Multistage nanoparticle delivery system for deep penetration into tumor tissue (original) (raw)
Abstract
Current Food and Drug Administration-approved cancer nanotherapeutics, which passively accumulate around leaky regions of the tumor vasculature because of an enhanced permeation and retention (EPR) effect, have provided only modest survival benefits. This suboptimal outcome is likely due to physiological barriers that hinder delivery of the nanotherapeutics throughout the tumor. Many of these nanotherapeutics are ≈100 nm in diameter and exhibit enhanced accumulation around the leaky regions of the tumor vasculature, but their large size hinders penetration into the dense collagen matrix. Therefore, we propose a multistage system in which 100-nm nanoparticles “shrink” to 10-nm nanoparticles after they extravasate from leaky regions of the tumor vasculature and are exposed to the tumor microenvironment. The shrunken nanoparticles can more readily diffuse throughout the tumor's interstitial space. This size change is triggered by proteases that are highly expressed in the tumor microenvironment such as MMP-2, which degrade the cores of 100-nm gelatin nanoparticles, releasing smaller 10-nm nanoparticles from their surface. We used quantum dots (QD) as a model system for the 10-nm particles because their fluorescence can be used to demonstrate the validity of our approach. In vitro MMP-2 activation of the multistage nanoparticles revealed that the size change was efficient and effective in the enhancement of diffusive transport. In vivo circulation half-life and intratumoral diffusion measurements indicate that our multistage nanoparticles exhibited both the long circulation half-life necessary for the EPR effect and the deep tumor penetration required for delivery into the tumor's dense collagen matrix.
Keywords: drug delivery, cancer therapy, nanomedicine
Nanoparticles (NPs) have offered new approaches to the delivery of cancer therapeutics (1–5). Doxil (≈100 nm PEGylated liposomal form of doxorubicin) and Abraxane (≈130 nm albumin-bound paclitaxel nanoparticle) are two examples of Food and Drug Administration-approved nanoparticle-based therapeutics for solid tumors; their large size compared with conventional cancer therapeutics allows them to preferentially accumulate in solid tumors by the enhanced permeation and retention (EPR) effect (6), thus reducing normal tissue toxicity. However, despite the improved pharmacokinetic properties (7) and reduced adverse effects, these drugs have provided only modest survival benefits (1, 8–10). This shortcoming is likely attributed to the physiological barriers imposed by the abnormal tumor vasculature and the dense interstitial matrix—a complex assembly of collagen, glycosaminoglycans, and proteoglycans—which hinder delivery of the drug throughout the entire tumor in sufficient concentration (11, 12).
Systemic delivery of therapeutics to the tumor is a three-step process: blood-borne delivery to different regions of the tumor, transport across the vessel wall, and passage through the interstitial space to reach the tumor cells (13). Abnormalities in the tumor vasculature lead to highly heterogeneous vascular perfusion throughout the tumor. The microvascular density is high at the invasive edge of the tumor, but sometimes the tumor center is unperfused, preventing delivery of therapeutics to this region. However, the tumor center's hostile microenvironment (low pH and low pO2) harbors the most aggressive tumor cells, and the tumor will regenerate if these cells are not eliminated. Moreover, exposure of the cancer cells to sublethal concentration of the therapeutic agent may facilitate the development of resistance.
Hyperpermeability of the abnormal vasculature and lack of functional lymphatics lead to elevated levels of interstitial fluid pressure (IFP) (14, 15). This interstitial hypertension, in turn, reduces convective transport across the vessel wall and into the interstitial space, leaving diffusion as the primary mode for drug transport to the poorly perfused regions. Large 100-nm nanoparticles are suitable for the EPR effect (6) but have poor diffusion in the dense collagen matrix of the interstitial space (16, 17), resulting in restrictive nanoparticle accumulation around tumor blood vessels and little penetration into the tumor parenchyma. In the case of Doxil for example, the liposomal particles are trapped close to the tumor vasculatures. Although the small size (≈400 MW) of doxorubicin, which is released from the liposomes, seemingly allow rapid diffusion, doxorubicin cannot migrate far from the particles because of avid binding to DNA and sequestration in acidic endosomes of perivascular tumor cells (18, 19), resulting in heterogeneous therapeutic effects.
Consequently, we propose a multistage approach in which nanoparticles change their size to facilitate transport by adapting to each physiological barrier. The original 100-nm nanoparticles preferentially extravasate from the leaky regions of the tumor vasculature. After extravasation into tumor tissue, the nanoparticles shrink to 10 nm, significantly lowering their diffusional hindrance in the interstitial matrix (20) and allowing penetration into the tumor parenchyma. These smaller nanoparticles can potentially be used as nanocarriers for therapeutics that are released as the particles penetrate deep into the tumor (Fig. S1). Surface PEGylation of the small nanoparticles allows them to diffuse smoothly in the interstitial matrix by reducing the binding, sequestration, and metabolism that hinder the transport of therapeutic agents (21, 22). Furthermore, 10-nm nanoparticles are not cleared from the tumor as rapidly as much smaller molecular species because of their larger size.
To achieve this size-shrinking property, a large nanoparticle should be triggered to release smaller nanoparticles after extravasation into the tumor. Several nanoparticles have been designed to release their contents remotely via an external stimulus (light, heat, ultrasound, magnetic field), but their use to date has been limited to local therapy. Systemic therapy is necessary to treat the metastases, which are the major cause of cancer mortality. Water hydrolysis- (23), diffusion-, or solvent-controlled release mechanisms can achieve systemic effects but do not give preferential release in the tumor, resulting in increased toxicity in normal tissues. To attain both systemic therapeutic effects and preferential release in tumor tissues, we aim to trigger the size change by using an endogenous stimulus characteristic of the tumor microenvironment, such as low pH, low partial oxygen pressure, or high concentrations of matrix metalloproteinases (MMPs). Acidic and hypoxic regions tend to be far from blood vessels (24), not in the perivascular regions where the large nanoparticles are trapped. MMPs, particularly gelatinases A and B (MMP-2 and MMP-9), are key effectors of angiogenesis, invasion, and metastasis including the epithelial-mesenchymal transition (EMT), a cell-biological program that executes many of the steps of the invasion-metastasis cascade. They cleave away the extracellular matrix (ECM), creating space for the cell to move and releasing sequestered growth factors (25, 26). Levels of MMP-2 and MMP-9 are high at the invasive edge of tumors and at the sites of angiogenesis—regions the large nanoparticles are likely to extravasate. These conditions make enzymatic degradation by MMPs a highly favorable trigger mechanism.
Because both MMP-2 and MMP-9 are extremely efficient at hydrolyzing gelatin (denatured collagen), we engineered a 100-nm nanoparticle with a core composed of gelatin and a surface covered with quantum dots (QDs), a model 10-nm nanoparticle (Fig. 1_A_). Gelatin nanoparticles have been shown to have a long blood circulation time and high accumulation in tumor tissues (27), properties necessary for the first-stage NP carrier. To assess the spatial and temporal distribution of the nanoparticles in the tumor milieu, we used ≈10 nm QDs as a stand-in for therapeutic nanocarriers, so that in vivo distribution of the nanoparticles can be imaged by using time-lapse multiphoton microscopy. Compared with traditional organic fluorophores, QDs have high resistance to photo and chemical degradation, narrow photoluminescence spectra, broad excitation spectral windows, and large two-photon absorption cross-sections—enabling the use of multiphoton microscopy to image deep into the tumor with high-spatial resolution (28).
Fig. 1.

QDGelNPs change their size in response to MMP-2. (A) Schematic of 100-nm QDGelNPs changing size to 10-nm QD NPs by cleaving away the gelatin scaffold with MMP-2, a protease highly expressed in tumor tissue. (B) GFC chromatograms of QDGelNPs at various times after incubation with MMP-2. Fluorescence signal at 565 nm is collected. (B Inset) Fluorescence spectrum of the peak at void volume for 2.2 h cleaving time shows that the signal originates from QDs on the QDGelNPs.
Results
The multistage quantum dot gelatin nanoparticles (QDGelNPs) are composed of a gelatin core with amino-PEG QDs conjugated to the surface using 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC)/sulfo-NHS coupling chemistry. Another layer of 5-kDa polyethylene glycol (PEG) is conjugated to the surface of the gelatin nanoparticles to confer long blood circulation time. X-ray photoelectron spectroscopy (XPS) was used to determine the presence of PEG chains on the QDGelNPs surface (Fig. S2). Dynamic light scattering (DLS) of the QDGelNPs before and after PEGylation (but before purification) indicate an increase in diameter from 78.3 ± 0.2 nm to 93.7 ± 0.5 nm. DLS of the final structure after purification and size selection using gel filtration chromatography (GFC) revealed a single particle distribution with a hydrodynamic diameter of 97.9 ± 2.1 nm and a polydispersity of 41.2% (Fig. S3). This value agreed well with the average diameter of 99 ± 1 nm estimated from scanning electron microscopy (SEM) (Fig. 2 C and D). Inductively coupled plasma optical emission spectroscopy (ICP-OES) determined a concentration of 15 pmol of QDs per mg of QDGelNPs. The QDGelNPs showed excellent colloidal stability; their diameter by DLS remained nearly unchanged while in storage at ≈4 °C over 48 d—from 95.7 ± 4.1 nm on day 1–101.1 ± 2.5 nm on day 48 (Fig. 2_B_).
Fig. 2.

QDGelNP physical and in vitro characterization. (A) Epifluorescence image of QDGelNPs on a silicon substrate at 100× magnification. (Scale bar: 5 μm.) (B) DLS distribution of QDGelNP on day 1 and day 48 after synthesis and storage at 4 °C. (C) SEM image of QDGelNPs at 15,000× magnification. (Scale bar: 1 μm.) (C Inset) SEM image of individual QDGelNP at 35,000× magnification. (Scale bar: 100 nm.) (D) Histogram of QDGelNPs’ size distribution from image analysis of SEM image. (E and F) Kinetics of MMP-2–induced QD release from QDGelNPs. (E) QD-release curve from incubation of 0.1 mg (0.16 μM) of QDGelNPs with 230 ng of MMP-2. (F) QD release from incubation of 0.1 mg of QDGelNPs for 12 h with varying amounts of MMP-2. (G and H) FCS cross-correlograms of QDGelNPs before (G) and after (H) incubation with MMP-2.
We designed both the initial 100-nm NPs and the released 10-nm NPs to have a neutral surface charge, ensuring the difference in transport before and after cleaving is only a result of size change. In addition, a previous report suggested neutral particles are optimal for diffusion in the interstitial matrix (29). The ζ potential of the ≈100-nm QDGelNPs at pH 7.5 was −6.29 ± 0.22 mV and at pH 6 was −5.00 ± 0.12 mV. The ζ potential of the ≈10 nm QDs used for the second stage NP was −5.13 ± 0.16 mV at pH 7.5 and −4.36 ± 0.17 mV at pH 6 (Fig. S3). These results confirm the charge neutrality of both particles in the pH range found in normal tissues and solid tumors.
We next investigated the ability of MMP-2 to change the size of QDGelNPs in vitro by using GFC. GFC chromatograms using fluorescence detection (ex: 250 nm, em: 565 nm) were obtained from incubation of 0.1 mg of QDGelNPs with 230 ng (0.16 μM) of MMP-2 at 37 °C (Fig. 1_B_).A previous report has estimated the extracellular concentration of MMP-2 in HT-1080 (human fibrosarcoma) xenograft tumor tissue in vivo to be ≈1 mM (30), significantly higher than the concentration used in our in vitro experiment. The QDGelNPs initially eluted at the GFC column's void volume but after incubation with MMP-2 for various times up to 12 h, the peak shifted to a longer elution time corresponding to the smaller size of individual QDs, whereas incubation with 50% FBS showed no such shift (Fig. S4). We assessed that 50% of the QDs were released in ≈1.5 h and the percent of freed QDs saturated at ≈90% (Fig. 2_E_), regardless of longer incubation times or addition of more MMP-2. We repeated this experiment with the incubation time kept constant at 12 h but the amount of MMP-2 was varied (Fig. 2_F_). Under this condition, only ≈25 ng of MMP-2 was necessary to release 50% of the QDs. These results demonstrated the MMP-2 triggered size change occurred in an efficient manner.
We then verified that the released QDs diffuse optimally in the interstitial matrix, that residual gelatin/glutaraldehyde on their surface, imparted by cleavage of the gelatin NPs, do not lead to extraneous binding interactions or significant size increase. To investigate this possibility, we used fluorescence correlation spectroscopy (FCS) to directly measure the hydrodynamic diameter/diffusion coefficient of the QDs before and after cleaving the gelatin core (Fig. 2 G and H). The hydrodynamic diameter by FCS before cleaving was 81.1 ± 2.3 nm (D = 5.6 ± 0.2 × 10−8 cm2·s−1), which is consistent with the DLS measurement of 90.9 ± 1.3 nm for this batch. After MMP-2 digestion, the hydrodynamic diameter decreased to 9.7 ± 0.3 nm (D = 4.7 ± 0.2 × 10−7 cm2·s−1), which is the size of individual QDs, indicating the size increase of the released QDs from gelatin/glutaraldehyde fragments was negligible.
We next evaluated whether the size change observed in GFC and FCS enhances diffusive transport in dense collagen environments resembling those in solid tumors. To simulate the interstitial matrix of a solid tumor, we prepared a collagen gel in a capillary tube at 0.74% (7.4 mg/mL) concentration, similar to the reported estimate of 9.0 ± 2.5 mg/(mL interstitial matrix) for interstitial collagen in both human colon adenocarcinoma (LS174T) and murine mammary carcinoma (MCalV) implanted in mouse dorsal chambers (17, 31). The collagen gel penetration of the QDGelNPs before and after cleaving with MMP-2 was compared with a noncleavable, PEGylated, and QD-coated silica nanoparticles (32) control (Diam. = 105.6 ± 0.8 nm, ζ potential at pH 7.5 = −3.9 ± 0.2 mV) designed to behave like QDGelNPs before cleaving. A mixture of SilicaQDs and QDGelNPs (before or after cleaving) were placed in contact with the gel and incubated for 12 h. Infiltration of both particles into the collagen was determined using multiphoton microscopy with simultaneous second-harmonic generation (SHG) imaging of fibrillar collagen (Fig. 3). The SilicaQDs and QDGelNPs before cleaving both had negligible penetration and were excluded from the collagen matrix (Fig. S5). However, after cleavage of QDGelNPs with MMP-2, the freed QDs were able to penetrate over a millimeter into the gel. By fitting the concentration profile of the cleaved QDGelNPs to a one-dimensional diffusion model (33, 34), we obtained a diffusion coefficient of 2.3 × 10−7 cm2·s−1, the same diffusion coefficient obtained for individual QDs in the collagen gel before conjugation to the gelatin NP. The resulting diffusion coefficient ratio (D/Do, where Do is diffusion coefficient of freed QDs in solution obtained by FCS) in the collagen matrix is 0.49. This value agrees well with the expected value for D/Do of ≈0.52 derived from previous reports (Materials and Methods) (29, 31). This result indicates that the diffusion coefficient of released QDs in dense collagen increases to that of ≈10 nm particles and any residual gelatin/glutaraldehyde fragments remaining on the surface do not impede their diffusion.
Fig. 3.

Diffusion of SilicaQDs and QDGelNPs (before and after MMP-2 cleavage) in a collagen gel. (A and B) Fluorescence images of SilicaQDs (A) and QDGelNPs before MMP-2 cleavage (B) penetrating into the collagen gel. (C) Second-harmonic generation (SHG) signal shows the corresponding location of the collagen matrix. (Scale bars: 125 μm.) (D) Normalized intensity profile of SilicaQDs and QDGelNPs in collagen gel. (E and F) Fluorescence images of SilicaQDs (E) and QDGelNPs after MMP-2 cleavage (F) penetrating into the collagen gel. (G) SHG signal shows the corresponding location of the collagen matrix. (Scale bars: 125 μm.) (H) Normalized intensity profile of SilicaQDs and QDGelNPs after MMP-2 cleavage in collagen gel. Black line displays theoretical intensity profile for particles with diffusion coefficient of 2.3 × 10−7 cm2·s−1.
To test whether tumor secreted MMP-2 can change the size of QDGelNPs in vivo, we intratumorally coinjected QDGelNPs and SilicaQDs in the HT-1080 tumor implanted in the dorsal skin-fold window chamber of severe combined immunodeficient (SCID) mice. The HT-1080 tumor model was selected because of its reported high MMP-2 activity, which we confirmed by in situ gelatin zymography on a tumor tissue section (Fig. S6). Multiphoton microscopy revealed a marked increase in QDGelNPs penetration into surrounding tumor tissue as compared with the noncleavable SilicaQDs control, confirming a substantial enhancement in interstitial transport associated with size change (Fig. 4 and Fig. S7). At 6 h postinjection, the QDGelNPs had penetrated up to ≈300 μm from the injection site while the SilicaQDs control exhibited little or no dissemination from its initial location. We fitted the concentration profile to a model for substances diffusing from a spherical source to obtain an effective diffusion coefficient of ≈2.2 × 10−8 cm2·s−1 inside the tumor (Fig. S8). This value is ≈10% the diffusion coefficient obtained in the collagen gel, which can be explained by the increased time needed to cleave the particles, the tortuosity of the interstitial space induced by cellular obstacles (35), and the possibly higher collagen concentration in the HT-1080 tumor than in the gel we prepared.
Fig. 4.

In vivo images of QDGelNPs and SilicaQDs after intratumoral coinjection into the HT-1080 tumor. QDGelNPs imaged 1 (A), 3 (B), and 6 h (C) after injection. SilicaQDs imaged 1 (D), 3 (E), and 6 h (F) after injection. (Scale bar: 100 μm.)
We next determined the QDGelNPs’ blood half-life (t1/2β) to show that the QDGelNPs are not rapidly removed from circulation by the reticuloendothelial system. We systemically administered to nontumor bearing mice a mixture of the QDGelNPs and SilicaQDs by retro-orbital injection and measured the decrease in fluorescence from both particles in the blood over time. The SilicaQDs exhibited a blood half-life of 12.9 ± 2.4 h, whereas the QDGelNPs had a half-life of 22.0 ± 3.4 h (Fig. S9). The difference in the half-lives may be due to variations in the QDGelNPs’ surface chemistry that make it less immunogenic compared with SilicaQDs. These results established that QDGelNPs possess both the long circulation half-life and large 100-nm size necessary for preferential extravasation from the leaky regions of the tumor vasculature as well as the deep interstitial penetration of a 10-nm particle required for delivery to the tumor's poorly accessible regions.
Discussion
We have established three main criteria for the multistage QDGelNPs: the size of nanoparticles must change from 100 nm to 10 nm, their surface before and after MMP-2 cleavage needs to be well PEGylated and neutral, and their sensitivity to cleavage should be at least as high as other reported MMP-2 probes (36–38). Satisfying these three criteria simultaneously presented several design and synthetic challenges. However, by optimizing the coupling scheme and the degree of glutaraldehyde cross-linking, we were able to meet the desired criteria for this system while preserving the simplicity in design to ensure scalability.
Because both the carrier and released nanoparticles need to be highly PEGylated and neutral, encapsulation strategies that rely on hydrophobic or charged interactions are unsuitable. Using QDs with a sticky surface (low coverage of PEG) allowed for encapsulation inside the gelatin core, but the particle after cleavage showed a broad size distribution by GFC, indicating binding to gelatin/glutaraldehyde fragments. An alternative strategy is to covalently attach the QDs onto the gelatin NP surface. Although this approach is susceptible to interparticle cross-linking, through careful optimization and using EDC/sulfo-NHS chemistry to conjugate amino-PEG QDs to the carboxylic acid groups on the gelatin NP surface, we produced nearly monodisperse QDGelNPs that released cleaved QDs without size increase.
By controlling the length and degree of glutaraldehyde polymerization used to cross-link the gelatin nanoparticles, we were able to optimize the size of the QDs after cleaving and their rate of release while maintaining particle stability. The method for gelatin nanoparticle synthesis developed in ref. 39 produced gelatin NPs with long extended networks of glutaraldehyde on their surface to maintain particle stability in aqueous solution. However, when the same scheme was applied to our design, the QDs remained covalently attached to this large glutaraldehyde polymer after MMP-2 degradation, resulting in QDs that were significantly larger upon release. To produce released QDs without augmenting their size, we constructed a network of numerous short cross-links on the gelatin particles instead. Because glutaraldehyde readily self-polymerizes in solution, we used nearly monomeric glutaraldehyde (grade I) for consistent formation of short glutaraldehyde polymer cross-links. In addition, using grade I glutaraldehyde improved the MMP-2 cleaving rate considerably such that the release is as sensitive as previously reported MMP-2 probes. By modifying the degree of cross-linking on the gelatin nanoparticle, we optimized the QDGelNPs to be highly responsive to MMP-2 degradation while preserving particle stability in storage for at least 48 d.
Our FBS incubation experiment suggested the QDGelNPs are stable in serum conditions, but additional studies will be needed to verify that the QDGelNPs are not degraded in the blood by circulating MMPs (40) and other proteases. Circulating MMPs, however, have been reported to be inhibited by serum proteins such as α2-macroglobulins that entrap the MMPs (36, 41). The degree of PEGylation on the surface of the QDGelNPs may need to be optimized such that there is minimal cleaving and opsonization in the blood while achieving the desired release rate in tumor tissue. However, MMP levels may vary in different tumor types/individuals, causing inconsistent behavior. A strength of our method is the potential for customized delivery of nanoparticles by using genomic and molecular data to achieve optimal delivery for a particular patient. For example, high levels of tumor-specific urinary MMPs measured noninvasively in an individual can be indicative of high levels of the MMPs in plasma and tumor tissues (42). This information can help us customize the QDGelNPs for lower sensitivity to proteases (e.g., by increasing the particles’ cross-linking) to minimize degradation in the blood while maintaining the same desired release rate in the tumor. In addition, we can design a series of customized nanoparticles that are activated by a variety of tumor-associated proteases—such as cathepsin B and urokinase-type plasminogen activator (uPA). By measuring and modeling the levels of various tumor-associated proteases in circulation, the primary tumor, and the metastatic sites, a personalized combination and dosage of protease-activated nanoparticles, may then be administered to achieve an optimal therapeutic effect.
Multistage nanoparticle systems provide additional tunability in the spatial control of delivery to solid tumors. Because of the multiple physiological barriers that a therapeutic agent must encounter, a multistage nanoparticle system can enhance penetration by changing its size, charge, shape, flexibility, and/or surface coating to suit the transport across each barrier. We can extend the utility of this approach by incorporating additional stages into the QDGelNPs to guide its delivery into tumor cells or even subcellular compartment(s). Such an approach may not only improve the efficacy of current anticancer drugs but also make available drugs that have been abandoned because of delivery problems. As interest in increasingly sophisticated delivery systems grows, we face a corresponding challenge to synthesize nanostructures of increasing complexity. Innovative schemes will be necessary to assemble nanocomponents in complex configurations that keep the synthesis simple yet functional so that production can be scalable and cost-effective.
We have provided a proof-of-principle demonstration that a size-changing nanoparticle can facilitate delivery into the dense collagen matrix of a tumor. Ultimately, the QDs will be replaced with a 10-nm nanocarrier of cancer therapeutics, and its antitumor efficacy and survival benefits will be compared with that of conventional drugs and other nanoparticle-based treatments. The loading capacity per particle for 10-nm nanocarrier is limited. Therefore, not only the penetration depth but also sufficient quantity of nanoparticles needs to be delivered.
Materials and Methods
Synthesis of Gelatin Nanoparticle.
Detailed information regarding synthesis of the QDGelNP can be found in SI Materials and Methods. Gelatin nanoparticles were prepared from a modification of the two-step desolvation method developed in ref. 39. Gelatin type A (0.625 g) was dissolved in 12.5 mL of deionized (DI) water at 40 °C. Acetone (12.5 mL) was added to the solution at 6 mL/min. Exactly 1 min after the addition was completed, the supernatant containing the low molecular weight gelatin fraction was removed. DI water (12.5 mL) was added to the remaining precipitate and heated again to 40 °C until dissolution. Half the solution was removed, and the pH of the remaining half was adjusted to 2.7. Under constant stirring at 600 rpm and 40 °C, 20.75 mL of acetone was added at 1 mL/min. After the acetone addition was completed, 30 μL of 50% glutaraldehyde solution (Grade I) diluted in 1 mL of acetone was added to the gelatin solution at 0.05 mL/min to cross-link the particles. Subsequently, the solution was kept at 40 °C and 600 rpm stir rate for 7.5 h. The acetone was then removed. The remaining solution was filtered through a 0.2-μm syringe filter. A 1 M glycine solution (0.2 mL) was added, and the solution was stored overnight at 4 °C. A 1-mL solution of the gelatin particles was injected into a Superose 6 GL 10/300 column (GE Healthcare) for GFC purification. The peak eluting at the void volume was collected with 0.5-mL fractions. This purification procedure was repeated once more, and the first concentrated fractions from both GFC runs were combined.
Synthesis of QDGelNP.
The 1-mL gelatin nanoparticle solution was combined with 20 μL of 8 μM PEG QDs. After stirring for 1 h and eventually changing the pH to 6, 0.4 mg (2.1 μmol) of EDC and 0.4 mg (1.9 μmol) of _N_-hydroxysulfosuccinimide (sulfo-NHS) was dissolved in 50 μL of DI water and then added to the gelatin nanoparticle/QD mixture. The reaction proceeded for 3 h. Afterward, a solution of mPEG amine 5 kDa (20 mg, ≈4 μmol) dissolved in 50 μL of DI water was added to the gelatin/QD solution. Then, an additional solution of EDC (0.4 mg) and sulfo-NHS (0.4 mg) was added. After 2 h, the pH was adjusted to 8 and stirring continued for 1 h. A 1 M glycine solution (50 μL) was added to quench the reaction. After 30 min, the resulting mixture was filtered through a 0.2-μm syringe filter and then purified by using GFC with the Superose 6 column. The peak eluting at the void volume was collected with 0.5-mL fractions and the first concentrated fraction was used for further experiments.
Collagen Gel Diffusion.
Collagen hydrogels were prepared by mixing the following components in order on ice: 141.75 μL of 8.6 mg/mL rat tail collagen I (354249; BD Biosciences), 3.8 μL of 1 M sodium hydroxide, and 19.5 μL of 0.17 M EDTA. The final concentration of collagen was 7.38 mg/mL and EDTA was 20 mM. After vortexing, the gel was added to partially fill a microslide capillary tube (Vitrocom no. 2540), then incubated overnight at 37 °C. QDGelNPs (0.1 mg) were incubated with 230 ng of activated MMP-2 for 12 h in 50 mM Hepes, 2 mM CaCl2. At the end of 12 h, EDTA was added to give a final concentration of 20 mM. A 20-μL mixture of the QDGelNPs either before or after incubation with MMP-2 and SilicaQDs was added into the capillary tube and placed in contact with the surface of the collagen gel. The concentration of the two particles and sensitivity of the avalanche photodiodes (APD) were adjusted so that both particles gave similar signal intensities. The sample was left in ≈295 K for 12 h and then imaged by using a multiphoton laser scanning microscope. Image analysis was performed by using ImageJ. The concentration profile for the QDGelNPs after cleaving was fitted to the following one-dimensional model to obtain the diffusion coefficient in the collagen gel (33):
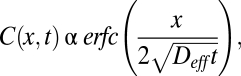
where erfc is the complementary error function. The nonlinear curve fitting was performed by using fminsearch in Matlab. The diffusion coefficient ratio (_D/D_o) was compared with reported values in refs. 31 and 29. In ref. 29, _D/D_o was found to be ≈0.35 for an 11.2 nm QD in 9.37 mg/mL collagen gel. In ref. 31, a value of ≈0.95 for _D/D_o was obtained for a ≈10-nm particle in 2.4 mg/mL collagen gel. Values for _D/D_o obtained for higher concentrations of collagen gel from ref. 31 were not used because these concentrations were prepared by centrifuging low-concentration gels, and these results did not match gels prepared directly from high-concentration solutions. By simple linear interpolation of these two values, we obtained a _D/D_o for ≈10 nm NP in 7.38 mg/mL collagen of 0.52.
Intravital Multiphoton Microscopy.
All animal procedures were done by following the guidelines of the Public Health Service Policy on Humane Care of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital. Human fibrosarcoma HT-1080 cells were implanted in the dorsal skin of SCID mice for in vivo imaging (35, 43). When tumors reached 5 mm in diameter, we injected a 1-μL mixture of QDGelNPs and SilicaQDs (≈0.05 μL/min) into the tumor at constant pressure by using a glass micropipette connected to a syringe filled with silicone oil.
Images were obtained with a custom-built multiphoton microscope by using a Ti:Sapphire laser (Mai-Tai Broadband; Spectra-Physics) at 900 nm, a 20× (0.5 N.A.; Olympus) water-immersion objective, and photon-counting photomultiplier tubes (H7421-40; Hamamatsu). Detection of QDGelNPs was performed via a 530DF100 emission filter and SilicaQDs via a 610DF75 emission filter. Collagen fibers were imaged with second-harmonic generation (16, 33, 44) via a 450DF100 emission filter. The laser power was set to 500 mW. Three-dimensional image stacks containing 21 images of 5-μm thickness were obtained wherever fluorescence intensity from the injected particles was detected. A maximum intensity z projection of each colored stack generated a 2D image. Images of consecutive adjacent regions in the x and y directions were combined into a montage, generating a single image of the entire injection site.
The intensity profiles along the dotted lines in Fig. S7 was extracted by using ImageJ and then normalized such that the backgrounds (a “dark” region from all three time-lapse images) had the same intensity. The background was subtracted, and the resulting profiles were fitted (Fig. S8) to the model for diffusing substance initially distributed uniformly through a sphere of radius a (34) to obtain the diffusion coefficient:
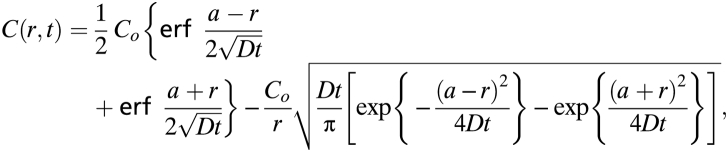
where _C_o is the initial concentration in the sphere. It should be noted that the diffusion coefficient obtained in collagen gel was obtained at ≈295 K, whereas the in vivo experiment was measured at the slightly higher body temperature of ≈310 K.
Supplementary Material
Supporting Information
Acknowledgments
We thank Patrick Boisvert for assistance with SEM imaging, Yves Boucher for help with zymography, Peigen Huang for the direction of gnotobiotic animal facility, Julia Kahn for animal model preparation, Eve Smith for cancer cell line preparation, Lisa Marshall for assistance with FCS, Li Miao for administrative support, Debby Pheasant for assistance with DLS, and Elisabeth Shaw for assistance with XPS. We thank Juwell Wu for assistance with image analysis and helpful discussions. This research was supported by US National Cancer Institute Grants R01-CA126642 (to R.K.J. and M.G.B.), R01-CA085140, R01-CA115767 (to R.K.J.), P01-CA080124 (to R.K.J. and D.F.), R01-CA096915 (to D.F.); MIT-Harvard NIH Center for Cancer Nanotechnology Excellence Grant 1U54-CA119349 (to M.G.B.); MIT DCIF Grants CHE-980806 and DBI-9729592; by ISN Grant W911NF-07-D-0004 (to M.G.B.); and The Susan G. Komen Foundation Grant KG091281 (to T.S.). R.K.J. is a recipient of Innovator Award W81XWH-10-1-0016 from the US Department of Defense Breast Cancer Research Program.
Footnotes
R.K.J. received commercial research grants from Dyax, AstraZeneca, and MedImmune; consultant fees from AstraZeneca/MedImmune, Dyax, Astellas-Fibrogen, Regeneron, Genzyme, MorphoSys, and Noxxon Pharma; and a speaker honorarium from Genzyme. R.K.J. owns stock in SynDevRx. No reagents or funding from these companies were used in these studies. There is no significant financial or other competing interest in the work.
References
- 1.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267:9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farokhzad OC, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 7.Choi HS, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winer EP, et al. Failure of higher-dose paclitaxel to improve outcome in patients with metastatic breast cancer: Cancer and leukemia group B trial 9342. J Clin Oncol. 2004;22:2061–2068. doi: 10.1200/JCO.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 9.Gradishar WJ, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 10.O'Brien MER, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 11.Jain RK. Delivery of molecular and cellular medicine to solid tumors. J Control Release. 1998;53:49–67. doi: 10.1016/s0168-3659(97)00237-x. [DOI] [PubMed] [Google Scholar]
- 12.Jain RK. Taming vessels to treat cancer. Sci Am. 2008;298:56–63. doi: 10.1038/scientificamerican0108-56. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 14.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988;48:7022–7032. [PubMed] [Google Scholar]
- 15.Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: Implications for therapy. Cancer Res. 1990;50:4478–4484. [PubMed] [Google Scholar]
- 16.McKee TD, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 17.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 18.Ouar Z, et al. Inhibitors of vacuolar H+-ATPase impair the preferential accumulation of daunomycin in lysosomes and reverse the resistance to anthracyclines in drug-resistant renal epithelial cells. Biochem J. 2003;370:185–193. doi: 10.1042/BJ20021411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11:8782–8788. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- 20.Pluen A, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc Natl Acad Sci USA. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain RK. Transport of molecules in the tumor interstitium: A review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 22.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 23.Gref R, et al. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 24.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 25.Roy R, Zhang B, Moses MA. Making the cut: Protease-mediated regulation of angiogenesis. Exp Cell Res. 2006;312:608–622. doi: 10.1016/j.yexcr.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Deryugina EI, Bourdon MA, Reisfeld RA, Strongin A. Remodeling of collagen matrix by human tumor cells requires activation and cell surface association of matrix metalloproteinase-2. Cancer Res. 1998;58:3743–3750. [PubMed] [Google Scholar]
- 27.Kaul G, Amiji M. Biodistribution and targeting potential of poly(ethylene glycol)-modified gelatin nanoparticles in subcutaneous murine tumor model. J Drug Target. 2004;12:585–591. doi: 10.1080/10611860400013451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stroh M, et al. Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo. Nat Med. 2005;11:678–682. doi: 10.1038/nm1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stylianopoulos T, et al. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys J. 2010;99:1342–1349. doi: 10.1016/j.bpj.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatakeyama H, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14:68–77. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]
- 31.Ramanujan S, et al. Diffusion and convection in collagen gels: Implications for transport in the tumor interstitium. Biophys J. 2002;83:1650–1660. doi: 10.1016/S0006-3495(02)73933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popović Z, et al. A nanoparticle size series for in vivo fluorescence imaging. Angew Chem Int Ed Engl. 2010;49:8649–8652. doi: 10.1002/anie.201003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clauss MA, Jain RK. Interstitial transport of rabbit and sheep antibodies in normal and neoplastic tissues. Cancer Res. 1990;50:3487–3492. [PubMed] [Google Scholar]
- 34.Crank J. The Mathematics of Diffusion. New York: Oxford Univ Press; 1979. [Google Scholar]
- 35.Chauhan VP, et al. Multiscale measurements distinguish cellular and interstitial hindrances to diffusion in vivo. Biophys J. 2009;97:330–336. doi: 10.1016/j.bpj.2009.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chau Y, Tan FE, Langer R. Synthesis and characterization of dextran-peptide-methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinase II and matrix metalloproteinase IX. Bioconjug Chem. 2004;15:931–941. doi: 10.1021/bc0499174. [DOI] [PubMed] [Google Scholar]
- 37.Harris TJ, Maltzahn GV, Derfus AM, Ruoslahti E, Bhatia SN. Proteolytic actuation of nanoparticle self-assembly. Angew Chem Int Ed Engl. 2006;45:3161–3165. doi: 10.1002/anie.200600259. [DOI] [PubMed] [Google Scholar]
- 38.Bremer C, Tung C-H, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7:743–748. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 39.Coester CJ, Langer K, van Briesen H, Kreuter J. Gelatin nanoparticles by two step desolvation—a new preparation method, surface modifications and cell uptake. J Microencapsul. 2000;17:187–193. doi: 10.1080/026520400288427. [DOI] [PubMed] [Google Scholar]
- 40.Zucker S, Lysik RM, Zarrabi MH, Moll U. M(r) 92,000 type IV collagenase is increased in plasma of patients with colon cancer and breast cancer. Cancer Res. 1993;53:140–146. [PubMed] [Google Scholar]
- 41.Woessner JF, Nagase H. Matrix Metalloproteinases and TIMPs. Oxford: Oxford Univ Press; 2000. [Google Scholar]
- 42.Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287–5297. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leunig M, et al. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 44.Brown EB, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med. 2001;7:864–868. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information