Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment (original) (raw)
. Author manuscript; available in PMC: 2011 Jan 21.
Published in final edited form as: Science. 2002 May 3;296(5569):920–922. doi: 10.1126/science.1068452
Abstract
Neurofibromatosis type 1 (NF1) is one of the most prevalent dominantly inherited genetic diseases of the nervous system. NF1 encodes a tumor suppressor whose functional loss results in the development of benign neurofibromas that can progress to malignancy. Neurofibromas are complex tumors composed of axonal processes, Schwann cells, fibroblasts, perineurial cells, and mast cells. Through use of a conditional (cre/lox) allele, we show that loss of NF1 in the Schwann cell lineage is sufficient to generate tumors. In addition, complete NF1-mediated tumorigenicity requires both a loss of NF1 in cells destined to become neoplastic as well as heterozygosity in non-neoplastic cells. The requirement for a permissive haploinsufficient environment to allow tumorigenesis may have therapeutic implications for NF1 and other familial cancers.
Approximately 1 in 3500 individuals is born with a mutation in the NF1 gene and, because afflicted individuals can be long-lived, most mutations are retained within the population (1–3). The NF1 gene encodes neurofibromin, a protein of 2818 amino acids that harbors a functional Ras-GAP (guanosine triphosphatase activating protein) domain in its central region (4, 5). Neurofibromin is highly conserved among vertebrate species and has a 60% protein-wide homology with the Drosophila homolog (6).
The plexiform neurofibroma, thought to be embryonic in origin, can physically impede normal neurologic function. In addition, plexiform neurofibromas undergo transformation into malignant peripheral nerve sheath tumors, the most common malignancy associated with NF1 (1–3, 7). It is unknown whether neurofibroma formation requires NF1 loss of heterozygosity (LOH) in a single cell type or in some specific complement of all the cell types commonly present in these tumors. A feature of NF1, as well as of other familial cancers, is that all nontumor tissue is heterozygous for the tumor suppressor. It is unknown whether tumor suppressor heterozygosity contributes to tumor formation or growth.
Chimeric analysis using NF1 homozygous (_NF1_−/−) embryonic stem cells has demonstrated that mice can develop tumors that resemble plexiform neurofibroma (8). These studies, however, did not address the identity of the cell type(s) that undergoes tumor initiation. To test the hypothesis that NF1 loss in Schwann cells is the genetic bottleneck for neurofibroma formation, we used a Cre transgene under control of the Krox20 promoter to ablate NF1 function in these cells (9, 10). Analysis of the Krox20-cre activity [Web figs. 1 and 2 (11)], confirms the specificity of the Cre transgene to Schwann cells in the peripheral nervous system. Among additional cell types that are normally present in neurofibromas, including neurons, fibroblasts, and mast cells, little or no Cre activity is found. Mice that bear the Krox20-cre transgene and are heterozygous for the NF1 locus (NF1flox/−; Krox20-cre) were bred to NF1flox/flox mice (12).
All progeny with the NF1flox/−;Krox20-cre genotype (n = 12) showed classic signs of illness after 10 to 12 months of age and were necropsied along with control littermates. In all cases, the peripheral nerves of the NF1flox/−; Krox20-cre mice were enlarged in diameter [Web fig. 3 (11)]. This feature has also been observed in NF1 patients who undergo surgery for plexiform neurofibromas (13). Histological, immunohistochemical, and ultrastructural analyses confirmed that the cause of enlarged nerve girth was increased cellularity between individual axons [Web fig. 3 (11)].
More detailed histological analysis of cranial nerves and spinal nerve roots revealed that each mouse with this genotype had extensive proliferation of delicate, elongated, bipolar cells identical to those seen in human neurofibromas (14) (Fig. 1, A to F). Tumor sections were evaluated immunohistochemically, and all NF1flox/−;Krox20-cre tumors exhibited S100 immunoreactivity (Fig. 1, C and F) and a series of neural crest, Schwann cell, perineurial fibroblast, and neuronal markers [Web table 1 (11)]. Human neurofibromas are composed of a mixture of cells that retain a functional NF1 allele and cells that have undergone LOH (15). Accordingly, DNA analysis of the isolated murine tumor tissues (Fig. 1, G and H) indicates the presence of the recombined NF1flox allele in a subpopulation of cells in conjunction with cells that retain the intact flox allele. Ultra-structural evaluation of the tumors revealed abundant collagen and prominent Schwann cell cytoplasmic processes between axons (Fig. 2, A and B), as well as concentric arrays of perineurial cells around individual axons (Fig. 2, C and D). We also observed mast cell infiltration into the tumors, as assessed by morphological, histological, and enzymatic criteria (Fig. 2, E and F). These findings indicate that Krox20-cre–mediated ablation of NF1 in Schwann cells is sufficient to initiate neurofibroma formation with 100% penetrance, in association with the presence of multiple additional cell types that retain NF1 function. The plexiform neurofibromas observed in NF1flox/−;Krox20-cre mice exhibit every discernible molecular and histological feature of the human counterpart.
Fig. 1.
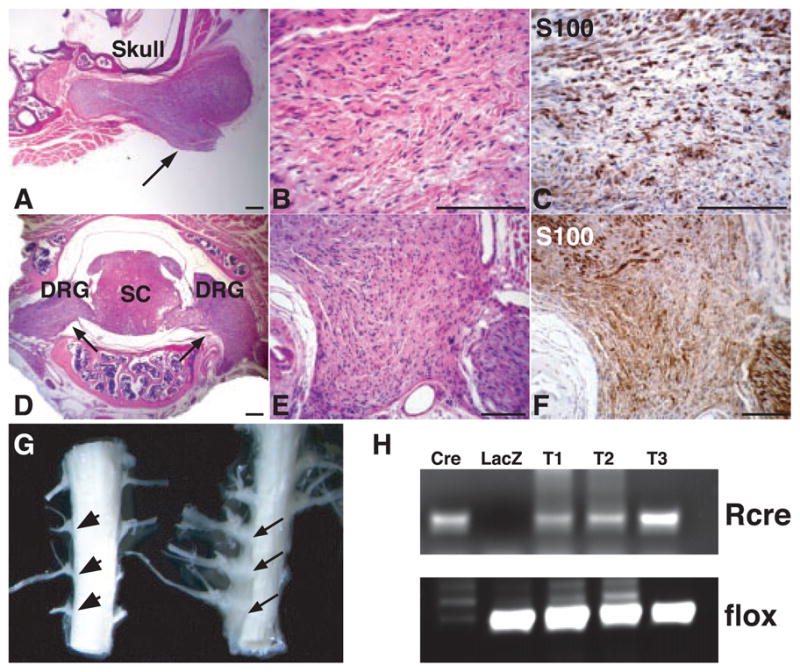
Histological and molecular analysis of neurofibromas from NF1 flox/−;Krox20-cre mice. Sections from a neurofibroma (arrow) associated with cranial nerves were stained with hematoxylin and eosin (H&E) (A and B) and S100 antibody (C). Sections from two neurofibromas (arrows) originating from dorsal root ganglia (DRG) were stained with H&E (D and E) and S100 (F). SC, spinal cord. (G) An example of three macroscopic neurofibromas from spinal roots (right, arrows), as compared to control spinal roots (left, arrowheads). (H) Genomic DNAs isolated from three neurofibromas in (G) (T1 to T3) were subjected to polymerase chain reaction analysis, as described in (11). Cre, genomic DNAs from a NF1flox/flox Schwann cell culture infected with Cre-expressing adenovirus. Of note, only the recombined allele (Rcre) remains in this culture. LacZ, genomic DNAs from similar Schwann cell culture infected with LacZ-expressing adenovirus. Scale bar, (A) to (F), 25 μm.
Fig. 2.
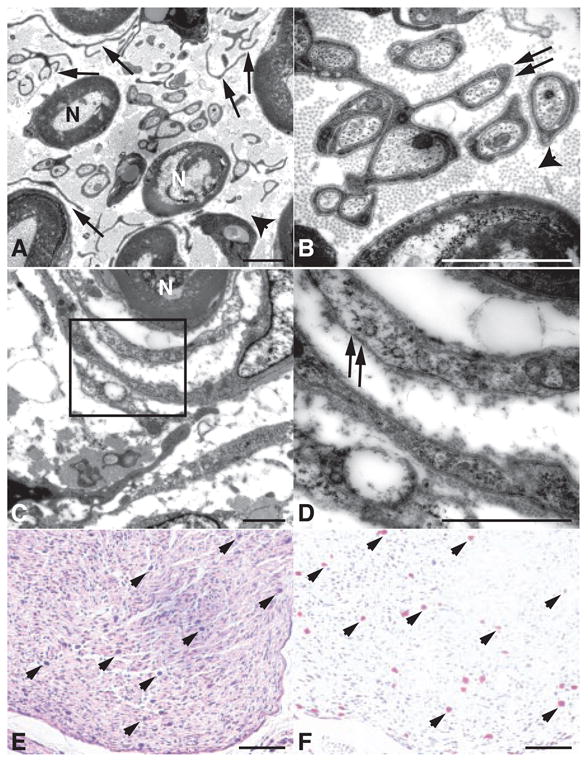
Neurofibromas from NF1flox/−;Krox20-cre mice contain multiple cell types. (A) Higher magnification of a neurofibroma demonstrates myelinated axons (N) separated by dissociated Schwann cells (arrows) and collagen bundles (arrowhead). (B) In addition to dissociated Schwann cells, the neurofibroma also contains differentiated Schwann cell processes surrounding individual unmyelinated axons (arrows), suggesting a continuum of dysplastic to overly neoplastic transformation. (C) Electron microscopy analysis demonstrates the presence of perineurial-like cells surrounding axons in a concentric fashion, reminiscent of the multilamellar arrays of perineurial cells seen in human perineuriomas (14). (D) In contrast to Schwann cells elsewhere in the tumor, these perineurial-like cells lack a continuous basal lamina (arrows). Sections from a neurofibroma were stained with H&E (E) and Leder staining, an enzymatic stain for Naphthol AS-D (3-hydroxy-2-naphoic acid-O-toluidine) choloroacetate esterase (F), displaying numerous mast cells (arrowheads) infiltrating the tumors. Scale bar, (A) to (D), 2 μm; (E) and (F), 100 μm.
Unlike sporadic forms of cancer, familial cancers are dominantly inherited and are triggered by the somatic loss of the one remaining functional allele at a tumor suppressor–encoding locus. To address whether the heterozygous state of NF1 in somatic cells other than the tumor-initiating Schwann cells plays an active role in neurofibroma formation, we constructed mice that have intact (wild-type) NF1 function in all cells except for those that express the Krox20-cre transgene (NF1flox/flox;Krox20-cre). We aged and necropsied three mice of the NF1flox/flox;Krox20-cre genotype. The NF1flox/flox;Krox20-cre mice did not exhibit enlarged peripheral nerves. However, microscopic examination revealed evidence of hyperplastic lesions in the cranial nerves. The frequency of lesions (Fig. 3A), as well as lesion size and collagen accumulation, was greatly reduced in NF1flox/flox;Krox20-cre mice as compared to NF1flox/−;Krox20-cre mice (Fig. 3B).
Fig. 3.

Phenotypic comparison of NF1flox/−;Krox20-cre (CKO×1+/−) and NF1flox/flox;Krox20-cre (CKO×1/×1) mice. (A) Quantification of neurofibromas associated with cranial nerves (CN) and spinal nerves (SN) from 12-month-old (n = 6) and 15-month-old (n = 1) CKO×1/− mutant mice and of hyperplastic lesions from similar areas of 12-month-old (n = 2) and 15-month-old (n = 1) CKO×1/×1 mice. Data are means ±SEM, n = 2 to 6, P < 0.01. (B) Neurofibromas (marked by dotted lines) that develop in the CKO×1/− mice are significantly larger than hyperplastic lesions from CKO×1/×1 mice. HB, hindbrain. (C) A substantial infiltration of mast cells (arrowheads), identified by Leder staining, found in 6-month-old CKO×1/− pre-neoplastic cranial and spinal nerves, whereas few mast cells are present in the hyperplastic nerves of 15-month-old CKO×1/×1 mice. Scale bar, (B), 50 μm; (C), 100 μm.
We also observed a marked reduction of mast cell infiltration in hyperplastic lesions from the wild-type background. We compared peripheral nerves from mice of three different genotypes: NF1+/− mice (containing +/− Schwann cells and +/− mast cells); NF1flox/−;Krox20-cre mice (containing −/− Schwann cells and +/− mast cells); and NF1flox/flox;Krox20-cre mice (containing −/− Schwann cells and +/+ mast cells). Six-month-old NF1flox/−;Krox20-cre mice exhibit Schwann cell hyperplasia with substantial infiltration of mast cells that persist as the lesions progress to frank neurofibromas. In contrast, the hyperplastic lesions found in the 12- to 16-month NF1flox/flox;Krox20-cre mice and peripheral nerves of 12-month NF1+/− mice had fewer mast cells (Fig. 3C), providing genetic evidence that the haploinsufficient state of the somatic tissue surrounding NF1 tumors has a functional contribution to tumor formation (initiation or progression).
The notion that tumor formation is a coordinated process in which incipient tumor cells recruit collaborating cells from the environment has established a firm foothold (16). For a normal cell to transform into a fully tumorigenic cell, many internal changes must take place. Among the requirements, it has been acknowledged that cell cycle suppressors must be shut down, growth factor requirements must be eluded, blood vessel formation must be induced, and apoptotic signals must be evaded (16). This cell autonomous process not only reconfigures the nature of the resident tumor cell but also reconfigures that of its cellular environment (17). The present study identifies a non–cell autonomous role for the development of tumors in NF1. The onset, growth potential, and multicellular nature of the _NF1_−/− neurofibromas is suppressed when the cellular environment retains both functional NF1 alleles. We have ruled out trivial explanations for the observed difference in tumor incidence that relate to the potential relative inefficiency of the Cre transgene to ablate two alleles of the floxed NF1 gene in the flox/flox configuration, versus one allele in the flox/− configuration. Cultured Schwann cells from newborn (P0) spinal nerves of NF1flox/flox;Krox20-cre mice exhibit a transformed morphology indistinguishable from that of the _NF1_−/− cells described previously [Web fig. 4 (11)] (18). Because _NF1_−/− Schwann cells have a growth disadvantage as compared with wild-type Schwann cells (18), we can rule out that the transformed cells may overtake any nontransformed cells in the cultures. Hence, a majority, if not all, of Schwann cells has undergone loss of NF1 in the context of two floxed alleles. Indeed, the requirement of a heterozygous NF1 state for plexiform neurofibroma formation may explain two clinical observations. (i) With extremely rare exceptions, the plexiform neurofibroma is found only in persons afflicted with NF1 (19). (ii) Tumors arising within spinal roots in the setting of NF1 are exclusively plexiform neurofibromas, whereas tumors in a similar location in the setting of NF2 or sporadic forms are predominantly Schwannomas (20). Finally, as elaborated below, we identify a specific cellular type that exhibits altered properties in the heterozygous versus wild-type environment.
The fact that NF1+/− mast cells invade pre-neoplastic nerves and remain present throughout the development of the tumor is in stark contrast to the absence of NF1+/+ mast cells in the NF1flox/flox;Krox20-cre hyperplasias that fail to form frank neurofibromas. Previous studies have described the enhanced proliferative properties of heterozygous mast cells from NF1 patients and from NF1+/− mice (21). Given the breadth of cytokine expression found in degranulating mast cells, it is tempting to speculate that these cells could play a central role in the initiation of neurofibroma formation (1). In this scenario, sensitized heterozygous (NF1+/−) mast cells homing to nullizygous (_NF1_−/−) NF1 Schwann cells in peripheral nerves would create a cytokine-rich microenvironment that is apparently permissive for tumor growth. This effect is presumably confined to heterozygous mast cells interacting with nullizygous Schwann cells, because in the original NF1 knockout mouse, the heterozygous Schwann cells in peripheral nerves were unable to attract heterozygous mast cells.
Our results suggest that it may be possible to prevent or delay tumor formation in NF1 by designing therapies that neutralize the effects of haploinsufficiency before the onset of tumorigenesis. Moreover, tumor formation in other familial cancers may merit similar scrutiny to determine the potential contribution of the heterozygous environment (22).
Supplementary Material
Science Zhu et al Supp Data
Acknowledgments
We thank P. Houston and F. Guignard for technical assistance; E. Rushing for pathology consultation; L. Klesse and S. Kernie for critically reading the manuscript; members of the Parada lab for support; M. Sliwkowski for providing heregulin; and members of the National Neurofibromatosis Foundation Consortium for stimulating discussions. Supported by grants from the National Institute of Neurological Disorders and Stroke and Department of Defense (L.F.P.). Y.Z. is a recipient of Young Investigator Award from the National Neurofibromatosis Foundation.
References and Notes
- 1.Riccardi VM. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. 2. Johns Hopkins Univ. Press; Baltimore, MD: 1992. [Google Scholar]
- 2.Cichowski K, Jacks T. Cell. 2001;104:593. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Parada LF. Exp Cell Res. 2001;264:19. doi: 10.1006/excr.2000.5138. [DOI] [PubMed] [Google Scholar]
- 4.Ballester R, et al. Cell. 1990;63:851. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 5.Xu GF, et al. Cell. 1990;63:835. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- 6.The I, et al. Science. 1997;276:791. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 7.Korf BR. Am J Med Genet. 1999;89:31. doi: 10.1002/(sici)1096-8628(19990326)89:1<31::aid-ajmg7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 8.Cichowski K, et al. Science. 1999;286:2172. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 9.Topilko P, et al. Nature. 1994;371:796. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 10.Voiculescu O, et al. Genesis. 2000;26:123. doi: 10.1002/(sici)1526-968x(200002)26:2<123::aid-gene7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Supplemental figures and details for methods are available on Science Online at www.sciencemag.org/cgi/content/full/296/5569/920/DC1.
- 12.Zhu Y, et al. Genes Dev. 2001;15:859. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martuza R. personal communication.
- 14.Kleihues P, Cavenee WK. Pathology and Genetics of Tumors of the Nervous System. IARC Press; Lyon, France: 2000. [Google Scholar]
- 15.Serra E, et al. Hum Mol Genet. 2000;9:3055. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 17.Coussens LM, et al. Cell. 2000;103:481. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HA, et al. Oncogene. 1995;11:325. [PubMed] [Google Scholar]
- 19.Woodruff JM. Am J Med Genet. 1999;89:23. doi: 10.1002/(sici)1096-8628(19990326)89:1<23::aid-ajmg6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Halliday AL, et al. J Neurosurg. 1991;74:248. doi: 10.3171/jns.1991.74.2.0248. [DOI] [PubMed] [Google Scholar]
- 21.Ingram DA, et al. J Exp Med. 2000;191:181. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudson AG. Annu Rev Genet. 2000;34:1. doi: 10.1146/annurev.genet.34.1.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Science Zhu et al Supp Data