The Hsp70 chaperone machinery: J-proteins as drivers of functional specificity (original) (raw)
. Author manuscript; available in PMC: 2011 Aug 1.
Published in final edited form as: Nat Rev Mol Cell Biol. 2010 Aug;11(8):579–592. doi: 10.1038/nrm2941
Abstract
Hsp70s, ubiquitous molecular chaperones, function in a myriad of biological processes, modulating polypeptides’ folding, degradation and translocation across membranes, as well as protein-protein interactions. This multitude of roles is not easily reconciled with the near conformity of biochemical activity of Hsp70s, an ATP-dependent client protein binding/release cycle. Much of the functional diversity of Hsp70s is driven by a diverse class of cofactors, J-proteins (also called Hsp40s). Often, multiple J-proteins function with a single Hsp70. Some target Hsp70 activity to clients at precise locations in cells; others bind client proteins directly, thereby delivering specific clients to Hsp70, directly determining their fate.
In their native cellular environment, polypeptides are constantly at risk of attaining conformations that prevent them from functioning properly and/or cause them to aggregate into large, potentially cytotoxic complexes. Molecular chaperones guide the conformation of proteins throughout their lifetime, preventing their aggregation by protecting interactive surfaces against non-productive interactions. Through such interactions, they aid in the folding of nascent proteins as they are synthesized by ribosomes, drive protein transport across membranes, and modulate protein-protein interactions by controlling conformational changes1, 2. In addition to these roles under optimal conditions, stresses can exacerbate protein conformational problems (for example, heat shock causing protein unfolding; oxygen radicals causing oxidation and nitrosylation). Although in some cases chaperones can facilitate (re)folding, often such rejuvenation is not possible. In such cases, chaperones can facilitate degradation, either by simply preventing aggregation and thus keeping clients susceptible to proteolysis or by actively facilitating their transfer to proteolytic systems. These diverse functions of molecular chaperones typically involve iterative client binding and release cycles until the client has reached its final active conformation, or has entered the proteolytic system (Figure 1).
Figure 1. Protein folding and degradation through client protein–chaperone binding and release cycle.
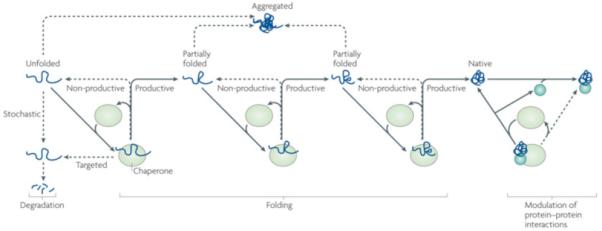
Chaperones were originally defined as “proteins that prevent improper interactions between potentially interactive surfaces and disrupt any improper liaisons that may occur”134. The Heat Shock Proteins (HSPs) family constitutes a large group of chaperones that interact with a wide variety of non-native proteins, facilitating acquisition of their native conformation, without being associated with their final functional structure. However, recent evidence indicates that chaperone function of HSPs is not restricted to assisting protein folding and assembly. It is also needed to facilitate client degradation via both proteasomal and autophagasomal pathways, as well as to stabilize or destabilize interactions between mature, folded proteins. In iterative cycles of client binding to and release from HSPs, which are often driven in an adenine nucleotide dependent manner, client aggregation is prevented (purple lines), productive folding occurs through a series of steps (bleu lines), and HSPs are re-cycled for client binding (black lines). If folding fails or in the case of non-foldable clients re-binding to the chaperone may occur, which in a stochastic manner, maintains client soluble for subsequent (proteasomal) degradation (red lines). Besides this passive, stochastic support of degradation, some chaperones can also ‘actively’ direct clients towards degradation (green line). In addition, chaperones can bind folded proteins (bleu circle and grey square) and induce conformational changes, thereby regulating protein:protein interactions and functionality of protein complexes.
Strikingly, Hsp70s, one of the most ubiquitous classes of chaperones, has been implicated in all of the biological processes mentioned above2, 3. This Review focuses on the means by which Hsp70 molecular chaperone machinery participates in such diverse cellular functions. Their functional diversity is remarkable considering that within and across species, Hsp70s have very high sequence identity. They share a single biochemical activity, an ATP-dependent substrate binding and release cycle combined with client protein recognition, which is typically rather promiscuous. The answer to this apparent conundrum lies in the fact that Hsp70s do not work alone, but rather as “Hsp70 machines”, collaborating with and regulated by a number of (co)chaperones and cofactors. Here, using examples from yeast and human, we discuss several such factors, particularly concentrating on how the array of J-proteins (also known as Hsp40s) orchestrates Hsp70 functions.
The core Hsp70 machinery
Hsp70s have never been found to function alone. Invariably they have been found to require a J-protein and, almost always, a nucleotide exchange factor (NEF) as partners. These co-factors are key, as they regulate Hsp70’s binding to client proteins by affecting Hsp70’s interaction with nucleotides.
The Hsp70 cycle
Hsp70 interaction with clients is profoundly affected by interaction with nucleotide. The 40kDa N-terminal adenine nucleotide-binding domain regulates the conformation of the 25 kDa C-terminal peptide-binding domain (PBD), which binds to a 5 amino acid segment of polypeptide clients that is enriched in hydrophobic residues4, 5 (Figure 2). Through ATP hydrolysis and nucleotide exchange reactions, Hsp70 cycles between ATP- and ADP-bound states, which differ dramatically in their interaction with client protein. On- and off-rates for client binding are very rapid in the ATP state and very slow in the ADP state. Thus engagement with clients is very fast in the ATP-state, but hydrolysis must occur to stabilize client interaction. However, the spontaneous transition between the two states is extremely slow, as Hsp70’s basal ATPase activity is low and typically nucleotides bind stably. Thus, Hsp70 function requires co-factors: ATPase activity is stimulated by J-proteins, facilitating client capture; dissociation of ADP is stimulated by NEFs, fostering client dissociation, consequently “recycling” Hsp70 molecules. We refer to this triad as the core machinery.
Figure 2. Canonical model of mode of action of core Hsp70 machinery in protein folding and Hsp70 structure.
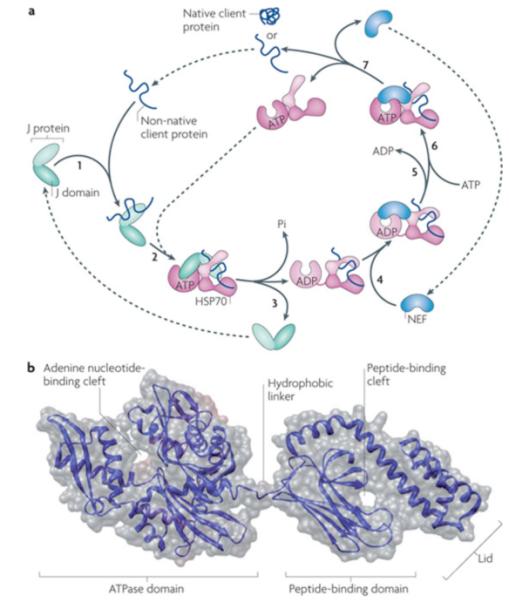
a|Canonical mode of functioning of the Hsp70 core machine based on in vitro refolding studies of denatured proteins. [1] J-protein binds to client protein via its peptide-binding domain and [2] interacts with Hsp70(ATP) via its J-domain (J). [3] The client rapidly, but transiently, interacts with the “open” peptide binding site of Hsp70. ATP hydrolysis is stimulated by both the J-domain and client causing a conformational change in Hsp70 closing the helical lid over the cleft, stabilizing client interaction. J-protein leaves the complex. [4] Nucleotide exchange factor (NEF), which has a higher affinity for Hsp70(ADP) than Hsp70(ATP), binds Hsp70; [5] ADP dissociates through distortion of the ATP binding domain, after which [6] ATP binds to Hsp70. [7] Client is released because of its low affinity for Hsp70(ATP). ATP-binding to Hsp70 is favored since cellular ATP concentrations are typically much higher than those of ADP. If the native state of the client is not attained upon release, J-protein rebinds to exposed hydrophobic regions and the cycle begins again. Typically such reiterative binding is required. b| Hsp70 structure with ADP bound to the nucleotide binding domain135 (PDB code #2KHO). The ATPase domain and peptide-binding domain are connected via a short flexible linker. These domains dock when in the ATP-bound state, which is also thought to displace the lid, allowing easy access and egress of the client protein from the cleft 17, 136.
Canonical model: J-protein delivery and NEF-driven release
Much of the early biochemical work on Hsp70 function focused on the ability of the core machinery to facilitate the folding of denatured protein (e.g. luciferase) in vitro. This body of work established the “canonical model” of function of Hsp70 machinery6-8. This model (Figure 2A) has two key features, in addition to those described above. First, initial binding of the unfolded client protein by the J-protein serves to prevent its aggregation and to “deliver” it to Hsp70, in addition stimulating Hsp70’s ATPase activity. Second, dissociation of the client caused by NEF action allows the client the opportunity to fold into its active conformation. If folding is not yet complete, clients can re-bind such that reiterative cycles of client binding and release occur. In the simplest scenario, this cycling allows productive folding merely because aggregation is prevented during the time the client is bound. This canonical mechanism of function for the core Hsp70 machine has clearly been shown to be valid in vivo in some cases. However, sometimes the mode of action of the machine is simpler. More often it is more complicated and diverse, as we describe in this Review.
Importance of “regulation” of the nucleotide cycle
It has become apparent that fine-tuning of the cycle of Hsp70 interaction with client proteins is crucial. Overstimulation of ATPase may prevent capture of clients; an excess of NEF activity may cause premature release of captured clients. In vivo both cofactors are typically present in sub-stochiometric amounts relative to their partner Hsp70. In vitro 10-20 times lower concentrations of J-protein than Hsp70 is most efficient for stimulation client protein folding9 and raising J-protein levels in cells without a concomitant rise in Hsp70s generally reduces refolding (Hageman and Kampinga, unpublished observations). Analogously, folding efficiency declines when NEF concentrations increase above optimal NEF:Hsp70 ratios in vitro10 and over expression of a NEF in cells reduces folding capacities of the Hsp70 machine11.
J-proteins drive Hsp70s’ multifunctionality
Whereas it is possible to imagine that the versatility of Hsp70 function could be achieved primarily through the amplification and diversification of Hsp70 genes during evolution3, this does not seem to be the case. The number of Hsp70s in each cell is rather limited. However, J-proteins often far outnumber the number of Hsp70s and NEFs in a cellular compartment12, 13. For example, in mammals, one type of Hsp70 in mitochondria and one type in the endoplasmic reticulum, while these organelles have 4 and 6 J-proteins, respectively12, 13. Overall, humans have only 11 Hsp70s and 13 NEFs, but 41 J-proteins (Figure 3: Supplemental Figure 1). In addition, and in contrast to Hsp70, J-proteins show a large degree of sequence and structural divergence (Figure 3), consistent with the idea that they play a major role in driving the multifunctionality of the Hsp70 machinery.
Figure 3. Diversity in domain architecture of J-proteins from the yeast S. cerevisiae and H. sapiens.

Members are clustered according to known or presumed client binding ability. Functional orthologs are connected by lines. For clarity, not all known domains are indicated and some differences between yeast and human orthologs are not indicated. For more detailed information see Supplementary Tables I, II, where information about the presumed localization and function of all members is also provided. Regions of Class I and II J-proteins: G/F regions, of which the functional relevance is disputed (see main text) are defined as segments containing more than 5 glycines or/and phenylanines in the first 25 amino acids C-terminal to J-domain. C-terminal domain 1 (CTD1) - canonical type I members have Zinc finger like region (ZFLR); type II members lack a ZFLR. However, type II often have cysteine rich stretches and/or binding site for Histone Deacetylases (HADCs). The indicated dimerization domain (DD) has been firmly established only for a few type I and II (Ydj1, DNAJA1, Scj1, Sis1); for the others this domain is presumed for simplicity. “X” in DNAJB13 indicates the lack of the canonical HPD motif in the J-domain. For reasons of space, some members are not drawn at full size (indicated by numbers); //..// indicate stretches not shown.
Diversity of J-protein structure
While the J-domain is both the defining domain and the key to functional interaction with Hsp70s by stimulating their ATPase activity12, 13. However, many J-proteins bear little if any sequence or structural similarity outside this defining domain (Figure 3 and Supplemental Table I,II). Accumulated data indicate that J-proteins serve as primary determinants of Hsp70 function via the action of their non-homologous domains.
J-domain – the common denominator
J-proteins, by definition, contain a conserved ~70 amino acid signature region, the remarkably conserved J-domain, which is named after the founding member, the Escherichia coli protein, DnaJ (Figure 4A). Particularly conserved are a histidine, proline and aspartic acid tripeptide (HPD) in a loop between the two main helices (II and III). This HPD motif is critical for J-domain function. The J-domain has the crucial role of stimulating Hsp70’s ATPase activity. The exact mechanism of J-domain stimulated ATPase activity and the ensuing conformational changes resulting in stabilization of client interaction remain a matter of debate. However, it is established that exposed residues of the J-domain form an Hsp70 interaction surface14, 15. Crucial interactions occur with both Hsp70s ATPase domain and the adjacent flexible region, which links it to the client protein-binding domain (Figure 2). These interactions are critical for transmitting the conformation change necessary for closing the peptide-binding pocket15-20.
Figure 4. J-domain and client protein binding domain structures.
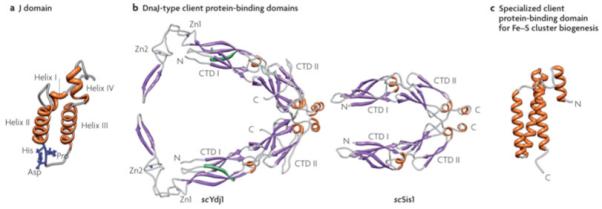
a| J-domains contain four alpha-helices, with the central ones forming a coiled-coil motif around a hydrophobic core137. The invariant HPD tripeptide located in the loop between helices II and III is critical for ATPase stimulation and in vivo function12. Residues in helix II and within the neighboring loop, including the HPD, form an Hsp70 interaction face. b| Class I Ydj1 and Class II Sis1 have similar client protein binding domains, the “DnaJ-type”. The structure of amino acids 102-350 of Ydj126, 29 and amino acids 180-343 of Sis130 are shown. Both have J-domains at their N-termini, followed by a glycine/phenyalanine-rich (G/F) region. No full-length structure of a Class I or Class II J-protein has been obtained, presumably because of the flexibility of the G/F regions. Both Ydj1 and Sis1 are dimers, with the dimerization domain at their C-termini (C). Each monomer of Ydj1 and Sis1 has two adjacent domains that are similar structure, being predominantly composed of beta-sheets. These are often referred to as “C-terminal domain” (CTD1) and CTD2. Ydj1 (like DnaJ) has two zinc fingers (Zn1 and Zn2), which extend out from CTD1. In addition, Ydj1 has a CAAX motif for farnesylation, a modification important for membrane localization and binding of some client proteins138 139. c|Jac1 (called HscB in E. coli), which is critical for Fe-S cluster biogenesis, has a specialized client protein binding domain that has neither sequence nor structural similarities to a DnaJ-type140; amino acids 63-171 of HscB are shown. The face pointing outwards interacts with the Fe-S cluster scaffold Isu141.
J-protein groups, structure and classification
Despite the omnipresent J-domain, J-proteins, as a group, are strikingly dissimilar, having a wide variety of additional domains (Figure 3, Supplemental Table I,II). Historically, J-proteins have been divided into three classes (I, II, and III; or A, B and C)13 21 22 23 with class I (A) designation based on the motifs/domains present in DnaJ of E. coli. Thus, by definition, Class I J-proteins have an N-terminal J-domain, followed by a glycine/phenylalanine (G/F)-rich region, four repeats of the CxxCxGxG type zinc finger, and a C-terminal extension, now known to bind client proteins24-26. This type of C-terminal region is composed of two barrel topology domains, CTDI and CTDII. CTDI has a hydrophobic pocket in which client protein is thought to bind, as well as the zinc-finger domain extruding from it, which also may be involved in substrate binding27, 28 (Figure 4B). The extreme C-terminus is a dimerization domain, and thus serves to increases affinity for clients29. Proteins were classified as Class II (B) members if they had N-terminal J-domain with an adjacent G/F region, but lacked the zinc-finger domain. Any J-protein that did not have a structure that fit the Class I or II classification were designated class III.
It must be emphasized that this historical classification does not edify the biochemical function or mechanism of action of group members. In fact, both within and between subclasses II and III large structural and functional diversities exist (Figure 3, Supplemental Tables I and II). In particular, the importance of client binding, a crucial feature of many J-proteins, is minimized by this classification system. Some class II J-proteins, such as yeast Sis1, have client-protein binding domains that, although they show little sequence identity, are strikingly structurally similar to that of the class I yeast protein Ydj126, 30 (Figure 4B). For clarity, we will refer to such client protein binding domains as “DnaJ-like” throughout. On the other hand, some class II proteins appear to have no client-binding domain at all, while many, likely the majority, of members of class III do have domains that bind clients. However, data to date reveal no structural similarity of any Class III J-proteins to the DnaJ-like domain. Rather, all appear to bind one, or perhaps at most a handful, of clients12 (Figure 4C). Finally, the presence (type II) or absence (type III) of the G/F region has led to ambiguity, as both the definition and function of this region is ill defined. Besides serving as a linker between domains, the functional importance of the glycine-rich region is in question. In cases where it has been shown to be important, as in the function DnaJ of E. coli and Sis1 of S. cerevisiae, the specific sequences found to be critical were either peripheral to or insertions in the glycine-rich segment31, 32.
In sum, whereas these classifications may be helpful for nomenclature purposes33, they should not be taken as informative regarding functionality. Indeed, the diversity of J-proteins have led to complicated and often confusing nomenclatures. Here, for human HSP we will use the recent NCBI accepted nomenclature33, indicating where appropriate commonly used (nick)names (in brackets) as well. The yeast protein nomenclature used is based on gene names established in the Saccharomyces Genome Database. To underscore whether discussing a yeast or a human protein we use the prefaces “sc” and “h”, respectively.
J-protein function without client binding
Despite the functional complexity of J-proteins, for perspective, it is important to note that the presence of only a J-domain may be sufficient for some cellular functions (Figure 5). Such is particularly the case, if the domain is localized to a particular site within a cellular compartment (Figure 5B). Such positioning serves to maintain a high local J-domain concentration, thus targeting Hsp70 to particular client proteins at these sites, without need for direct J-protein interaction with the client itself (Figure 5A,B).
Figure 5. J-protein function with or without client binding.
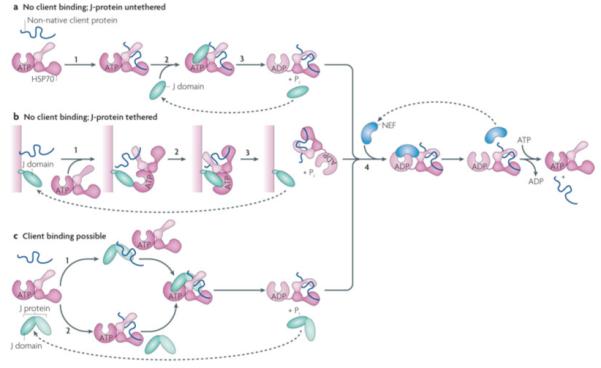
J-proteins can act without binding to clients, either untethered (a) or tethered to a particular site in the cell (b). In the case of J-proteins having client protein binding domains, clients may bind either bind the J-protein before interaction with Hsp70 or only serve to stimulate Hsp70’s ATPase activity (c) a| The simplest J-protein function is the action of a J-domain in the absence of a client protein binding domain to stimulate the ATPase activity, allowing it to capture a client protein that has transiently entered the open peptide-binding cleft of Hsp70(ATP) (i), by binding Hsp70 (ii) and stimulating ATP hydrolysis (iii). In such cases, Hsp70 is the driving force of client protein interaction, as there is no facilitation by the J-protein, either through direct binding or by subcellular localization. The in vivo function of a J-domain lacking other sequences has been demonstrated experimentally44. b|J-domains either lacking (shown) or having (not shown) client protein binding domains are often tethered to a site rich in client proteins. In this way, upon initiation of client protein binding by Hsp70(ATP) (1), a high concentration of J-domains is present (2) facilitating ATP hydrolysis and thus client capturing by Hsp70 (3). c|In the case of J-proteins having client protein binding domains, two modes of J-protein function can occur: top: as in the canonical model of J-protein:Hsp70 function J-protein binds client first (x) and targets it to Hsp70 (y,z); bottom: binding occurs directly to Hsp70 (x’) as described in a); in such cases J-proteins stimulate Hsp70 ATPase activity only even though a client binding domain is present (y’,z). Evidence for such an alternative pathway has been found in the mitochondrial Fe-S cluster biogenesis pathway in yeast with the specialized J-protein Jac1 and Hsp70 Ssq1142. In all cases, release of client is facilitated by NEFs (iv).
In the simplest cases, a J-protein consists of little besides the J-domain and sequences required for localization. For example, via a single transmembrane domain the J-domain of scHlj1 is positioned at the cytosolic face of the ER membrane, where it recruits soluble cytosolic Hsp70s to assist in the degradation of proteins exiting the ER (that is ER-associated degradation, ERAD)34. J-proteins tethered near the polypeptide exit site of the ribosome35 are another example of Hsp70 recruitment to a site having a high concentration of clients (Figure 6). In this case positioning ensuring prevention of nascent chain aggregation and facilitating folding. Fungi have a specialized ribosome-associated Hsp70, independently tethered to the ribosome36; however, higher eukaryotes rely on the ribosome associated J-protein (DNAJC2) for ribosomal recruitment of the soluble hHSPA8 (also known as Hsc70), which itself has no intrinsic affinity for the ribosome37, 38(Figure 6).
Figure 6. Example of tethering of J-proteins to site of action.
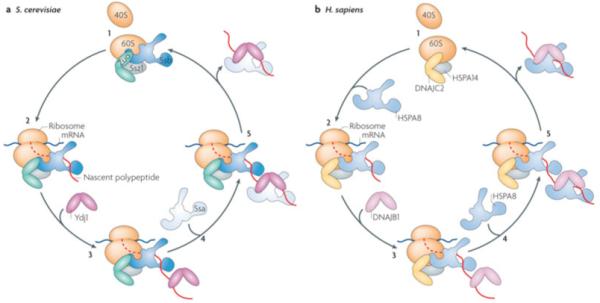
Ribosome associated chaperones, an example of tethering to a site within a cellular compartment. All eukaryotes, illustrated by budding yeast (left) and humans (right), have a ribosome associated J-protein that binds near the exit site of the 60S subunit, regardless of whether translation is occurring or not37 (i). Fungi, though not other eukaryotes, also have a specialized ribosome-associated Hsp70 (Ssb) that independently associates with the 60S subunit. Zuo1 and Ssb function as a J-protein:Hsp70 pair upon emergence of a nascent polypeptide from the ribosome36 (ii). Later events in polypeptide folding, include binding of soluble J-proteins (iii) and recruitment of soluble Hsp70 (iv) prior to completion of translation and after nascent chain release (v). Herefore, yeast utilizes the abundant soluble J-protein (Ydj1) and Hsp70 (Ssa). In humans, the ribosome-associated J-protein (DNAJC2) recruits HSPA8 as a partner (ii), which partners with the soluble DNAJB1 in downstream folding events (iii-iv). Note, both Zuo and DNAJC2 form a stable heterodimer with the unusual Hsp70 (scSsz1 or hsHSP70A14) (gray oval). The function of this Hsp70, which is not known to have client protein binding activity, beyond being important for the ability of Zuo1 to stimulate the ATPase activity of its Hsp70 partner Ssb20 is not known.
A J-domain positioned at the translocon of the inner mitochondrial membrane responsible for translocating polypeptides from the cytosol into the matrix is another example of a “minimal J-protein”. The Hsp70 machinery, of which this J-protein (scPam18 or hDNAJC19) is a part, is not involved in protein folding, rather it drives the movement of polypeptides through the translocon. However, the core Hsp70 machinery, scPam18/hDNAJC19, the mitochondrial Hsp70 (scSsc1/hHSPA9) and the NEF (scMge1/hBAP), that forms this “import motor” follows the basic biochemical rules for Hsp70 machines described above. In the process of driving polypeptide import39: (1) Hsp70 binds to exposed hydrophobic sequences in unfolded translocating polypeptide; (2) the J-protein stimulating its ATPase activity to enhance interaction with the client and (3) the NEF effecting nucleotide release, and thus dissociation of the client. In addition to the matrix-localized J-domain, scPam18/hDNAJC19 consists of only a transmembrane domain, which serves to localize it in the mitochondrial inner membrane, and a short intermembrane space domain, which directly interacts with the translocon40, 41. Ssc1/HSPA9 is independently localized to the translocon42, 43. Thus in this case, and others in which the Hsp70 partner is localized (such as fungal ribosome-associated Hsp70s) the tethered J-domain is not necessary for recruitment of Hsp70 per se. Beyond resulting in high local concentrations of both components, dual localization also serves to precisely position the J-domain juxtaposition to Hsp70 (Figure 5B) for optimal ATPase stimulation. This precise positioning likely results in exquisite modulation of the interaction of Hsp70 with client protein (i.e. translocating polypeptide in the case of the import motor).
Remarkably, for some cellular functions, the J-domain - that is the ability to stimulate Hsp70’s ATPase activity - may be sufficient, even without sub-compartment localization (Figure 5A,C). This functional robustness is illustrated by the ability of solely the J-domain of several cytosolic J-proteins to rescue the severe defects caused by the absence of Class I scYdj1, the most abundant J-protein of the yeast cytosol44. Surprisingly, this rescue occurs even when a J-domain is expressed at normal scYdj1 levels. This and other observations, also underscore the idea that little if any specificity resides in J-domains themselves45. Perhaps, facilitation of folding of some newly synthesized proteins by cytosolic Hsp70 needs a J-protein only for stimulation of ATP hydrolysis, but not the direct binding to prevent aggregation or increase the probability of its interaction with Hsp70. However, it should be noted that this J-domain rescue of the effects of the absence of Ydj1 is not complete. As discussed below, client protein binding by J-proteins serves many important functions.
J-protein function with client binding
In many other functions of the core-machine, J-protein client binding is crucial (Figure 3). These client protein-binding functions are not restricted to preventing aggregation or supporting folding, but include protein degradation and remodeling of folded proteins as well. As illustrated below, J-proteins play a directive role in these cases.
Client protein binding for folding
Both in vitro and in vivo results strongly indicate an important role for J-proteins having the DnaJ-like fold in de novo protein folding (Figure 2, 5C) 25, 46. However, some results appear to challenge the importance of client protein binding per se in the function of these proteins in vivo: 1) a J-domain alone can substantially substitute for scYdj1 as described above44 and 2) complete deletion of the client protein binding domains of either scYdj1 or scSis1, the other abundant J-protein of the yeast cytosol having a DnaJ-like client protein binding domain (Figure 3, Supplemental Table I), had little phenotypic effect47. However, mutants lacking the C-terminus of both scYdj1 and scSis1 are non-viable47, implying that the client binding ability of these cytosolic Hsp40s is an essential function in vivo. Thus, functional overlap between J-proteins can mask the importance of client protein binding. This overlap is particularly remarkable, as the peptide binding domains have limited sequence homology (29% identity).
Another demonstration of client binding as an essential feature of J-protein function comes from studies on mammalian hDNAJB11 (also known as ERdj3), an ER luminal J-protein with a DnaJ-like peptide-binding domain. Even in the absence of an active J-domain, hDNAJB11 can bind directly to several nascent, unfolded and mutant secretory pathway proteins, implying that this binding is Hsp70-independent. As predicted from the canonical folding model, after client binding, Hsp70 joined the complex, which led to hDNAJB11 client dissociation before protein folding was completed48. So, hDNAJB11 seems to prevent substrate aggregation and ‘presents’ the client for Hsp70-dependent folding. For its own release, hDNAJB11 must recruit HSPA5 (also known as BiP) and stimulate its ATPase activity to convert HSPA5 into its high-affinity state for clients49.
Client protein binding for degradation
Besides facilitating protein folding, several J-proteins have been found to have specific functions in preventing aggregation and/or shunting clients towards degradative pathways. For example, hDNAJB6 and hDNAJB8, two closely related J-proteins, were identified as very potent inhibitors of aggregation and the associated toxicity of polyglutamine containing proteins50-53. Both client binding and prevention of aggregation was completely dependent on a serine-rich stretch in the C-terminus named the SSF-SST region50. This region shows no obvious sequence similarity to the canonical DnaJ-type client protein-binding domain, and thus is likely a novel client binding domain. The SSF-SST region serves at least two other functions as well. First, it is involved in forming higher ordered structures. In contrast to J-proteins having the canonical DnaJ-like client-binding domain, hDNAJB6 and hDNAJB8 do not form dimers, but rather exist in vivo as polydispersed complexes. Their formation is dependent upon this region. Second, the SSF-SST region is required for interaction with several histone deacetylases (HDACs). Intriguingly, deacetylation of two C-terminal lysines in hDNAJB8 is functionally important in vivo50. How clients bound to hDNAJB6 or hDNAJB8 are handled remains to be elucidated; intriguingly, however, besides acting on polyglutamine containing proteins, both proteins are also able to inhibit aggregation of poly-Q peptides (E. Reits, personal communication). Such peptides, which may arise from proteasomal cleavage of full-length polyglutamine containing proteins, are highly aggregation-prone and have been implied as possible seeds in poly-Q aggregation. Together, this suggests the possibility that hDNAJB6 or hDNAJB8 maintain aggregation-prone peptides in a form competent for peptidase degradation.
Some J-proteins contain well-defined domains that specifically drive their clients toward degradation. hDNAJB2 (also known as Hsj1) is closely related to hDNAJB6 and hDNAJB850, but contains two ubiquitin-interacting motifs (UIMs) C-terminal to its putative client-binding domain, which are not present in the other proteins54 (Figure 3). hDNAJB2 not only efficiently targets misfolded targets, such as poly-glutamine containing proteins, for degradation55, but also antagonizes refolding of heat denatured luciferase mediated by hHSPA1A (also known as the stress-inducible Hsp70) and hDNAJB1 (also know as Hsp40)56 by increasing luciferase ubiquitylation. Both the UIMs and the J-domain are required for this action of hDNAJB2, indicating an Hsp70-dependent function57(Figure 7A). This observation clearly illustrates that the fate of clients can be primarily determined by the J-protein, rather than by the Hsp70 component of a core machinery, as the same clients may either fold or be degraded, depending on whether they interact with hDNAJB1 or hDNAJB2. In these studies, hDNAJB8 could also prevent aggregation of heat-unfolded luciferase, but could not support refolding; unlike hDNAJB2, however, refolding was not, or only marginally, inhibited by hDNAJB8, further suggesting functional differentiation based on J-protein diversity. However, it must be stressed that -besides directed targeting-fates also depend on the client itself. For example, in the case of non-foldable clients such as polyglutamine rich proteins, hDNAJB1 can support client degradation50, 58, 59. However, in such cases, degradation is likely facilitated in a stochastic manner via perpetual cycles of client loading onto and release from Hsp70. That is, the client is simply keep competent for eventual degradation (Figure 1).
Figure 7. Examples of J-protein function beyond protein refolding.
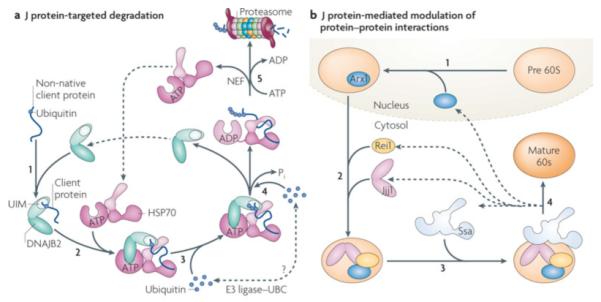
a| J-protein targeted degradation. The ubiquitin-interacting motifs (UIMs) in DNAJB2 (HSJ1) recognizes clients that contain a mono- or poly-ubiquitin moiety (indicated by small yellow circle) (i). After transfer of the client to Hsp70 (ii), E3 ligases (such as CHIP) and the Ubc ubiquitin conjugation machinery can associate with the Hsp70/DNAJB2 complex (the precise manner and specificity of this associations remains to be understood) leading to further ubiquitination of the bound client (iii; iv). After the canonical ATP hydrolysis step (iv) and NEF-mediated nucleotide exchange (v), the poly-ubiquitinated client released from Hsp70 (iii,iv) is transferred to the proteasome for degradation54 55. b| J-protein mediated modulation of protein:protein interactions. Alteration of interactions between mature, folded proteins typically are part of complex biological processes. Illustrated here is the role of the J-protein Jjj1 in the biogenesis of the 60S ribosome subunit, the destabilization of the biogenesis factor Arx1 in the biogenesis of 60S65-67. Arx1 is loaded on the pre60S subunit (beige) in the nucleus (i). Jjj1 binds directly to the ribosome, as does Rei1, another cytosolic factor required for Arx1 destabilization, and with which Jjj1 interacts (ii). Jjj1 partners with soluble Ssa (another example of targeting of Hsp70 by J-protein localization, see Figures 5c and 6). Once the Arx1:pre60S subunit interaction is destabilized, a step needed to generate the mature subunit (red), Arx1 is transported back into the nucleus (iv), where it engages in another cycle of subunit biogenesis.
More complex examples than simply the presence of UIMs also exist. hDNAJC10 (also known as ERdj5) of the ER lumen not only has a putative peptide binding domain, but also protein-disulfide isomerase and thioredoxin domains60, 61 (Figure 3, Supplemental Table I). In addition, hDNAJC10 is also able to interact with EDEM (ER degradation–enhancing a-mannosidase–like protein). It cleaves aberrant disulfide bonds in EDEM clients, in an Hsp70-independent manner. Recruitment of the ER-resident Hsp70 (hHSPA5, also known as BiP) via the J-domain is needed for client release and transfer to components of the ER-associated degradation pathway (P97/VCP) for retrotranslocation to the cytosol and subsequent degradation by the proteasome62.
Working on folded client proteins
It is becoming apparent over time that mature, folded proteins are J-protein clients much more commonly than previously appreciated. Such functions often involve the “remodeling” of large multiprotein complexes, affecting the stability of protein–protein interactions. Ironically, this was the first identified function for the DnaK-DnaJ machinery in E. coli: for initiation of lambda phage DNA replication in E. coli, DnaK and DnaJ mediate the destabilization of the lambda P protein from the initiation complex, causing activation of the DNA helicase DnaB63, 64. While lambda replication involves the multifunctional DnaJ, in eukaryotes similar roles are often carried out by J-proteins that specialize in a single function. scJjj1 (hDNAJC21) and Swa2 (hDNAJ6, also known as auxillin), are both conserved, structurally complex J-proteins of the eukaryotic cytosol that destabilize protein–protein complexes. scJjj1 is required for one of the final steps in formation of the 60S ribosomal subunit65, 66. In collaboration with the soluble constitutive Hsp70 (scSsa or hHSPA8) and scRei 1, another cytosolic protein with which it interacts, scJjj1 facilitates the destabilization of two biogenesis factors, allowing them to recycle back to the nucleus67(Figure 7B). Auxilin (scSwa2 or hDNAJC6), working with the same Hsp70 family member, is required for the uncoating of clathrin-coated vesicles, carrier of lipids and proteins between the plasma membrane and Golgi network. These vesicles are encased in an intricate “lattice” made up of clathrin trimers68, which interdigitate with one another, forming a very stable structure. Thus, this lattice must be destabilized, that is uncoated, which entails dissociation of trimers from the lattice, likely by rotation of one trimer relative to another68. Both scSwa2/hDNAJC6 and scSsa/hHSPA8 specifically bind clathrin, but at different sites69-71, and as a J-protein:Hsp70 pair facilitate trimer release from the lattice72.
Specialized J-proteins having folded proteins as clients can also play roles other than destabilization of multimeric complexes. In one case, chaperones appear to facilitate partial unfolding of the client. The ubiquitous mitochondrial J-protein scJac1 or hDNAJC20 is required for the biogenesis of Fe-S clusters73, 74, moieties critical for function of many proteins, including those in the electron transport chain. In yeast, clusters are assembled on a scaffold protein, scIsu, and then transferred to recipient proteins75. scIsu is the client protein of both scJac1 and the Hsp70 with which it functions, with chaperone action being required for the transfer, but not the assembly, of the cluster76-78. In the simplest model, chaperone binding serves to facilitate partial unfolding of scIsu, thus “releasing” the cluster. Such a model is supported by recent observations that Isu from E. coli attains several conformations in vitro, with one being significantly disordered79. Thus binding of the J-protein:Hsp70 machinery may favor the disordered over the ordered conformation of Isu, and in so doing favor cluster transfer. This system also provides one of the very few examples of the evolution of a specialized Hsp70. A small subset of fungi have evolved a second, specialized mitochondrial Hsp70 that functions with Jac1; like Jac1, this specialized Hsp70 (scSsq1) functions only in cluster biogenesis, having scIsu as its only client protein80. But interestingly, the vast majority of eukaryotes, including humans, utilize the multifunctional HSPA9 (also known as GRP75) of the mitochondrial matrix in Fe-S cluster biogenesis80. In these organisms, the specificity for the system is driven by the J-protein.
Hsp70-independent J-protein function?
Besides functioning within the context of the core machine, some J-proteins have evolved functions that largely do not require their J-domains and are thus independent of Hsp70s. Such cases uncovered so far involve either prevention of aggregation or modulation of stability of protein complexes. For example, the ability of hDNAJB6 and hDNAJB8 to prevent accumulation of aggregated polyglutamine proteins, as described above, does not depend on their J-domains; variants having alterations in or complete deletion of the J-domain were only slightly impaired in preventing aggregation. The slight impairment was merely due to a defect in supporting degradation of the unfolded, soluble polypeptides that required interaction with Hsp70, leading to the conclusion that aggregation prevention did not require Hsp7050.
scCwc23 serves as an extreme example of J-domain dispensability in the modulation of protein:protein interactions. scCwc23, an essential protein with an N-terminal J-domain (table I), is required for disassembly of the spliceosome81, 82. Dissasembly is an essential process, as the components must be reassembled for each pre-mRNA that is spliced. While alterations in the extreme C-terminus of scCwc23 causes a global defect in pre-mRNA splicing; complete deletion of the J-domain has no effect. The J-domain is functional, but its role is revealed only when the interaction between two other components required for spliceosome disassembly, scPrp43 and scNtr1, are affected. As scCwc23 interacts directly with scNtr183 via its C-terminus81, it is thought that this region is sufficient for its essential role in destabilization of the spliceosome. Likely the J-domain fine-tunes destabilization of spliceosomal components, but its absence is only noticeable when interaction amongst other proteins involved in the disassembly process are functionally compromised.
How did such J-domain/Hsp70 independent functions of J-proteins evolve? One can envision two extreme possibilities: (1) J-proteins have gained domains, making them more complex, and in some cases the added domains surpass the J-domain in functional importance and/or (2) proteins that independently functioned in certain cellular processes gained a J-domain, bringing the hsp70 system to fine-tune an existing function. In at least one instance, J-domain function may have been lost during evolution. The J-protein RSP16 plays a role in flagellar stroke movement in Chlamydomonas84, but a fragment lacking the J-domain appears to function as well as full-length protein in regulation of flagellar beating85. Interestingly, although RSP16 has the defining HPD motif, its orthologs from human (hDNAJB13), zebrafish, mouse and mosquito do not, raising the possibility of an increase in functional importance of non-J-domain portions of the protein during evolution, with a concomitant loss or decrease in J-domain function.
NEF and Hsp70 multifunctionality
Although we argue above that J-proteins are the prime drivers of the Hsp70 functional diversity, NEFs may play a role as well. Unlike J-proteins, which have a common domain responsible for effects on Hsp70’s nucleotide cycle, four different types of NEFs have been identified (Supplemental Figure 1B). No sequence similarity exists among them. Although they all interact with Hsp70’s ATPase domain, destabilization of nucleotide is accomplished in mechanistically distinct ways86-88. Some appear to function only in nucleotide release, but others have additional domains. Clear insight into how these other domains affect Hsp70 function and relate to NEF activity is still lacking.
Acting only as NEFs
Two types of NEFs, the GrpE- and HspBP1/Bap-type, appear to have no domains in addition to those involved in nucleotide release. Bacteria and mitochondria have a single NEF, the GrpE-like type, which functions with resident Hsp70s in functions from protein folding to protein translocation89-92. The cytosol and ER contain hHspBP1 (scFes1) and hBap (scSls1p/scSil1p), respectively93-96. As with GrpE-types, the HspBP1/Bap-type appears to support ‘classical’ chaperone actions of Hsp70 machines, from stress-related protein refolding reactions to ER associated protein degradation (ERAD)93, 97.
The Hsp70-like NEFs
The Hsp110 (hHSPH1-4/scSse1-2 and scLhs1) proteins were initially grouped as Hsp70 family members because of similarities in sequence. Like Hsp70s, they consist of an N-terminal ATPase domain that is connected to peptide binding domain (in this case, a nine-stranded β sandwich) via a flexible linker similar to that of Hsp70’s98, 99(Supplemental figure 1). Interestingly, at least some Hsp110s, like Hsp70s, can bind unfolded proteins and prevent their aggregation99, raising the possibility that the interacting Hsp70 and Hsp110 chaperone pair act in concert, both binding client proteins. However, although Hsp110 proteins do possess an ATPase activity, unlike the canonical Hsp70s, they cannot not employ a nucleotide-dependent, peptide-binding release cycle86. Therefore, in terms of their chaperone activity, their interaction with client proteins cannot be modulated, that is Hsp110’s can only act as “holdases”. Both yeast and humans contain stress-inducible Hsp110 members (scSse2 and hHSPH1, respectively), raising the possibility that they may “store” partially denatured clients under conditions of stress and then, using their NEF activity, help Hsp70 facilitate their refolding86, 87, 98, 100-102.
The Bag family of NEFs
The Bag-type NEFs are the most complex, both in terms of the numbers and diversity of sequence. All contain a so-called “Bag” domain, an ~85 amino acid region that can interact with Hsp70’s ATPase domain103. While yeast has a single Bag-type NEF (scSnl1), humans have six members (hBAG-1, hBAG-2, hBAG-3 (also known as CAIR-1/Bis), hBAG-4 (also known as SODD), hBAG-5, and hBAG-6 (also known as Scythe/BAT3)(Supplemental Figure 1B). All are found in the cytosol and/or nucleus. Specific interaction of four of these (1, 2, 3, and 6) with Hsp70 has been experimentally verified103. But how the different members function in relation to Hsp70 beyond their NEF activity remains unclarified. Intriguing hints exist, but no clear picture emerges. The most obvious domain, besides the defining Bag domain, found in some family members (hBAG-1 and hBAG-6) is an ubiquitin-like domain (Ubl). The Ubl may serve to sort them to and associate with the proteasome104, 105. Another member, hBAG-3, has also been implicated in protein degradation. However, hBAG-3 has no Ubl and seems to support protein degradation via (macro)autophagy rather than via the proteasome. This action does not appear to be Hsp70-specific. Rather, it is dependent on the small HSP family member HSPB8, with which it forms a stoichiometric complex106, 107. Therefore, although hBAG-3 also associates with Hsp70s in vivo, the precise role of hBAG-3 as NEF for Hsp70 is not yet understood.107. In summary, there is yet limited, if any, evidence that NEF activity or the mechanism by which nucleotides are released drive the specificity of Hsp70 machines.
Beyond the core Hsp70 machinery
It is important to keep in mind that, in part, the functional diversity of Hsp70 machinery depends on partnerships with other chaperone systems and fine-tuning by a set of co-factors, as outlined below.
Chaperone Partnerships
Often Hsp70 machines do not act alone, but rather in concert with other chaperone machines (Supplemental Figure 2). Such partnering does not necessarily entail physical interactions between the different machineries; rather, “networks” of chaperone activity exist, driven by the affinity of partially folded proteins for particular chaperone systems. For example, in the folding of nascent polypeptides, Hsp70s act upstream, binding the more extensively unfolded clients, prior to their interaction with chaperonins108, 109. But, adaptor proteins that interact directly with different chaperone machines and act as physical bridges between them also exist. Such is the case with the Hsp70:Hsp90 chaperone network, which is critical for final maturation steps of certain clients, for example hormone receptors and some transcription factors. The transfer of clients from Hsp70 to Hsp90 is facilitated by a TRP-repeat protein, hHOP (also known as scSti), which binds to both chaperones110, 111. These and other partnerships, such as those with small Hsps112, 113, serve to expand the repertoire of Hsp70 function.
Nucleotide cycle regulation beyond the core-machine
Several factors have been identified, which, although not universal and thus not part of the core Hsp70 machinery, affect the Hsp70 ATP/ADP hydrolysis and release cycle. The best studied, hHIP (Hsp70-interacting protein, also known as p48) and CHIP (carboxy terminus of Hsp70 interacting protein), were identified over a decade ago. But, whether or how they may functionally direct Hsp70 dependent reactions in vivo remains elusive. HIP was identified as a protein that preferentially binds to and stabilizes the ADP-bound state of Hsp70114-116. HIP competes with the NEF BAG-1 for binding to Hsp70’s ATPase domain117, thereby slowing down the nucleotide cycle and extending the time during which clients are bound. Consistent with these biochemical properties, increasing Hip concentration generally enhances folding in vitro114 and in vivo118. HIP also facilitates the assembly of Hsp70s into multichaperone complexes with Hsp90119. However, this effect appears independent of interaction with Hsp70, underscoring both the complexity and need for more information to understand Hip function.
CHIP has three distinct domains, a N-terminal tetratricopeptide repeat (TPR) domain and an adjacent charged domain, both of which interact with Hsp70, as well as a C-terminal U-box domain with E3-ubiquitin ligase activity120, 121. This combination of domains lends itself to the hypothesis that CHIP:Hsp70 interaction serves to shunt Hsp70-bound clients to the proteolytic pathway. However, CHIP’s biochemical properties and the results of in vivo experiments suggest a more specific role in targeted degradation of proteins that are clients of Hsp90 122-124 and perhaps a more antagonistic role regarding Hsp70s. First, most clients (e.g. steroids receptors, CFTR, c-ErbB2) that showed accelerated degradation in the presence of elevated CHIP expression are clients of Hsp90. Second, CHIP has chaperone-like activity itself, as it can bind unfolded proteins and prevent their aggregation125, which, depending of the client, may either lead to folding126 or degradation125. Third, CHIP, binding to the ATP-bound form of Hsp70, can act as an inhibitor of the J-protein-stimulated ATPase activity of Hsp70120. This activity neither favors Hsp70’s binding to clients nor stabilization of the client-bound form. So, rather than directing Hsp70-bound clients towards degradation, these data suggest that CHIP, by inhibiting the ability of J-proteins to stimulate Hsp70’s ATPase activity, may inhibit Hsp70’s binding to its own clients, thus promoting their degradation. Alternatively, CHIP’s action on Hsp70 could recruit and maintain Hsp70 in a form able to rapidly interact with CHIP-bound clients, once they have been ubiquitylated, to facilitate their transfer to the proteasome. Clearly, much more work is needed before CHIP function can be discussed with certainty.
Perspectives
Whereas Hsp70 provides the “horse-power” to the core machine, exquisite fine-tuning by co-factors, particularly J-proteins, provides the machine with functional and client specificities. Despite the progress reported here, there is still much to learn at many levels. Understanding the means by which J-proteins, NEFs and other proteins mechanistically alter the functioning of the core machinery, requires insight that can only come from further structural and biochemical work. For example, determination of whether individual J-proteins bind client proteins and if so, their binding specificity, is paramount for a global understanding of this chaperone machinery. Also, more detailed biochemical, cell free experiments using (unfolded) clients and sequential addition of J-proteins (with and without intact J-domains), Hsp70s and NEFs will be needed to gain better insights in the mode of action of the core machine and its dependence of its specific composition.
How to practically manipulate the machinery for clinical intervention is a future challenge. Because of their diversity, J-proteins may be better, more specific targets than Hsp70s. Indeed, manipulations of Hsp70 in mice have had some desirable effects (such as cardioprotection127, 128 and delay in progression of neurodegenerative diseases129-131), but negative effects as well (such as carcinogenesis132, 133). Screening for individual J-proteins that specifically interact with disease-associated misfolded proteins or protein aggregates may be one possible approach to find candidates for selective manipulation of the core machine in a specific disease. In cases where good cellular models of folding diseases are available, comparison of the effects of manipulating expression of the various J-protein on the fate of the disease-associated clients could also lead to productive approaches. Clearly, in addition, more emphasis will need to be placed on generating in vivo transgenic and/or (conditional) knockout models targeting specific J-proteins. In addition, basic information about many human J-proteins is only rudimentary. Thus many challenges and opportunities exist for insights into the fundamental specificities of J-proteins that drive composition and function of the (core) machines, which hopefully will lead to practical, medical applications.
Supplementary Material
Supplementary Material
Acknowledgements
Harm H. Kampinga’s work on J-proteins was funded by Senter Novem (IOP genomics Grant IGE03018), the Prinses Beatrix Foundation (WAR05-0129) and High Q foundation (Grant #0944). Elizabeth A. Craig’s work was funded by National Institutes of Health Grants (GM27870 and GM31107) and the Muscular Dystrophy Association. The authors wish to thank Jure Hageman for his detailed work on the human J-proteins and help with the bioinformatics and Michael Cheetham (UK) for valuable discussions on the functionality and nomenclature of the Human J-proteins.
References
- 1.Ellis J. Proteins as molecular chaperones. Nature. 1987;328:378–9. doi: 10.1038/328378a0. [DOI] [PubMed] [Google Scholar]
- 2.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 3.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–7. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szabo A, et al. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc Natl Acad Sci U S A. 1994;91:10345–9. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarty JS, Buchberger A, Reinstein J, Bukau B. The role of ATP in the functional cycle of the DnaK chaperone system. J Mol Biol. 1995;249:126–37. doi: 10.1006/jmbi.1995.0284. [DOI] [PubMed] [Google Scholar]
- 8.Laufen T, et al. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–7. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamant S, Goloubinoff P. Temperature-controlled activity of DnaK-DnaJ-GrpE chaperones: protein-folding arrest and recovery during and after heat shock depends on the substrate protein and the GrpE concentration. Biochemistry. 1998;37:9688–94. doi: 10.1021/bi980338u. [DOI] [PubMed] [Google Scholar]
- 10.Gassler CS, Wiederkehr T, Brehmer D, Bukau B, Mayer MP. Bag-1M accelerates nucleotide release for human Hsc70 and Hsp70 and can act concentration-dependent as positive and negative cofactor. J Biol Chem. 2001;276:32538–44. doi: 10.1074/jbc.M105328200. [DOI] [PubMed] [Google Scholar]
- 11.Nollen EA, Brunsting JF, Song J, Kampinga HH, Morimoto RI. Bag1 functions in vivo as a negative regulator of Hsp70 chaperone activity. Mol Cell Biol. 2000;20:1083–8. doi: 10.1128/mcb.20.3.1083-1088.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craig EA, Huang P, Aron R, Andrew A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev Physiol Biochem Pharmacol. 2006;156:1–21. doi: 10.1007/s10254-005-0001-0. [DOI] [PubMed] [Google Scholar]
- 13.Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14:1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene MK, Maskos K, Landry SJ. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc Natl Acad Sci U S A. 1998;95:6108–13. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang J, et al. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–33. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281:38705–11. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 17.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suh WC, et al. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–8. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gassler CS, et al. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–34. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang P, Gautschi M, Walter W, Rospert S, Craig EA. The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat Struct Mol Biol. 2005;12:497–504. doi: 10.1038/nsmb942. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham ME, Caplan AJ. Structure, function and evolution of DnaJ: conservation andadaptation of chaperone function. Cell Stress Chaperones. 1998;3:28–36. doi: 10.1379/1466-1268(1998)003<0028:sfaeod>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtsuka K, Hata M. Mammalian HSP40/DNAJ homologs: cloning of novel cDNAs and a proposal for their classification and nomenclature. Cell Stress Chaperones. 2000;5:98–112. doi: 10.1379/1466-1268(2000)005<0098:mhdhco>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hennessy F, Cheetham ME, Dirr HW, Blatch GL. Analysis of the levels of conservation of the J domain among the various types of DnaJ-like proteins. Cell Stress Chaperones. 2000;5:347–58. doi: 10.1379/1466-1268(2000)005<0347:aotloc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goffin L, Georgopoulos C. Genetic and biochemical characterization of mutations affecting the carboxy-terminal domain of the Escherichia coli molecular chaperone DnaJ. Mol Microbiol. 1998;30:329–40. doi: 10.1046/j.1365-2958.1998.01067.x. [DOI] [PubMed] [Google Scholar]
- 25.Lu Z, Cyr DM. The conserved carboxyl terminus and zinc finger-like domain of the co-chaperone Ydj1 assist Hsp70 in protein folding. J Biol Chem. 1998;273:5970–8. doi: 10.1074/jbc.273.10.5970. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Qian X, Sha B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure. 2003;11:1475–83. doi: 10.1016/j.str.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278:44457–66. doi: 10.1074/jbc.M307491200. [DOI] [PubMed] [Google Scholar]
- 28.Kota P, Summers DW, Ren HY, Cyr DM, Dokholyan NV. Identification of a consensus motif in substrates bound by a Type I Hsp40. Proc Natl Acad Sci U S A. 2009;106:11073–8. doi: 10.1073/pnas.0900746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Li J, Jin Z, Fu Z, Sha B. The crystal structure of the C-terminal fragment of yeast Hsp40 Ydj1 reveals novel dimerization motif for Hsp40. J Mol Biol. 2005;346:1005–11. doi: 10.1016/j.jmb.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 30.Sha B, Lee S, Cyr DM. The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure. 2000;8:799–807. doi: 10.1016/s0969-2126(00)00170-2. [DOI] [PubMed] [Google Scholar]
- 31.Lopez N, Aron R, Craig EA. Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+] Mol Biol Cell. 2003;14:1172–81. doi: 10.1091/mbc.E02-09-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cajo GC, et al. The role of the DIF motif of the DnaJ (Hsp40) co-chaperone in the regulation of the DnaK (Hsp70) chaperone cycle. J Biol Chem. 2006;281:12436–44. doi: 10.1074/jbc.M511192200. [DOI] [PubMed] [Google Scholar]
- 33.Kampinga HH, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–11. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–12. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan W, et al. Zuotin, a ribosome-associated DnaJ molecular chaperone. EMBO J. 1998;17:4809–17. doi: 10.1093/emboj/17.16.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell. 1992;71:97–105. doi: 10.1016/0092-8674(92)90269-i. [DOI] [PubMed] [Google Scholar]
- 37.Hundley HA, Walter W, Bairstow S, Craig EA. Human Mpp11 J protein: ribosome-tethered molecular chaperones are ubiquitous. Science. 2005;308:1032–4. doi: 10.1126/science.1109247. [DOI] [PubMed] [Google Scholar]
- 38.Otto H, et al. The chaperones MPP11 and Hsp70L1 form the mammalian ribosome-associated complex. Proc Natl Acad Sci U S A. 2005;102:10064–9. doi: 10.1073/pnas.0504400102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–44. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mokranjac D, Berg A, Adam A, Neupert W, Hell K. Association of the Tim14.Tim16 subcomplex with the TIM23 translocase is crucial for function of the mitochondrial protein import motor. J Biol Chem. 2007;282:18037–45. doi: 10.1074/jbc.M701895200. [DOI] [PubMed] [Google Scholar]
- 41.D’Silva PR, Schilke B, Hayashi M, Craig EA. Interaction of the J-protein heterodimer Pam18/Pam16 of the mitochondrial import motor with the translocon of the inner membrane. Mol Biol Cell. 2008;19:424–32. doi: 10.1091/mbc.E07-08-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slutsky-Leiderman O, et al. The interplay between components of the mitochondrial protein translocation motor studied using purified components. J Biol Chem. 2007;282:33935–42. doi: 10.1074/jbc.M704435200. [DOI] [PubMed] [Google Scholar]
- 43.Schiller D, Cheng YC, Liu Q, Walter W, Craig EA. Residues of Tim44 involved in both association with the translocon of the inner mitochondrial membrane and regulation of mitochondrial Hsp70 tethering. Mol Cell Biol. 2008;28:4424–33. doi: 10.1128/MCB.00007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahi C, Craig EA. Network of general and specialty J protein chaperones of the yeast cytosol. Proc Natl Acad Sci U S A. 2007;104:7163–8. doi: 10.1073/pnas.0702357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higurashi T, Hines JK, Sahi C, Aron R, Craig EA. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci U S A. 2008;105:16596–601. doi: 10.1073/pnas.0808934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agashe VR, et al. Function of trigger factor and DnaK in multidomain protein folding: increase in yield at the expense of folding speed. Cell. 2004;117:199–209. doi: 10.1016/s0092-8674(04)00299-5. [DOI] [PubMed] [Google Scholar]
- 47.Johnson JL, Craig EA. An essential role for the substrate-binding region of Hsp40s in Saccharomyces cerevisiae. J Cell Biol. 2001;152:851–6. doi: 10.1083/jcb.152.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol Biol Cell. 2005;16:40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin Y, Awad W, Petrova K, Hendershot LM. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J. 2008;27:2873–82. doi: 10.1038/emboj.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hageman J, et al. A DNAJB Chaperone Subfamily with HDAC-dependent Activities Suppresses Toxic Protein Aggregation. Mol Cell. 2010;37:355–69. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–40. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 52.Fayazi Z, et al. A Drosophila ortholog of the human MRJ modulates polyglutamine toxicity and aggregation. Neurobiol Dis. 2006;24:226–44. doi: 10.1016/j.nbd.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Chuang JZ, et al. Characterization of a brain-enriched chaperone, MRJ, that inhibits Huntingtin aggregation and toxicity independently. J Biol Chem. 2002;277:19831–8. doi: 10.1074/jbc.M109613200. [DOI] [PubMed] [Google Scholar]
- 54.Chapple JP, van der Spuy J, Poopalasundaram S, Cheetham ME. Neuronal DnaJ proteins HSJ1a and HSJ1b: a role in linking the Hsp70 chaperone machine to the ubiquitin-proteasome system? Biochem Soc Trans. 2004;32:640–2. doi: 10.1042/BST0320640. [DOI] [PubMed] [Google Scholar]
- 55.Westhoff B, Chapple JP, van der Spuy J, Hohfeld J, Cheetham ME. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr Biol. 2005;15:1058–64. doi: 10.1016/j.cub.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 56.Michels AA, et al. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem. 1997;272:33283–9. doi: 10.1074/jbc.272.52.33283. [DOI] [PubMed] [Google Scholar]
- 57.Howarth JL, et al. Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol Ther. 2007;15:1100–5. doi: 10.1038/sj.mt.6300163. [DOI] [PubMed] [Google Scholar]
- 58.Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–23. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 59.Rujano MA, Kampinga HH, Salomons FA. Modulation of polyglutamine inclusion formation by the Hsp70 chaperone machine. Exp Cell Res. 2007;313:3568–78. doi: 10.1016/j.yexcr.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 60.Cunnea PM, et al. ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J Biol Chem. 2003;278:1059–66. doi: 10.1074/jbc.M206995200. [DOI] [PubMed] [Google Scholar]
- 61.Hosoda A, Kimata Y, Tsuru A, Kohno K. JPDI, a novel endoplasmic reticulum-residentprotein containing both a BiP-interacting J-domain and thioredoxin-like motifs. J Biol Chem. 2003;278:2669–76. doi: 10.1074/jbc.M208346200. [DOI] [PubMed] [Google Scholar]
- 62.Ushioda R, et al. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–72. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- 63.Zylicz M, Ang D, Liberek K, Georgopoulos C. Initiation of lambda DNA replication with purified host- and bacteriophage-encoded proteins: the role of the dnaK, dnaJ and grpE heat shock proteins. EMBO J. 1989;8:1601–8. doi: 10.1002/j.1460-2075.1989.tb03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoffmann HJ, Lyman SK, Lu C, Petit MA, Echols H. Activity of the Hsp70 chaperone complex--DnaK, DnaJ, and GrpE--in initiating phage lambda DNA replication by sequestering and releasing lambda P protein. Proc Natl Acad Sci U S A. 1992;89:12108–11. doi: 10.1073/pnas.89.24.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer AE, Hung NJ, Yang P, Johnson AW, Craig EA. The specialized cytosolic J-protein, Jjj1, functions in 60S ribosomal subunit biogenesis. Proc Natl Acad Sci U S A. 2007;104:1558–63. doi: 10.1073/pnas.0610704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Demoinet E, Jacquier A, Lutfalla G, Fromont-Racine M. The Hsp40 chaperone Jjj1 is required for the nucleo-cytoplasmic recycling of preribosomal factors in Saccharomyces cerevisiae. RNA. 2007;13:1570–81. doi: 10.1261/rna.585007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meyer AE, Hoover LA, Craig EA. The cytosolic J-protein, Jjj1, and Rei1 function in the removal of the pre-60S subunit factor Arx1. J Biol Chem. 2009 doi: 10.1074/jbc.M109.038349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fotin A, et al. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–9. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- 69.Scheele U, Kalthoff C, Ungewickell E. Multiple interactions of auxilin 1 with clathrin and the AP-2 adaptor complex. J Biol Chem. 2001;276:36131–8. doi: 10.1074/jbc.M106511200. [DOI] [PubMed] [Google Scholar]
- 70.Heymann JB, et al. Visualization of the binding of Hsc70 ATPase to clathrin baskets: implications for an uncoating mechanism. J Biol Chem. 2005;280:7156–61. doi: 10.1074/jbc.M411712200. [DOI] [PubMed] [Google Scholar]
- 71.Rapoport I, Boll W, Yu A, Bocking T, Kirchhausen T. A motif in the clathrin heavy chain required for the Hsc70/auxilin uncoating reaction. Mol Biol Cell. 2008;19:405–13. doi: 10.1091/mbc.E07-09-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Braell WA, Schlossman DM, Schmid SL, Rothman JE. Dissociation of clathrin coats coupled to the hydrolysis of ATP: role of an uncoating ATPase. J Cell Biol. 1984;99:734–41. doi: 10.1083/jcb.99.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voisine C, et al. Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2001;98:1483–8. doi: 10.1073/pnas.98.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lutz T, Westermann B, Neupert W, Herrmann JM. The mitochondrial proteins Ssq1 and Jac1 are required for the assembly of iron sulfur clusters in mitochondria. J Mol Biol. 2001;307:815–25. doi: 10.1006/jmbi.2001.4527. [DOI] [PubMed] [Google Scholar]
- 75.Vickery LE, Cupp-Vickery JR. Molecular chaperones HscA/Ssq1 and HscB/Jac1 and their roles in iron-sulfur protein maturation. Crit Rev Biochem Mol Biol. 2007;42:95–111. doi: 10.1080/10409230701322298. [DOI] [PubMed] [Google Scholar]
- 76.Chandramouli K, Johnson MK. HscA and HscB stimulate [2Fe-2S] cluster transfer from IscU to apoferredoxin in an ATP-dependent reaction. Biochemistry. 2006;45:11087–95. doi: 10.1021/bi061237w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dutkiewicz R, et al. The Hsp70 chaperone Ssq1p is dispensable for iron-sulfur cluster formation on the scaffold protein Isu1p. J Biol Chem. 2006;281:7801–8. doi: 10.1074/jbc.M513301200. [DOI] [PubMed] [Google Scholar]
- 78.Bonomi F, Iametti S, Morleo A, Ta D, Vickery LE. Studies on the mechanism of catalysis of iron-sulfur cluster transfer from IscU[2Fe2S] by HscA/HscB chaperones. Biochemistry. 2008;47:12795–801. doi: 10.1021/bi801565j. [DOI] [PubMed] [Google Scholar]
- 79.Kim JH, et al. Structure and dynamics of the iron-sulfur cluster assembly scaffold protein IscU and its interaction with the cochaperone HscB. Biochemistry. 2009;48:6062–71. doi: 10.1021/bi9002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schilke B, et al. Evolution of mitochondrial chaperones utilized in Fe-S cluster biogenesis. Curr Biol. 2006;16:1660–5. doi: 10.1016/j.cub.2006.06.069. [DOI] [PubMed] [Google Scholar]
- 81.Sahi C, Lee T, Inada M, Pleiss JA, Craig EA. Cwc23, an essential J-protein critical for pre-mRNA splicing with a dispensable J-domain. Mol Cell Biol. 2009 doi: 10.1128/MCB.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pandit S, et al. Spp382p interacts with multiple yeast splicing factors, including possible regulators of Prp43 DExD/H-Box protein function. Genetics. 2009;183:195–206. doi: 10.1534/genetics.109.106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pandit S, Lynn B, Rymond BC. Inhibition of a spliceosome turnover pathway suppresses splicing defects. Proc Natl Acad Sci U S A. 2006;103:13700–5. doi: 10.1073/pnas.0603188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang C, Comptom MM, Yang P. Dimeric novel HSP40 is incorporated into the radial spoke complex during the assembly process in flagella. Mol Biol Cell. 2005;16:637–48. doi: 10.1091/mbc.E04-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang C, Owen HA, Yang P. Dimeric heat shock protein 40 binds radial spokes for generating coupled power strokes and recovery strokes of 9 + 2 flagella. J Cell Biol. 2008;180:403–15. doi: 10.1083/jcb.200705069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–79. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 87.Schuermann JP, et al. Structure of the Hsp110:Hsc70 nucleotide exchange machine. Mol Cell. 2008;31:232–43. doi: 10.1016/j.molcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cyr DM. Swapping nucleotides, tuning Hsp70. Cell. 2008;133:945–7. doi: 10.1016/j.cell.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schroder H, Langer T, Hartl FU, Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–44. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ang D, Chandrasekhar GN, Zylicz M, Georgopoulos C. Escherichia coli grpE gene codes for heat shock protein B25.3, essential for both lambda DNA replication at all temperatures and host growth at high temperature. J Bacteriol. 1986;167:25–9. doi: 10.1128/jb.167.1.25-29.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laloraya S, Dekker PJ, Voos W, Craig EA, Pfanner N. Mitochondrial GrpE modulates the function of matrix Hsp70 in translocation and maturation of preproteins. Mol Cell Biol. 1995;15:7098–105. doi: 10.1128/mcb.15.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Westermann B, Prip-Buus C, Neupert W, Schwarz E. The role of the GrpE homologue, Mge1p, in mediating protein import and protein folding in mitochondria. EMBO J. 1995;14:3452–60. doi: 10.1002/j.1460-2075.1995.tb07351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyson JR, Stirling CJ. LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 2000;19:6440–52. doi: 10.1093/emboj/19.23.6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kabani M, Beckerich JM, Brodsky JL. Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol. 2002;22:4677–89. doi: 10.1128/MCB.22.13.4677-4689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boisrame A, Kabani M, Beckerich JM, Hartmann E, Gaillardin C. Interaction of Kar2p and Sls1p is required for efficient co-translational translocation of secreted proteins in the yeast Yarrowia lipolytica. J Biol Chem. 1998;273:30903–8. doi: 10.1074/jbc.273.47.30903. [DOI] [PubMed] [Google Scholar]
- 96.Chung KT, Shen Y, Hendershot LM. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J Biol Chem. 2002;277:47557–63. doi: 10.1074/jbc.M208377200. [DOI] [PubMed] [Google Scholar]
- 97.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–58. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 98.Mukai H, et al. Isolation and characterization of SSE1 and SSE2, new members of the yeast HSP70 multigene family. Gene. 1993;132:57–66. doi: 10.1016/0378-1119(93)90514-4. [DOI] [PubMed] [Google Scholar]
- 99.Oh HJ, Easton D, Murawski M, Kaneko Y, Subjeck JR. The chaperoning activity of hsp110. Identification of functional domains by use of targeted deletions. J Biol Chem. 1999;274:15712–8. doi: 10.1074/jbc.274.22.15712. [DOI] [PubMed] [Google Scholar]
- 100.Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006;25:2519–28. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–8. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shaner L, Sousa R, Morano KA. Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry. 2006;45:15075–84. doi: 10.1021/bi061279k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:E237–41. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
- 104.Alberti S, et al. Ubiquitylation of BAG-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J Biol Chem. 2002;277:45920–7. doi: 10.1074/jbc.M204196200. [DOI] [PubMed] [Google Scholar]
- 105.Luders J, Demand J, Hohfeld J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J Biol Chem. 2000;275:4613–7. doi: 10.1074/jbc.275.7.4613. [DOI] [PubMed] [Google Scholar]
- 106.Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283:1437–44. doi: 10.1074/jbc.M706304200. [DOI] [PubMed] [Google Scholar]
- 107.Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem. 2009;284:5523–32. doi: 10.1074/jbc.M807440200. [DOI] [PubMed] [Google Scholar]
- 108.Teter SA, et al. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;97:755–65. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 109.Kerner MJ, et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–20. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 110.Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008;283:18473–7. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- 111.Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22:2229–40. doi: 10.1210/me.2008-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–6. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 113.Liberek K, Lewandowska A, Zietkiewicz S. Chaperones in control of protein disaggregation. EMBO J. 2008;27:328–35. doi: 10.1038/sj.emboj.7601970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hohfeld J, Minami Y, Hartl FU. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell. 1995;83:589–98. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 115.Prapapanich V, Chen S, Toran EJ, Rimerman RA, Smith DF. Mutational analysis of the hsp70-interacting protein Hip. Mol Cell Biol. 1996;16:6200–7. doi: 10.1128/mcb.16.11.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ziegelhoffer T, Johnson JL, Craig EA. Chaperones get Hip. Protein folding. Curr Biol. 1996;6:272–5. doi: 10.1016/s0960-9822(02)00476-1. [DOI] [PubMed] [Google Scholar]
- 117.Gebauer M, Zeiner M, Gehring U. Proteins interacting with the molecular chaperone hsp70/hsc70: physical associations and effects on refolding activity. FEBS Lett. 1997;417:109–13. doi: 10.1016/s0014-5793(97)01267-2. [DOI] [PubMed] [Google Scholar]
- 118.Nollen EA, et al. Modulation of in vivo HSP70 chaperone activity by Hip and Bag-1. J Biol Chem. 2001;276:4677–82. doi: 10.1074/jbc.M009745200. [DOI] [PubMed] [Google Scholar]
- 119.Nelson GM, et al. The heat shock protein 70 cochaperone hip enhances functional maturation of glucocorticoid receptor. Mol Endocrinol. 2004;18:1620–30. doi: 10.1210/me.2004-0054. [DOI] [PubMed] [Google Scholar]
- 120.Ballinger CA, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–45. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang M, et al. Chaperoned ubiquitylation--crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell. 2005;20:525–38. doi: 10.1016/j.molcel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 122.Connell P, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 123.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–5. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 124.Xu W, et al. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99:12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rosser MF, Washburn E, Muchowski PJ, Patterson C, Cyr DM. Chaperone functions of the E3 ubiquitin ligase CHIP. J Biol Chem. 2007;282:22267–77. doi: 10.1074/jbc.M700513200. [DOI] [PubMed] [Google Scholar]
- 126.Kampinga HH, Kanon B, Salomons FA, Kabakov AE, Patterson C. Overexpression of the cochaperone CHIP enhances Hsp70-dependent folding activity in mammalian cells. Mol Cell Biol. 2003;23:4948–58. doi: 10.1128/MCB.23.14.4948-4958.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marber MS, et al. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;95:1446–56. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Radford NB, et al. Cardioprotective effects of 70-kDa heat shock protein in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:2339–42. doi: 10.1073/pnas.93.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cummings CJ, et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–8. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 130.Hansson O, et al. Overexpression of heat shock protein 70 in R6/2 Huntington’s disease mice has only modest effects on disease progression. Brain Res. 2003;970:47–57. doi: 10.1016/s0006-8993(02)04275-0. [DOI] [PubMed] [Google Scholar]
- 131.Adachi H, et al. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–11. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jaattela M. Over-expression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int J Cancer. 1995;60:689–93. doi: 10.1002/ijc.2910600520. [DOI] [PubMed] [Google Scholar]
- 133.Nylandsted J, et al. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002;62:7139–42. [PubMed] [Google Scholar]
- 134.Ellis RJ, Hemmingsen SM. Molecular chaperones: proteins essential for the biogenesis of some macromolecular structures. Trends Biochem Sci. 1989;14:339–42. doi: 10.1016/0968-0004(89)90168-0. [DOI] [PubMed] [Google Scholar]
- 135.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–6. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vogel M, Bukau B, Mayer MP. Allosteric regulation of Hsp70 chaperones by a proline switch. Mol Cell. 2006;21:359–67. doi: 10.1016/j.molcel.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 137.Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wuthrich K. NMR structure of the J-domain and the Gly/Phe-rich region of the Escherichia coli DnaJ chaperone. J Mol Biol. 1996;260:236–50. doi: 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- 138.Caplan AJ, Tsai J, Casey PJ, Douglas MG. Farnesylation of YDJ1p is required for function at elevated growth temperatures in Saccharomyces cerevisiae. J Biol Chem. 1992;267:18890–5. [PubMed] [Google Scholar]
- 139.Flom GA, Lemieszek M, Fortunato EA, Johnson JL. Farnesylation of Ydj1 is required for in vivo interaction with Hsp90 client proteins. Mol Biol Cell. 2008;19:5249–58. doi: 10.1091/mbc.E08-04-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cupp-Vickery JR, Vickery LE. Crystal structure of Hsc20, a J-type Co-chaperone from Escherichia coli. J Mol Biol. 2000;304:835–45. doi: 10.1006/jmbi.2000.4252. [DOI] [PubMed] [Google Scholar]
- 141.Fuzery AK, et al. Solution structure of the iron-sulfur cluster cochaperone HscB and its binding surface for the iron-sulfur assembly scaffold protein IscU. Biochemistry. 2008;47:9394–404. doi: 10.1021/bi800502r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Andrew AJ, Dutkiewicz R, Knieszner H, Craig EA, Marszalek J. Characterization of the interaction between the J-protein Jac1p and the scaffold for Fe-S cluster biogenesis, Isu1p. J Biol Chem. 2006;281:14580–7. doi: 10.1074/jbc.M600842200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material