi: 10.1073/pnas.041593198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR. Dramatic amplification of a rice transposable element during recent domestication. Proc. Natl Acad. Sci. USA. 2006;103:17620–17625. doi: 10.1073/pnas.0605421103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR. An active DNA transposon family in rice. Nature. 2003;421:163–167. doi: 10.1038/nature01214. [DOI] [PubMed] [Google Scholar]
- 18.Feschotte C, Swamy L, Wessler SR. Genome-wide analysis of mariner-like transposable elements in rice reveals complex relationships with stowaway miniature inverted repeat transposable elements (MITEs) Genetics. 2003;163:747–758. doi: 10.1093/genetics/163.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Feschotte C, Zhang Q, Jiang N, Eggleston WB, Wessler SR. P instability factor: an active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proc. Natl Acad. Sci. USA. 2001;98:12572–12577. doi: 10.1073/pnas.211442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moreno-Vazquez S, Ning J, Meyers BC. hATpin, a family of MITE-like hAT mobile elements conserved in diverse plant species that forms highly stable secondary structures. Plant Mol. Biol. 2005;58:869–886. doi: 10.1007/s11103-005-8271-8. [DOI] [PubMed] [Google Scholar]
- 21.Lerat E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2009;104:520–533. doi: 10.1038/hdy.2009.165. [DOI] [PubMed] [Google Scholar]
- 22.Saha S, Bridges S, Magbanua ZV, Peterson DG. Computational approaches and tools used in identification of dispersed repetitive DNA sequences. Tropical Plant Biol. 2008;1:85–96. [Google Scholar]
- 23.Bergman CM, Quesneville H. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 2007;8:382–392. doi: 10.1093/bib/bbm048. [DOI] [PubMed] [Google Scholar]
- 24.Santiago N, Herraiz C, Goni JR, Messeguer X, Casacuberta JM. Genome-wide analysis of the Emigrant family of MITEs of Arabidopsis thaliana. Mol. Biol. Evol. 2002;19:2285–2293. doi: 10.1093/oxfordjournals.molbev.a004052. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Zhou F, Li G, Xu Y. MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene. 2009;436:1–7. doi: 10.1016/j.gene.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 26.Jiang N, Feschotte C, Zhang X, Wessler SR. Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs) Curr. Opin. Plant Biol. 2004;7:115–119. doi: 10.1016/j.pbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Bureau TE, Ronald PC, Wessler SR. A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proc. Natl Acad. Sci. USA. 1996;93:8524–8529. doi: 10.1073/pnas.93.16.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka T, Antonio BA, Kikuchi S, Matsumoto T, Nagamura Y, Numa H, Sakai H, Wu J, Itoh T, Sasaki T, et al. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36:D1028–1033. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 30.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han Y, Burnette JM, III, Wessler SR. TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res. 2009;37:e78. doi: 10.1093/nar/gkp295. [DOI] [PMC free article] [PubMed] [Google Scholar]