The structure and function of the β2-adaptin appendage domain (original) (raw)
Abstract
The heterotetrameric AP2 adaptor (α, β2, µ2 and σ2 subunits) plays a central role in clathrin-mediated endocytosis. We present the protein recruitment function and 1.7 Å resolution structure of its β2-appendage domain to complement those previously determined for the µ2 subunit and α appendage. Using structure-directed mutagenesis, we demonstrate the ability of the β2 appendage alone to bind directly to clathrin and the accessory proteins AP180, epsin and eps15 at the same site. Clathrin polymerization is promoted by binding of clathrin simultaneously to the β2-appendage site and to a second site on the adjacent β2 hinge. This results in the displacement of the other ligands from the β2 appendage. Thus clathrin binding to an AP2–accessory protein complex would cause the controlled release of accessory proteins at sites of vesicle formation.
Keywords: adaptins/clathrin/endocytosis/eps15/vesicle-formation
Introduction
Clathrin-mediated endocytosis is the process whereby clathrin-coated vesicles (CCVs) are formed by the pinching off of a portion of the plasma membrane into which cargo molecules have been sorted (reviewed in Schmid, 1997; Mellman and Warren, 2000; Pearse et al., 2000). It is involved in many cellular processes including the internalization and subsequent downregulation of activated growth factor receptors, regulating the number and type of small molecule receptors, channels and transporters in the plasma membrane, recycling of synaptic vesicles, and the maintenance of membrane identity. CCVs have three layers: an inner membrane layer containing transmembrane cargo; an outer protein layer, which serves as a mechanical scaffold formed from a lattice of clathrin triskelia; and linking them, a middle layer composed predominantly of clathrin adaptors (AP complexes and arrestins) (Smith et al., 1998).
The AP2 adaptors are heterotetramers composed of two large ∼110 kDa subunits (α and β2), one medium ∼50 kDa subunit (µ2) and one small ∼17 kDa subunit (σ2) (reviewed in Traub, 1997; Hirst and Robinson, 1998). The large subunits can each be divided into a 60–70 kDa trunk domain separated from a 25–30 kDa appendage domain (sometimes referred to as an ‘ear’ domain) by an ∼100-residue, protease-sensitive linker (Kirchhausen et al., 1989). AP2 adaptors play a central role in CCV formation (reviewed in Kirchhausen, 1999). They link the endocytic cargo to the clathrin coat, and are also able to interact with a number of proteins believed to regulate various aspects of CCV formation. AP2 adaptors bind to the short linear internalization motifs on cargo molecules, YxxΦ (where Φ is a bulky hydrophobic amino acid and x represents any amino acid) via the µ2 subunit (Ohno et al., 1995; Owen and Evans, 1998), and D/ExxxLL probably via the β2 trunk domain (Rapoport et al., 1998). The interaction of clathrin with the AP adaptors is through the binding of the clathrin N-terminal β-propeller domain to a canonical clathrin box motif (LΦxΦD/E) (Dell’Angelica et al., 1998; Ter Haar et al., 2000) in the linker or hinge region of the β2-adaptin subunit (Shih et al., 1995). Similar sequences are found in many other clathrin-binding proteins including β1-, β2- and β3-adaptin subunit hinge domains, the amphiphysins and members of the arrestin family (Dell’Angelica et al., 1998). The interaction of the adaptor complex with CCV formation regulatory/accessory proteins such as AP180, auxilin, amphiphysin, eps15 and epsin is via the appendage domain of the α subunit (Owen et al., 1999; Traub et al., 1999).
In this work we describe the structure of the β2- appendage domain determined by X-ray crystallography at a resolution of 1.7 Å. The β2-appendage domain has a similar overall bilobal structure to the α-appendage domain (Owen et al., 1999; Traub et al., 1999), with which it shows only low sequence identity. They both have a single patch of high hydrophobic surface potential on their C-terminal platform subdomains. In the α appendage this site mediates interactions with a number of proteins that play accessory/regulatory roles in CCV formation. We demonstrate that the hydrophobic patch in the β2 appendage binds to a subset of DΦF/W motif-containing proteins that are bound by the α-adaptin appendage domain (epsin, AP180, eps15). In addition to binding these proteins, the β2-appendage domain alone shows significant direct clathrin binding. The strength of binding is increased when the hinge domain is added to the N-terminus, suggesting that the binding site for clathrin on β2 adaptin is bipartite. This increase in strength of binding to clathrin is reflected in the ability of the β2 appendage+hinge, unlike the β2-appendage domain on its own, to block transferrin uptake in vivo. The bipartite nature of the interaction may serve to orientate domains of clathrin triskelia correctly, which would explain its ability to drive the clathrin cage formation at physiological conditions. The binding of clathrin to the β2 appendage+hinge is shown to displace AP180, epsin and eps15 bound to the β2 appendage. The implications of these findings for the mechanism of CCV formation are discussed.
Results
Structure of the β2 appendage domain
The structure of the appendage domain of β2 adaptin (residues 705–937) determined by X-ray crystallography was solved by single isomorphous replacement using a mercury derivative and solvent flattening, to a resolution of 1.7 Å. Surprisingly, given the low homology (see Figure 2), the β2-appendage structure is similar to that of the α appendage (Owen et al., 1999; Traub et al., 1999). The structures of the β2- and α-appendage domains are shown in Figure 1 as both ribbon and topological representations. The β2 appendage consists of two subdomains, which contact each other in a tightly packed interface that buries 1340 Å2 of accessible surface, indicating that little motion is possible between the subdomains. The relative orientation of the subdomains is significantly different in the two structures, with the angle between the two subdomains differing by 46°. The N-terminal subdomain (residues 705–825) is an 8-stranded β sandwich reminiscent of an immunoglobulin fold, lacking the first β-sheet strand found in the α-adaptin appendage domain. The C-terminal or ‘platform’ subdomain (residues 826–937) is made up of a 5 stranded β-sheet flanked on the outer side by helix α1 and by helices α2 and α3 on the inner side. It differs from the α domain around the putative binding site (see below) in two important ways: β-sheet strand 12 is elongated in β2 to replace the missing short β10 strand; and the loop between β12 and β13 is shorter. In the N-terminal subdomains, residues conserved between the two appendages are largely internal, playing a structural role in the hydrophobic core. In contrast on the platform subdomain there is a large surface patch of residues conserved between the appendages (Figure 2), suggesting a functional (protein–protein interaction) rather than structural role.

Fig. 2. Sequence conservation between the β2- and α-appendage domains. (A) Structurally homologous residues, defined as those whose Cαs are within 3 Å in the two structures, are boxed in grey. The alignment was performed using each subdomain separately to allow for the relative rotation of C-terminal subdomains. Identities between the two sequences are coloured dark purple and conservation is indicated by pale purple. (B) Two views of the surface representation of the β2-appendage domain coloured for conservation between β2 and α in the same colour scheme as (A). The loop containing Leu747 from an adjacent molecule and the oxidized DTT molecule are shown as in Figure 3.

Fig. 1. Structural comparisons of β2- and α-appendage domains. (A) Comparison of the two Cα backbone traces of β2- (blue and red) and α-appendage (green and gold) domains. Ribbon diagrams, aligned on the N-terminal domains, coloured from N- (blue or green) to C- (red or yellow) termini. This figure, and Figures 2B and 3 were prepared using Aesop (M.E.M.Noble, unpublished). (B) Topological diagrams comparing the β2- and α-appendage domains, coloured using the same colour scheme as above.
In order to locate potential protein binding sites on β2 appendage, its surface hydrophobic potential was analysed: this exploits the fact that as two proteins come into contact, the majority of water molecules at the interface between the two will be displaced. If the water molecules sit on a hydrophobic surface, their removal will have a beneficial effect on the energy of interaction. The energy of a hydrophobic probe (which does not form hydrogen bonds) can be evaluated over a protein molecule’s entire surface using the program GRID (Goodford, 1996) and the values plotted on a Connolly surface representation of the protein, which is then coloured by hydrophobic potential. Previous hydrophobic potential analysis of the α- appendage domain showed it to possess a single good candidate site for protein–protein interaction, located on the platform subdomain and centred on W840 (Owen et al., 1999; Traub et al., 1999); this was subsequently confirmed by mutagenesis. A similar structural analysis was performed on the β2-appendage domain. The results of the hydrophobic surface analysis (shown in Figure 3A, left panel) were similar to those for the α-appendage domain (Figure 3A, right panel), showing a single site of favourable hydrophobic potential that would therefore be a strong candidate for a site of protein–protein interaction. The site is centred on W841, which is spatially homologous to W840 in the α-appendage domain. However, the putative binding site is larger than that on α due to the exposure of neighbouring hydrophobic residues Y888, M886 and F837 on the surface of β2 (see Figure 3C). The site is more accessible in β2 because of the absence of the α-appendage domain 879–884 loop: in the β2-appendage domain, strand β12 is longer with only a short loop into strand β13.


Fig. 3. ‘Top’ views of the C-terminal subdomains of β2- and α-appendages. The same views are used in each case for β2- (left) and α- (right) appendage domains. (A) Surfaces coloured such that the sites of favourable hydrophobic interaction are coloured dark green, sites of moderate hydrophobic interaction are coloured pale green, and sites of neutral or disfavoured hydrophobic interaction are coloured grey. The outstanding features are the hydrophobic pockets centred on the homologous tryptophan residues, occupied in β2 by Leu747 from the neighbouring molecule in the crystal and an oxidized DTT molecule. Inset: electron density for oxidized DTT as an example of overall electron density. (B) Surfaces coloured for electrostatic potential [positive (+10 kT e–1), blue, to negative (–10 kT e–1), red]. Surface created with GRASP (Nicholls et al., 1991). (C) Molecular detail of the surface-exposed residues on the ‘top’ of the C-terminal subdomains. (D) Effects of point mutations on protein ligand binding. Space-filling representations with colours according to the effect on protein ligand binding: red, large effect; yellow, moderate effect.
Several other observations also suggested that this hydrophobic patch is a good candidate site for protein–protein interaction. It is located in a pocket that in the crystal is occupied by two hydrophobic moieties: the sidechain of L747 from the N-terminal subdomain of another β2-appendage molecule in the crystal lattice, and an oxidized dithiothreitol (DTT) molecule that sits against Y888 and makes hydrogen bonds with R879. In keeping with the binding site in the α appendage and many other protein–protein interaction sites there are charged residues adjacent to the hydrophobic pocket such as R834, K842, E849, R879, E902, R904 and K917, which could provide an element of specificity to an interaction whose strength was mainly derived from a hydrophobic component.
The pattern of charged residues around the site of high hydrophobic potential in the two appendage domains is, however, quite different (see Figure 3B). While both preserve a basic patch on the surface due to the conservation of K842 and K917 in β2 as K841 and R920 in α, the rest of the platform subdomain surfaces have different electrostatic properties. E902 and R904 in the platform subdomain of the β2 appendage are exchanged for R905 and E907, respectively, in the α appendage and the positive potential due to R834 and R879 in β2 is replaced by negative potential due to D881 in α.
In the light of the structural similarity between the binding sites we investigated whether the β2 appendage could also act as a protein recruitment domain.
Function of β2-appendage domain
The β2 appendage and β2 appendage+hinge domains were produced as N-terminal glutathione _S_-transferase (GST) fusion proteins and used in pull-down experiments from rat brain cytosol to determine whether they had any protein binding partners. The results are shown alongside those for the α appendage, together with a control GST fusion protein (the SH3 domain of amphiphysin2) (Figure 4A and B). The β2-appendage domain binds a subset of the proteins that also bind to the α-appendage domain (AP180, epsin and eps15). The interactions with the two appendage domains are of approximately equal affinity, as estimated from the similar strengths of signal seen when equimolar amounts of each appendage domain are used in the assays. On the other hand, the amphiphysins, which bind strongly to the α-appendage domain, bind either very weakly or not at all to the β2-appendage domain. The β2-appendage+hinge domain binds the same three proteins as the β2-appendage domain but in lower amounts, despite there being equimolar amounts of folded appendage domain in each sample. The β2 appendage and the β2 appendage+hinge showed significant binding to clathrin (presence confirmed by comparison with purified clathrin, data not shown), whereas the α-appendage domain bound clathrin only very weakly. Clathrin binding was weaker to the β2 appendage than to the β2 appendage+hinge (∼4-fold more binding on average to the appendage+hinge than to the appendage, as estimated from five separate experiments).
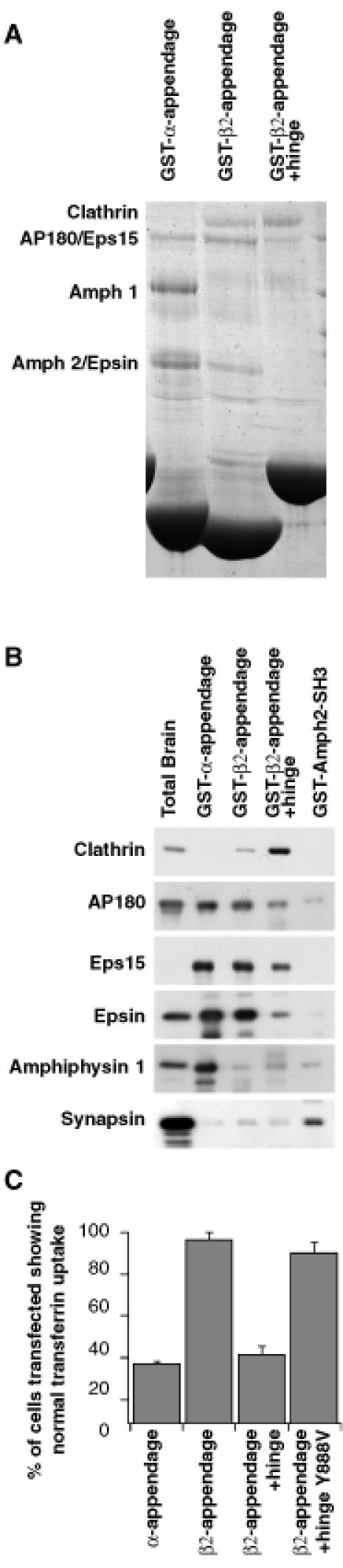
Fig. 4. Binding partners of β2 appendage and β2 appendage+hinge. Proteins bound to GSTβ2 appendage, β2 appendage+hinge, α appendage and Amph2SH3 [control in (B)] were analysed by Coomassie Blue staining (A) and immunoblotting with antibodies (B) against clathrin, AP180, epsin, eps15, amphiphysin1 and synapsin (an abundant protein in brain that has been proposed to bind to the SH3 domains of the amphiphysins) as a control to show that ligand binding to the appendages is specific. (C) Effects of transfection of constructs expressing the α appendage, β2 appendage, β2 appendage+hinge, and β2 appendage+hinge containing the mutation Y888V domains on transferrin endocytosis in COS7 fibroblasts. Expression levels of each construct were equal as judged by the intensity of immunofluorescence.
Constructs overexpressing the β2 appendage, β2 appendage+hinge and β2 appendage+hinge containing the mutation Y888V (which abolishes ligand binding to the appendage, see later) under the control of the cytomegalovirus promoter were transfected into COS7 fibroblasts and the effect on endocytosis was examined by studying the uptake of biotinylated transferrin. Unlike the α-appendage domain, the β2-appendage domain does not inhibit endocytosis (see Figure 4C). However, when the hinge domain was included, this construct inhibited endocytosis to a similar extent as the α-appendage domain: ∼60% cells blocked for transferrin uptake. The β2 appendage+hinge containing the Y888V mutation was, however, unable to inhibit transferrin uptake. Since the only protein that is bound more by the β2 appendage+ hinge than the β2 appendage alone is clathrin, we conclude that the block of endocytosis caused by the β2 appendage +hinge domain is due to it binding clathrin in the cytoplasm, making it unavailable for recruitment to the plasma membrane and its subsequent use in forming CCVs. This is supported by the observation that overexpression of the β2 appendage+hinge causes an alteration in the distribution of clathrin within the cell (data not shown).
Confirmation of the location of the protein–protein interaction site
Mutations in the β2-appendage domain were designed on the basis of the structure and produced as N-terminal GST fusion proteins. Mutations were made both in surface-exposed hydrophobic (W841A, M886Q, Y888V, L838E) and in charged residues (R834E, K842, R879E, R904E, K917Q) in the putative protein–protein interaction site. Many of these mutations were spatially homologous to mutations of the α-appendage domain, which had a significant effect on binding. Mutations were also made in residues distant from the proposed binding site both in the platform domain (N895R/I897S, Q906S) and in the sandwich domain (H735S/Y737S; identified as a small surface-accessible hydrophobic patch). One mutation, N895R/I897S, caused the protein to be expressed in an insoluble unfolded form. Circular dichroism and the yield of the other mutants indicated that there were no significant changes in secondary structure (data not shown). Attempts were made to crystallize the mutants W841A and Y888V but these proved unsuccessful, presumably since the hydrophobic patch of which they form the major part is an essential site of molecule–molecule contact in the crystal (via L747 and its spatially adjacent residues in another molecule). GST fusion proteins of the soluble mutants were used in ‘pull down’ experiments from brain cytosol and the results assayed by immunoblotting for the proteins previously shown to bind to the β2- and α-appendage domains (Figure 5A). The effects of the various mutations on binding are different for different ligands. Mutation of residues distant from the putative binding site (such as Q906S, H735S/Y737S) had no effect on binding of any of the ligands. Mutation of Y888, which sits at the centre of the pocket, has the largest effect on ligand binding. It strongly inhibits the binding of clathrin, AP180 and epsin but has only a slight effect on the binding of eps15 (providing further indication that this mutant is folded). Mutations of the other surface-exposed hydrophobic residues (W841, M886) have less effect. By way of contrast, in the α appendage mutation of the tryptophan spatially homologous to W841 (W840 in the α appendage) abolished binding of all ligands. Mutation of the charged residues R879, which packs against, and forms a hydrogen bond with, the oxidized DTT molecule, and R904 and K917, which are in the centre of the platform, causes a significant reduction in strength of binding to ligands, suggesting that they play a role in mediating the interaction between the β2-appendage domain and its binding partners. A summary of the effects of mutations from multiple experiments are given in Table I and their locations on the surface on the β2 appendage are shown in Figure 3C and D. The Y888V mutation has also been incorporated into the β2 appendage+hinge. As in the β2 appendage alone, this mutation abolishes binding of epsin and AP180, and reduces that of Eps15. However, binding to clathrin was reduced only by ∼50% (data not shown), compared with its complete abolition by the Y888V mutation in the β2 appendage alone (Table I).
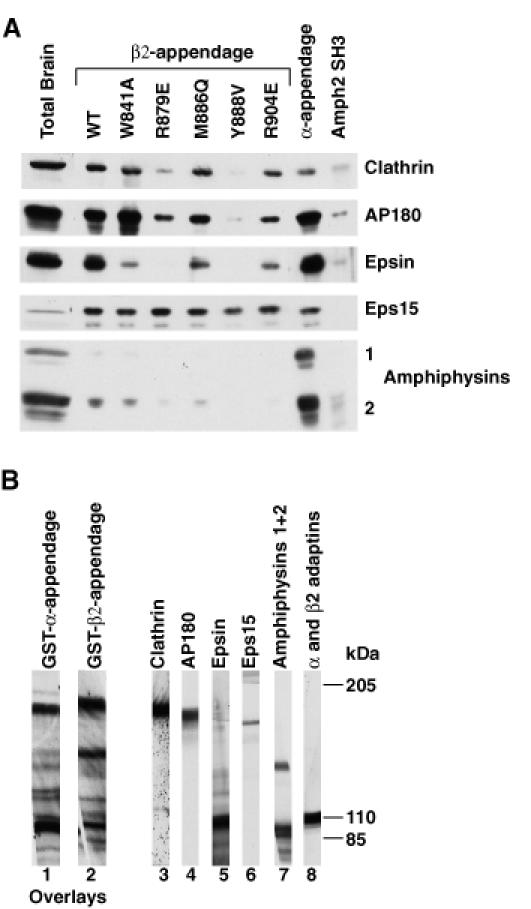
Fig. 5. The protein–protein interaction site on the β2-appendage domain. (A) GSTβ2-appendage domain point mutants, GSTα appendage and GSTAmph2 SH3 (negative control) were tested for their ability to bind proteins in ‘pull down’ experiments from brain cytosol detected by immunoblotting. (B) Overlay assays. Lanes 1 and 2 show overlays of rat brain cytosolic proteins with the α- and β2-appendage domains. Appendage domains were detected with Ra5.2 (α) or Ra2.2 (β). Direct detection of proteins in rat brain cytosol with specific antibodies are shown in the remaining lanes.
Table I. Summary of data from ‘pull down’ experiments using GST fusion proteins of wild type (WT) β2-appendage domain and mutant forms of the domain that affected ligand binding, α appendage and the Amph2 SH3.
| | Clathrin | AP180 | Epsin | Eps15 | | | ----------- | ----- | ----- | ----- | --- | | WT β2 | +++ | +++ | +++ | +++ | | W841A | ++ | ++++ | + | +++ | | R879E | + | + | – | +++ | | M886Q | ++ | ++ | ++ | +++ | | Y888V | – | – | – | ++ | | R904E | ++ | ++ | ++ | +++ | | H735S/Y737S | +++ | +++ | +++ | +++ | | R834E | +++ | +++ | +++ | +++ | | L838E | +++ | +++ | ++ | +++ | | E902R | +++ | +++ | +++ | +++ | | Q906S | +++ | +++ | +++ | +++ | | K917Q | – | – | + | ++ | | K842E | + | ++ | ++ | +++ | | α appendage | – | +++ | +++ | +++ | | Amph2 SH3 | – | – | – | – | | GST | – | – | – | – |
The question remains whether or not these interactions are direct, since some of these proteins are known to be able to bind to each other [e.g. epsin to eps15 (Salcini et al., 1997; Chen et al., 1998) and clathrin to AP180 (Morris et al., 1993) and to epsin (Drake et al., 2000)]. Several observations suggest that the interactions are indeed direct. First, different mutations have different effects on the relative ligand binding. For instance, mutation of Y888, R879 and K917 causes little change in eps15 binding but a significant reduction in the binding of the other ligands; mutation of M886 causes a reduction in the binding of epsin but less change in the binding of other ligands and mutation of W841 causes a decrease in the binding of epsin, a slight decrease in the binding of eps15 but an increase in the binding of AP180. Secondly, the affinity of these interactions, which are often only mediated by peptide–protein contacts, are relatively weak with _K_ds in the 10–1000 µM range (De Beer et al., 1998; Owen et al., 1999). Thus the amount of a protein that appeared on a ‘pull down’ experiment due to a weak secondary interaction would be so low as to be virtually undetectable. Finally, the α-appendage domain does not bind significantly to clathrin, but all the proteins that bind significantly to the β2-appendage domain also bind to the α-appendage domain at least as strongly, so clathrin binding must be direct (see Figures 4 and 5). Supporting evidence that the interactions of the β2-appendage domain with its ligands are direct comes from overlay assays (Figure 5B). These showed that the β2-appendage domain is able to bind to proteins of the same apparent molecular weight as some of those that bind to the α-appendage domain. In addition, the fact that the β2 appendage+hinge containing the Y888V mutation still binds appreciable amounts of clathrin but no AP180 or epsin indicates that the interactions of these two latter ligands are not via clathrin.
Clathrin cage formation
Since clathrin was able to bind to both the appendage and the appendage+hinge domain of β-adaptin, we investigated whether the appendage site on its own or both the appendage and hinge sites together were capable of driving clathrin polymerization at physiological conditions. Clathrin cage formation was assayed in the presence and absence of GSTβ2 appendage or GSTβ2 appendage+hinge. Figure 6A shows that β2 appendage+hinge but not the appendage alone drives clathrin into a polymeric state, which can be pelleted at 100 000 g. The electron micrographs of the negatively stained samples (representative fields of view shown in Figure 6B and C) showed that the polymeric state was discrete cages and not non-specific aggregates and that the GSTβ2 appendage+hinge stimulated cage formation ∼10-fold. The GSTβ2 appendage+hinge pellets with the clathrin (Figure 6A) and appears as an additional layer of material within the clathrin polyhedral cages in the micrographs, seen as a solid pale interior to the cages in Figure 6B as compared with the dark interior in the empty cages in Figure 6C. This result implies that the clathrin ‘pulled down’ by GSTβ2 appendage+hinge from cytosol is polymeric and that ‘pulled down’ by the GSTβ2-appendage domain alone is monomeric. Thus, we propose that the ability of clathrin to compete with other ligands (eg. AP180, epsin, eps15) for the β2 appendage+hinge is a combination of (i) the higher affinity between clathrin and the β2 appendage+hinge, and (ii) the steric competition due to the clathrin forming polymers, which would prevent the simultaneous binding of clathrin to the hinge and another protein to the appendage domain.
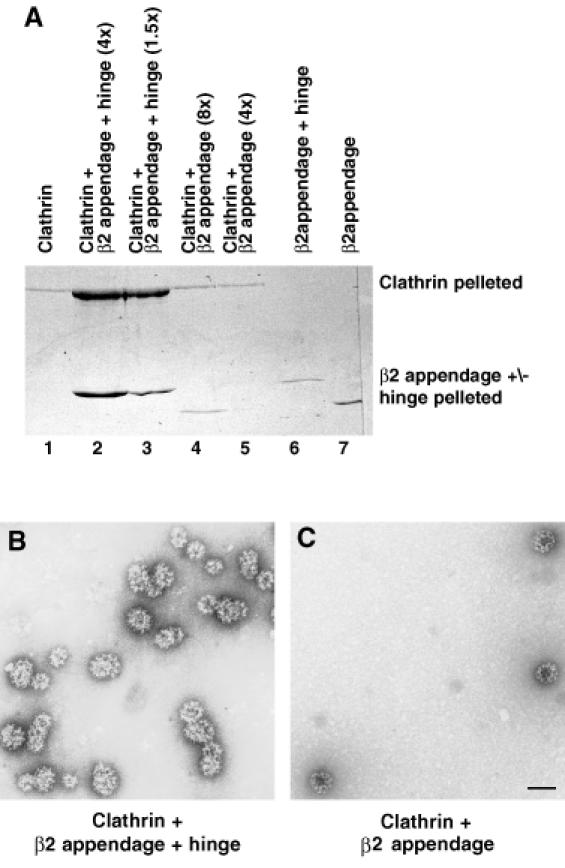
Fig. 6. GSTβ2 appendage+hinge but not GSTβ2 appendage can drive clathrin cage formation. Clathrin at a final concentration of 0.7 µM was incubated on its own (lane 1) or in the presence of a 4-fold (lane 2) or 1.5-fold (lane 3) molar excess of GSTβ2 appendage+hinge, or an 8-fold (lane 4) or 4-fold (lane 5) molar excess of GSTβ2 appendage. Lanes 6 and 7 contained the same amounts of the GST proteins as in lanes 2 and 4, respectively, but no clathrin. Samples were centrifuged at 100 000 g and pelletable material assayed by SDS–PAGE followed by Coomassie Blue staining (A) or by negative stain electron microscopy (B and C). GSTβ2 appendage+hinge but not GSTβ2 appendage can drive clathrin cage formation, as can be seen by the increased amount of pelletable clathrin in lanes 2 and 3 compared with lanes 1, 4 and 5 (A). The electron micrographs in (B) and (C) show the pelletable material to be cages and not non-specific aggregates.
Discussion
The structure of the appendage domain of β2 adaptin is similar to that of α-adaptin in that it has two subdomains with similar topology, but with a difference of ∼46° in the orientation between the two subdomains. Like the α-appendage, the β2-appendage domain binds AP180, epsin and eps15, and therefore is likely to be involved in recruiting proteins involved in controlling CCV formation to sites of coated pit formation in vivo. The two proteins possess a partly conserved binding site of high hydrophobic potential on the C-terminal platform domain, suggesting that they bind similar motifs i.e. DΦF/W. The binding site residues are also conserved in β1-adaptin from the adaptor AP1, so this homologue would be predicted to bind the same ligands as β2. Indeed, a GSTβ1 appendage construct does bind to eps15, and weakly to clathrin and epsin (data not shown). Although the α and β2 appendages share some of the same ligands, they bind to them with different relative affinities: the α appendage binds appreciable amounts of the amphiphysins and has epsin as its highest affinity ligand (Owen et al., 1999;Traub et al., 1999), whereas the highest affinity ligand for β2 appendage is eps15, and the amphiphysins show no significant binding. The differences in specificity of the two appendages are probably due to the differences in the chemical properties of the residues surrounding the conserved hydrophobic/basic patch on the platform subdomain (see Figure 3). The β2-appendage domain hydrophobic patch is larger and more open than that in α and this is likely to account for its additional ability to bind to clathrin. β2 adaptin thus has a bipartite site for binding to clathrin, with elements on the hinge (the LΦxΦD clathrin box motif known to bind to the N-terminal β propeller) (Dell’Angelica et al., 1998; Ter Haar et al., 2000) and the platform domain of the appendage. That there are two separate binding sites for β2 adaptin on clathrin has also been alluded to in previous work (Murphy and Keen, 1992). The observation that only the β2 appendage+hinge is able to drive clathrin polymerization suggests that the role of the two binding sites is to orientate correctly the different parts of the clathrin molecule with respect to each other such that they can easily form a clathrin lattice. This ability to bind and drive clathrin polymerization in solution is presumably responsible for the ability of the β2 appendage+hinge but not the β2 appendage alone to inhibit transferrin endocytosis in vivo. Mutation of Y888 in the β2 appendage+hinge construct prevents this inhibition of transferrin uptake, showing the importance of both clathrin binding sites, on the β2 appendage and on the hinge. The observation that the α-appendage domain but not the β2 appendage can block clathrin-mediated endocytosis in vivo is presumably due to the α appendage being able to ‘titrate out’ sufficient amounts of a protein that the β2 appendage binds only weakly. The most likely candidates for this are the amphiphysins, which have been implicated in recruiting dynamin and synaptojanin to sites of vesicle formation (Shupliakov et al., 1997; Wigge et al., 1997), implying that it is their level in COS7 fibroblasts that is limiting for CCV formation.
There is no sequence homology between the proteins that are ligands of both appendage domains except that they all possess multiple copies of the sequence DΦF/W, where Φ is a hydrophobic residue e.g. proline, alanine or leucine. This motif has been implicated as binding to the α-appendage domain (Iannolo et al., 1997; Owen et al., 1999). Indeed a short 57 amino acid fragment of AP180 that contains the sequence DLFxDAF has been shown to bind to the α-appendage domain and whole β2-adaptin (Hao et al., 1999). A possible model would place the phenylalanine in the place of the oxidized cyclical DTT molecule, the Φ residue in the place of the leucine from an adjacent molecule and the aspartate close to conserved basic residues. It is interesting to note that in the first clathrin heavy chain repeat (CHCR1) (Ybe et al., 1999), the one nearest to the N-terminal propeller, there is a conserved DΦF motif that could be the appendage domain binding site on the clathrin heavy chain distal portion. The presence of an AP2 binding site in the distal portion of the clathrin heavy chain leg is supported by the observation by Brodsky and co-workers (Greene et al., 2000) that this part of the clathrin molecule is required for cage formation, in addition to the N-terminal clathrin domain, which binds the β2 hinge.
CD measurements of the region of epsin that contains the multiple DPW motifs indicate that this region has little secondary structure (data not shown). Secondary structure predictions of other multiple DΦF/W motif-containing regions suggest that they will have a similar extended structure. The hinge regions of the adaptins are extended and contain small amounts of 310 helix (as determined by CD measurements; T.Dafforn, personal communication). Lack of restraint on the relative orientation of DΦF/W motifs in regulatory/accessory proteins and of the appendage domains relative to the rest of the adaptor complexes would allow both appendages to bind simultaneously to different DΦF/W motifs on a single target molecule. This would result in increasing the relative strength of the interaction between the AP2 adaptor complex and that target protein, and may explain why it is possible to coimmunoprecipitate AP2s with some of these regulatory molecules from cells (Benmerah et al., 1995; Cupers et al., 1998; Hao et al., 1999).
In the light of the data presented here and elsewhere, we propose the following model for CCV formation. In the cytosol and away from regions of active endocytosis (Gaidarov et al., 1999; Roos and Kelly, 1999), individual AP2 adaptors are bound via both their appendage domains to a single regulatory/accessory protein containing multiple DΦF/W motifs such as AP180, epsin or eps15. In these regions the clathrin concentration is low compared with that at the membrane (Goud et al., 1985; Wilde and Brodsky, 1996; Gaidarov et al., 1999) and so is unable to compete effectively with these regulatory/accessory molecules for binding to AP2 adaptors. The preference for binding to an accessory/regulatory molecule via both appendages, rather than to a clathrin molecule on only one appendage, may be enhanced by phosphorylation of serine residues in the hinge region of β2 adaptin with a consequent inhibition of clathrin binding (Wilde and Brodsky, 1996). In order for clathrin-mediated endocytosis to take place, the AP2 adaptors must be recruited to the plasma membrane via interactions of their various domains with other protein molecules including cargo (Owen and Evans, 1998) and phospholipid headgroups (Rapoport et al., 1997; Gaidarov and Keen, 1999). The consequence of high clathrin concentrations at the membrane and dephosphorylation of the β2-adaptin hinge (Morris et al., 1990) is that clathrin can compete effectively with the DΦF/W motif-containing molecules for binding to the AP2 complex. When bound to the β2-adaptin appendage and hinge, clathrin is able to polymerize to form a lattice and can itself recruit more clathrin. This continues to raise the effective clathrin concentration and enhances clathrin’s ability to compete with the regulatory/accessory molecules for AP2. Clathrin polymerization will also be favoured by the high AP2 concentration in a coated pit since there will be many correctly orientated adjacent clathrin binding sites (Smith et al., 1998). The phenomenon of a high local concentration of AP2 adaptors driving clathrin polymerization may be reflected in the ability of the partially dimeric GSTβ2 appendage+hinge to drive clathrin cage formation at near physiological conditions. The amphiphysins are involved in the final stage of CCV formation (i.e. scission) through their ability to recruit dynamin to the sites of CCV formation (Shupliakov et al., 1997; Wigge et al., 1997; Owen et al., 1998, 1999). It is interesting to note that they are the only proteins that bind exclusively to the α appendage and so would be relatively less affected by competition from clathrin binding/polymerization in their binding to AP2 adaptors.
Clathrin binding to AP2 adaptors and its subsequent polymerization displaces accessory/regulatory proteins such as eps15, epsin and AP180 from the appendage domains of AP2s to which they are bound. AP180 and epsin are subsequently incorporated into CCVs via direct interactions with clathrin (Hao et al., 1999; Drake et al., 2000), while eps15 is restricted to the edges of coated pits (Cupers et al., 1998). This would result in the controlled liberation from AP2 adaptors of proteins, which are needed to regulate CCV formation at forming coated pits.
Materials and methods
Constructs
The cDNA encoding residues 700–937 of human β2 adaptin (Ponnambalam et al., 1990) (the appendage domain) and residues 616–937 (the appendage+hinge domains) were cloned into the vector pGEX 4T2 for production as an N-terminal GST fusion protein, into pET15b or pMW172H6 for expression as an N-terminal His6-tagged fusion protein and into pCMV-MYC for eukaryotic expression as an N-terminal myc tag fusion protein under the control of a CMV promoter. Mutants of β2 appendage and β2 appendage+hinge were made by PCR using primers incorporating the changed bases.
Protein expression and purification
GST fusion protein for biochemical studies were expressed in DH5α at 25°C overnight and produced as described (Owen et al., 1999). Clathrin was purified from pig brain by the standard protocol (Smith et al., 1998). N-terminal His6 tagged fusion proteins for crystallization trials were grown in bacterial strain BL21DE3 pLysS at 25°C overnight. The proteins were purified on a Ni-NTA-agarose column (Qiagen) and bound protein was eluted with bufferA (0.2 M NaCl, 20 mM HEPES pH 7, 4 mM β-mercaptoethanol) containing 0.3 M imidazole. The proteins were further purified on S200 gel filtration and subsequently dialysed into 5 mM HEPES, 50 mM NaCl, 4 mM dithiothreitol (DTT) and concentrated to 40 mg/ml.
Crystallization and structure determination of β2-appendage domain
Crystals were grown by hanging drop vapour diffusion against a reservoir containing 1.6–2.0 M MgCl2, 0.1 M Bicine pH 8.7–9.0, 15% v/v glycerol, 10 mM DTT over a period of 2 weeks with final dimensions 0.4 × 0.4 × 0.1 mm. Crystals were of space group C2 with two molecules in the asymmetric unit, unit cell dimensions a = 97.0 Å, b = 128.6 Å, c = 67.6 Å, β = 124.4). X-ray diffraction data were collected at 100 K at SRS Daresbury station 9.6 (Table II). A single mercury derivative was made by soaking a crystal in cryoprotected buffer containing 1 mM ethylmercury thiosalicylate (EMTS) for 2 h. Data were recorded on a ADSC Quantum4 CCD detector, integrated with MOSFLM (Leslie, 1992), and scaled using CCP4 programs (Collaborative Computational Project, 1994). Mercury sites were determined from difference Pattersons, and heavy-atom refinement and phasing were performed with SHARP (de la Fortelle and Bricogne, 1997), followed by solvent flipping and flattening with SOLOMON (Abrahams and Leslie, 1996), leading to an excellent electron density map at 1.7 Å resolution. The model was built with O (Jones et al., 1991) and refined with REFMAC (Murshudov et al., 1997). The final model consists of 466 residues (705–937), 801 water molecules, four Mg2+ ions, one Ni2+ ion, two Cl– ions, two glycerols and two molecules of oxidized DTT. The DTT molecules were clear in the electron density (see Figure 3, inset), and one of them for each protein molecule is located in the putative protein binding site. The metal ions showed clear octahedral coordination by water molecules: one of them was interpreted as a Ni2+ ion, since it is bound by a histidine side chain rather than by an oxygen atom, as would be more likely for a Mg2+ ion. The coordinates and structure factors have been deposited in the Protein Data Bank, accession code 1E42. Hydrophobic interaction surface potentials were calculated as in Owen et al. (1999) and displayed using Aesop (M.E.M.Noble, unpublished).
Table II. Statistics on data collection and phasing.
| | Native | EMTS | | | --------------------------------------------------- | ------------- | ------------- | | Data collectiona | | | | resolution (Å) (outer bin) | 16–1.7 (1.79) | 2.19 (2.40) | | _R_mergeb | 0.157 (0.747) | 0.076 (0.222) | | _R_measc | 0.170 (0.842) | 0.105 (0.302) | | </σ()> | 11.2 (1.5) | 11.9 (3.1) | | completeness (%) | 98.3 (98.3) | 97.6 (98.6) | | multiplicity | 6.9 (4.8) | 3.4 (2.8) | | Wilson plot B (Å2) | 33 | 29 | | MIR phasing | | | | No. of sites | | 6 | | _R_derivd | | 0.211 | | _R_cullise | | 0.902 | | phasing power: isomorphous (anomalous)f | | 0.88 (0.94) | | mean figure of merit | 0.26 | | | figure of merit after solvent flattening (all data) | 0.77 | | | Refinement | | | | R (_R_free)g | 0.196 (0.246) | | | (Å2) | 32 | | | _N_reflections (_N_free) | 71 601 (3621) | | | _N_atoms (_N_water) | 4559 (801) | | | r.m.s.d. bond length (Å) | 0.020 | | | r.m.s.d. angle (°) | 1.9° | | | No. of Ramachandran violations | 0 | |
Protein–protein binding assays
Binding assays were performed by incubating 20 µg GST fusion protein with 0.5 ml of 0.1% Triton X-100 brain extract in buffer A in the presence of 20 µl 50% slurry of glutathione–Sepharose at 4°C for 1 h. The beads were washed three times for 3 min with buffer A and the proteins bound analysed by SDS–PAGE followed by staining with Coomassie Blue or immunoblotting. Antibodies used were: Ra3 (anti-Amphiphysin 1), Ra1.2 (anti-Amphiphysin 2), Ra2.2 (anti β2-adaptin appendage domain), Ra5.2 (anti α-adaptin appendage domain), Ra14 (anti-Epsin), Ra24 (anti-AP180) anti-eps15 (Santa Cruz Laboratories) and anti-Clathrin Heavy Chain (Transduction Laboratories).
Protein overlay assay
Brain extract was separated on a 7% SDS polyacrylamide gel and transferred onto nitrocellulose. After blocking with 5% milk, the blots were incubated overnight with 1 mg/ml appendage domain in 1% goat serum. After washing extensively, bound appendage domain was detected with an appropriate antibody raised against an appendage domain. On the 7% SDS polyacrylamide gels used in the overlay assays, clathrin and AP180 migrate with similar apparent molecular weights.
Clathrin cage formation assay
To 100 µl of 1.5 µM clathrin (25 mM HEPES, 125 mM potassium acetate, 5 mM magnesium acetate pH 6.9) were added the protein whose cage-forming ability was to be tested (in buffer A) and buffer A to a final volume of 200 µl, giving a final pH of ∼7. The reaction was incubated at 15°C for 10 min. Clathrin cages were pelleted by centrifugation at 100 000 g for 15 min at 4°C. The supernatant was removed and the cages resuspended in 20 µl buffer. Samples of the resuspended cages were analysed by SDS–PAGE and negative stain (uranyl acetate) electron microscopy to determine the degree of clathrin cage formation that had occurred.
Endocytosis assay and immunocytochemistry
Clathrin-mediated endocytosis was measured by assaying transferrin uptake into COS-7 fibroblasts as described (Wigge et al., 1997). For quantitation, blocked cells were defined as all transfected cells in which transferrin uptake was <20% of normal. Blocked cells were then expressed as a percentage of the total number of transfected cells in multiple fields.
Acknowledgments
Acknowledgements
We thank A.J.McCoy and C.J.Smith for critical reading of the manuscript, M.E.M.Noble for hydrophobic potential calculations and H.M.Kent and M.G.Ford for technical assistance.
References
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Benmerah A., Gagnon,J., Begue,B., Megarbane,B., Dautry-Varsat,A. and Cerf-Bensussan,N. (1995) The tyrosine kinase substrate eps15 is constitutively associated with the plasma membrane adaptor AP2. J. Cell Biol., 131, 1831–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Fre,S., Slepnev,V.I., Capua,M.R., Takei,K., Butler,M.H., Di Fiore,P.P. and De Camilli,P. (1998) Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature, 394, 793–797. [DOI] [PubMed] [Google Scholar]
- Collaborative Computing Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cupers P., Jadhav,A. and Kirchhausen,T. (1998) Assembly of clathrin coats disrupts the association between Eps15 and AP2 adaptors. J. Biol. Chem., 273, 1847–1850. [DOI] [PubMed] [Google Scholar]
- De Beer T., Carter,R., Lobel-Rice,K.E., Sorkin,A. and Overduin,M. (1998) Structure and asn-pro-phe binding pocket of the eps15 homology domain. Science, 281, 1357–1360. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica E., Klumperman,J., Stoorvogel,W. and Bonifacino,J. (1998) Association of the AP-3 adaptor complex with clathrin. Science, 280, 431–434. [DOI] [PubMed] [Google Scholar]
- Drake M., Downs,M. and Traub,L. (2000) Epsin binds to clathrin by associating directly with the clathrin-terminal domain. J. Biol. Chem., 275, 6479–6489. [DOI] [PubMed] [Google Scholar]
- Gaidarov I. and Keen,J. (1999) Phosphoinisitide-AP2 interactions required for targetting to plasma membrane clathrin-coated pits. J. Cell Biol., 146, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I., Santini,F., Warren,R. and Keen,J. (1999) Spatial control of coated-pit dynamics in living cells. Nature Cell Biol., 1, 1–7. [DOI] [PubMed] [Google Scholar]
- Goodford P. (1996) Multivariate characterisation of molecules for QSAR. J. Chemometrics, 10, 107–117. [Google Scholar]
- Goud B., Huet,C. and Louvard,D. (1985) Assembled and unassembled pools of clathrin: a quantitative study using an enzyme immunoassay. J. Cell Biol., 100, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene B., Liu,S.-H., Wilde,A. and Brodsky,F.M. (2000) Complete reconstitution of clathrin basket formation with recombinant protein fragments: Adaptor control of clathrin self-assembly. Traffic, 1, 69–75. [DOI] [PubMed] [Google Scholar]
- Hao W., Luo,Z., Zheng,L., Prasad,K. and Lafer,E.M. (1999) AP180 and AP2 interact directly in a complex that cooperatively assembles clathrin. J. Biol. Chem., 274, 22785–22794. [DOI] [PubMed] [Google Scholar]
- Hirst J. and Robinson,M.S. (1998) Clathrin and adaptors. Biochim. Biophys. Acta, 1404, 173–193. [DOI] [PubMed] [Google Scholar]
- Iannolo G., Salcini,A.E., Gaidarov,I., Goodman,O.B., Baulida,J. and Carpenter,G. (1997) Mapping of the molecular determinants involved in the interaction between eps15 and AP2. Cancer Res., 57, 240–245. [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T. (1999) Adaptors for clathrin-mediated traffic. Annu. Rev. Cell. Dev. Biol., 15, 705–732. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T., Nathanson,K.L., Matsui,W., Vaisberg,A., Chow,E.P., Burne,C., Keen,J.H. and Davis,A.E. (1989) Structural and functional division into two domains of the large (100–115kDa) chains of the clathrin-associated protein complex AP2. Proc. Natl Acad. Sci. USA, 86, 2612–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A.G. W. (1992) Recent changes to the MOSFLM package for processing film and image plate data. In, Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography No. 26. SERC, Daresbury Laboratory, Warrington, UK. [Google Scholar]
- Mellman I. and Warren,G. (2000) The road taken: past and future foundations of membrane traffic. Cell, 100, 99–112. [DOI] [PubMed] [Google Scholar]
- Morris S., Schroder,S., Plessmann,U., Weber,K. and Ungewickell,E. (1993) Clathrin assembly protein AP180: primary structure, domain organization and identification of a clathrin binding site. EMBO J., 12, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.A., Mann,A. and Ungewickell,E. (1990) Analysis of 100–180kDa phosphoproteins in clathrin-coated vesicles from bovine brain. J. Biol. Chem., 265, 3354–3357. [PubMed] [Google Scholar]
- Murphy J. and Keen,J. (1992) recognition sites for clathrin-associated protein AP2 and protein AP3 on clathrin triskelia. J. Biol. Chem., 267, 10850–10855. [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Ohno H. et al. (1995) Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science, 269, 1872–1875. [DOI] [PubMed] [Google Scholar]
- Owen D.J. and Evans,P.R. (1998) A structural explanation for the recognition of tyrosine based endocytic signals. Science, 282, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.J., Wigge,P., Vallis,Y., Moore,J.D.A., Evans,P.R. and McMahon,H.T. (1998) Crystal structure of the amphiphysin2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J., 17, 5273–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.J., Vallis,Y., Noble,M.E.M., Hunter,J.B., Dafforn,T.R., Evans,P.R. and McMahon,H.T. (1999) A structural explanation for the binding of multiple ligands by the α-adaptin appendage domain. Cell, 97, 805–815. [DOI] [PubMed] [Google Scholar]
- Pearse B., Smith,C. and Owen,D. (2000) Clathrin coat construction in endocytosis. Curr. Opin. Struct. Biol., 10, 220–228. [DOI] [PubMed] [Google Scholar]
- Ponnambalam S., Robinson,M., Jackson,A., Peiperl,L. and Parham,P. (1990) Conservation and diversity in families of cpated vesicle adaptins: structural analysis of β-adaptin. J. Biol. Chem., 265, 4814–4820. [PubMed] [Google Scholar]
- Rapoport I., Miyazaki,M., Boll,W., Duckworth,B., Cantley,L.C., Shoelson,S. and Kirchhausen,T. (1997) Regulatory interactions in the recognition of endocytic sorting signals by AP-2 complexes. EMBO J., 16, 2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport I., Chen,Y.,C., Cupers,P., Shoelson,S.E. and Kirchhausen,T. (1998) Dileucine-based sorting signals bind to the β chain of AP-1 at a site distinct and regulated differently form the tyrosine-based motif-binding site. EMBO J., 17, 2148–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J. and Kelly,R. (1999) The endocytic machinery of nerve terminals surrounds sites of exocytosis. Curr. Biol., 9, 1411–1414. [DOI] [PubMed] [Google Scholar]
- Salcini A., Confalonieri,S., Doria,M., Santolini,E., Tassi,E., Minenkova,O., Cesareni,G., Pelicci,P. and DiFiore,P. (1997) Binding specificty and in vivo targets of the EH domain, a novel protein–protein interaction module. Genes Dev., 11, 2239–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S.L. (1997) Clathrin-coated vesicle formation and protein sorting. Annu. Rev. Biochem., 66, 511–548. [DOI] [PubMed] [Google Scholar]
- Shih W., Gallusser,A. and Kirchhausen,T. (1995) A clathrin-binding site in the hinge region of the β2 chain of mammalian AP2 complexes. J. Biol. Chem., 270, 31083–31090. [DOI] [PubMed] [Google Scholar]
- Shupliakov O., Low,P., Grabs,D., Gad,H., Chen,H., David,C., Takei,K., De Camilli,P. and Brodin,L. (1997) Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science, 276, 259–263. [DOI] [PubMed] [Google Scholar]
- Smith C.J., Grigorieff,N. and Pearse,B.M.F. (1998) Clathrin coats at 21 Å resolution: a cellular assembly deigned to recycle multiple membrane receptors. EMBO J., 17, 4943–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Haar E., Harrison,S.C. and Kirchhausen,T. (2000) Peptide-in-groove interactions link target proteins to the β-propeller of clathrin. Proc. Natl Acad. Sci. USA, 97, 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub L.M. (1997) Clathrin-associated adaptor proteins putting it all together. Trends Cell Biol., 7, 43–46. [DOI] [PubMed] [Google Scholar]
- Traub L.M., Downs,M.A., Westrich,J.L. and Fremont,D.H. (1999) Crystal structure of the α appendage of AP2 reveals a recruitment platform for clathrin-coat assembly. Proc. Natl Acad. Sci. USA, 96, 8907–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P., Vallis,Y. and McMahon,H. (1997) Inhibition of receptor-mediated endocytosis by the amphiphysin SH3 domain. Curr. Biol., 7, 554–560. [DOI] [PubMed] [Google Scholar]
- Wilde A. and Brodsky,F.M. (1996) In vivo phosphorylation of adaptors regulates their interaction with clathrin. J. Cell Biol., 135, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybe J.A., Brodsky,F.M., Hofmann,K., Lin,K., Liu,S.-H., Chen,L., Earnest,T.N., Fletterick,R.J. and Hwang,P.K. (1999) Clathrin self-assembly is mediated by a tandemly repeated superhelix. Nature, 399, 371–375. [DOI] [PubMed] [Google Scholar]