Comprehensive Analysis of Missense Variations in the BRCT Domain of BRCA1 by Structural and Functional Assays (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 15.
Abstract
Genetic screening of the breast and ovarian cancer susceptibility gene BRCA1 has uncovered a large number of variants of uncertain clinical significance. Here, we use biochemical and cell-based transcriptional assays to assess the structural and functional defects associated with a large set of 117 distinct BRCA1 missense variants within the essential BRCT domain of the BRCA1 protein that have been documented in individuals with a family history of breast or ovarian cancer. In the first method, we used limited proteolysis to assess the protein folding stability of each of the mutants compared with the wild-type. In the second method, we used a phosphopeptide pull-down assay to assess the ability of each of the variants to specifically interact with a peptide containing a pSer-X-X-Phe motif, a known functional target of the BRCA1 BRCT domain. Finally, we used transcriptional assays to assess the ability of each BRCT variant to act as a transcriptional activation domain in human cells. Through a correlation of the assay results with available family history and clinical data, we define limits to predict the disease risk associated with each variant. Forty-two of the variants show little effect on function and are likely to represent variants with little or no clinical significance; 50 display a clear functional effect and are likely to represent pathogenic variants; and the remaining 25 variants display intermediate activities. The excellent agreement between the structure/function effects of these mutations and available clinical data supports the notion that functional and structure information can be useful in the development of models to assess cancer risk.
Introduction
Since the cloning of BRCA1 in 1994, a large effort has been made to sequence the BRCA1 genes of women who are at increased risk for early-onset breast and ovarian cancers (1). This effort has revealed several mutations that are strongly linked to cancer. Unfortunately, a large number of rare BRCA1 variants have been uncovered in the human population for which risk assessment has been problematic, due to a lack of informative pedigree data linking each mutation with disease risk. BRCA1 encodes a large, 1,863–amino acid nuclear protein that plays a critical role in the response of cells to genotoxic stress (2–4). BRCA1 is thought to act as an essential mediator protein in DNA damage–induced nuclear signaling events, in which it interacts with phosphorylated partner proteins such as the DNA helicase, BACH1, the nuclease CtIP, and another signaling protein, Abraxas, to relay signals generated from chromatin surrounding the damage to downstream targets such as DNA repair proteins and factors involved in cell cycle regulation (5–11). Interactions with phosphorylated partner proteins are mediated by a tandem pair of repeats at the COOH terminus of BRCA1, termed BRCT repeats (12–14). The BRCT repeat region can also act as a transcriptional activation domain when linked to a sequence-specific DNA binding module, and this activity may contribute to the ability of BRCA1 to regulate the expression of genes such as p21 and GADD45 (15, 16) and modulate the activity of other transcription factors such as p53 and ER (17, 18). The NH2-terminal region of the protein contains a RING domain, which forms a heterodimeric complex with the RING domain of BARD1 to form a ubiquitin ligase (19–21). Cancer-associated mutations tend to cluster in the RING and BRCT repeat regions, showing the critical role of these domains in BRCA1 tumor suppression.
The BRCA1 BRCT domain specifically interacts with phosphorylated protein targets containing the motif pSer-x-x-Phe. Structural studies have revealed that recognition involves a conserved phosphoserine recognition pocket in the NH2-terminal BRCT repeat, composed of Ser1655, Gly1656, and Lys1702, which each supply ligands to recognize the phosphate (12–14). The phenylalanine residue at the +3 position of the peptide target is recognized by a largely hydrophobic groove formed at the interface between the repeats. Phosphopeptide recognition is likely a common function of BRCT domains in many proteins associated with DNA damage–mediated signal transduction. For example, the phosphorylated form of the histone variant, H2AX, which serves as a critical chromatin mark of DNA double-strand breaks, is specifically bound by the tandem BRCT repeats of MDC1 to initiate DNA double-strand break signaling (22, 23).
Several studies have attempted to predict the cancer risks associated with unclassified missense variants in BRCA1 through bioinformatics analysis based on multiple sequence alignment data and protein structure prediction, as well as analyses of the clinical and family history data available for some of these mutations (1, 24–28). These studies indicate that the RING and BRCT domains, which are the most highly conserved regions of BRCA1, likely contain the vast majority of cancer-associated mutations. Here, we use in vitro protein folding, phosphopeptide-binding, and cell-based transcriptional assays to assess the structural and functional consequences of a set of 117 distinct missense variants that have been uncovered in the BRCA1 BRCT domain in the human population. Our results indicate that nearly one half of the BRCT missense variants result in significant structural and/or functional defects that are likely associated with an increased cancer risk.
Materials and Methods
Proteolysis assays
pLM1 plasmid (0.25 μg) encoding the BRCT variants was used directly as template for protein synthesis reactions with the TNT-Quick in vitro transcription/translation system (Promega). Both wild-type and mutant proteins were produced by in vitro transcription/translation because many mutants could not be expressed in soluble form in Escherichia coli. The stability of the protein fold for the BRCA1 BRCT mutants was tested using proteolysis assays, as previously reported (29). The measurements were normalized against the internal background, and the control measurements taken in the same day. The missense variants were tested in triplicate.
Phosphopeptide-binding assays
The BRCT variants were translated and labeled as in proteolysis assays. The ability of the BRCA1 BRCT mutants to bind pSer-containing peptides was tested using peptide-binding assays, as previously reported (14).
Bead-immobilized peptide affinity resin was prepared by incubating a 10-fold molar excess of a biotinylated phosphopeptide (SRSTpSPTFNK) and the corresponding unphosphorylated peptide (SRSTSPTFNK) with streptavidin agarose beads (Sigma-Aldrich). Seven mutants, with the wild-type BRCA1-BRCT (1,646–1,858) as a positive control, and M1775R and A1708E as negative controls were examined at a time. The intensities of the bands were measured and the backgrounds were corrected using ImageQuaNT (Amersham Biosciences), and the measurements were normalized against the wild-type measurement taken in the same day. The missense variants were tested in triplicate.
Transcriptional assays
The construct containing wild-type BRCA1 amino acids 1,646 to 1,859 (wt) was used as a positive control and variants M1775R and Y1853X as negative controls. HEK293T cells were seeded in 24-well tissue culture plates, 1 × 105 cells per well, and 24 hours later were transfected in triplicates with Fugene 6 according to the manufacturer's instructions (Roche). The vectors containing the variants were cotransfected with pG5Luc, which contains a firefly luciferase reporter gene driven by GAL4-responsive binding sites, and phGR-TK, which contains a Renilla luciferase gene driven by a constitutive TK basal promoter used as an internal control. Cells were harvested 24 hours after transfection, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer's instructions. Activity was plotted as a percentage of the wt BRCA1 activity. Thirty-four of 117 variants had already been assessed in previous articles, albeit in different amino acid contexts (amino acids 1,396–1863 or aa 1,560–1863; refs. 30–32). In only one case (G1706A), the qualitative assessment differed because it was considered of moderate effect previously and of no effect in the present study. This shows the high reproducibility of the transcriptional assays.
Align-GVGD classification of BRCA1 BRCT variants
The 117 BRCT variants were analyzed with the Align-GVGD algorithm at three depths of alignment. Nine mammalian BRCA1 sequences, chicken and one additional sequence (either frog, puffer fish, or sea urchin), were used for the alignment (for more detail, see ref. 33). A-class scores were assigned for variants in which the scores from at least two of three alignments agreed.
Definition of a validation set
To compile our validation set (Supplementary Table S1; Table 1), we identified variants that had been previously analyzed and classified by genetic (1) or integrative methods (25, 34–40). Integrative methods use a combination of data from different sources, including cooccurrence with known deleterious mutations, personal and family history of patients carrying the variant, and cosegregation of the variant with disease within pedigrees, to assign an overall risk score. Sufficient data were available to assign 14 of the BRCA1 BRCT variants as associated with disease, whereas 10 of the variants were predicted to be neutral (Table 1). Where possible, the probability of pathogenicity of each of these variants was classified according to the IARC scheme (Supplementary Table S1; ref. 41). We calculated specificity, sensitivity, and 95% confidence interval measures for each of the structure and functional assays based on a comparison with this variant set (Supplementary Table S2).
Table 1. Cross validation of functional assays with genetic data.
A description of how the BRCA1 BRCT variant set was obtained is given in Supplementary Materials and Methods.
Results
Generation of a large set of BRCA1 BRCT missense variants
Many of the BRCA1 variants that have been uncovered through worldwide sequencing efforts are now deposited within the Breast Cancer Information Core database (http://research.nhgri.nih.gov/bic/) as well as the Kathleen Cuningham Foundation Consortium for research into Familial Breast Cancer database (http://www.kconfab.org). To facilitate the assessment of the structural and functional defects associated with these mutations, we cloned all the currently available missense variants (Fig. 1) into an E. coli expression vector that we used previously for our structural and functional work of the BRCA1 BRCT domain. This vector places the BRCA1 BRCT coding sequence (corresponding to residues 1,646–1,859) under control of a T7 promotor. Using this plasmid system, recombinant protein can be expressed in E. coli and purified using established procedures for detailed biochemical and structural studies. The T7 promoter can also be used for in vitro transcription/translation of the recombinant protein. The in vitro transcription/translation system is rapid and allows the generation of multiple proteins in parallel and, because the recombinant protein is specifically labeled with 35S-methionine, no purification is required. All variants can be successfully made in this system, regardless of their folding stability. This is a further advantage over E. coli expression, in which we have found that only well-folded variants can be expressed in soluble form and purified. Thus, we have used in vitro transcribed/translated material for our structural stability and peptide-binding assays described below. We also evaluated the possibility that some of these variants could result in splicing abnormalities. We used the NNSplice 0.9 algorithm (42) to predict the effects of these changes on the efficiency with the BRCT coding region (see Supplementary Materials).
Figure 1.
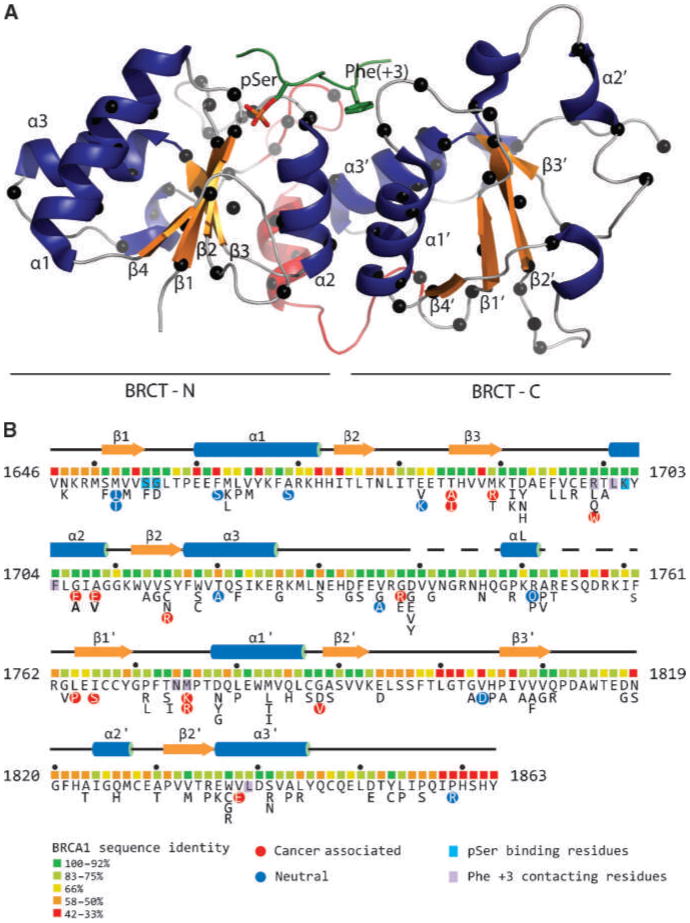
A, ribbon diagram representation of the BRCA1 BRCT tandem repeats (PDB ID 1T15; ref. 13). Blue, α-helices; orange, β-strands. Red, the linker between the N-BRCTs and C-BRCTs. Green, the bound BACH1 decapeptide; sticks, the pSer and Phe(+3) residues. Black spheres along the polypetidic chain, the positions of the studied mutations. B, the complete BRCA1 BRCT missense variant set. Shown is the sequence of the human BRCA1 BRCT domain with the protein secondary structure indicated. Below the sequence are colored boxes indicating the level of sequence identity of each residue within a 13 species alignment (http://agvgd.iarc.fr/alignments.php). Neutral and disease-associated variants (Table 1) are highlighted as are residues that contact the pSer and Phe at the +3 position of the phosphopeptide target.
Determination of the protein folding defects associated with BRCT missense variants
We used limited proteolysis to determine the stability of the protein fold in the set of BRCA1 BRCT missense variants using in vitro transcribed/translated material (Supplementary Fig. S1). The wild-type protein is highly resistant to digestion by trypsin, elastase, or chymotrypsin, indicating that the in vitro produced BRCT protein is stably folded (43). On the other hand, a pilot study involving 25 of the BRCA1 BRCT missense variants showed that certain single amino acid substitutions in the BRCT domain of BRCA1 can destabilize its structure, leading to increased susceptibility to trypsin-mediated proteolysis (29). The assay is sufficiently sensitive to discriminate wild-type protein from the strongly destabilized variant A1708E as well as the moderately destabilized variant M1775R. Proteolysis of each of the missense variants was carried out at a series of trypsin concentrations, and the amount of full-length protein remaining was visualized by SDS-PAGE. The results were compared with those of the wild-type protein as well as the M1775R and A1708E variants carried out in parallel. We quantified the percentages of full-length protein remaining following digestion at different concentrations of trypsin and determined the severity of the destabilizing effects by comparing the percentage of protein remaining following digestion at 10 μg/mL trypsin to the controls (Supplementary Fig. S2A). We defined highly destabilizing mutations as those with similar rates of digestion to the A1708E control (<5% of the protein remaining after digestion with 10 μg/mL of trypsin). Moderately destabilizing mutations were defined as those with a similar stability to M1775R (between 5% and 65% of the protein remaining after digestion with 10 μg/mL of trypsin). Finally, structurally stable variants were defined as those with a stability similar to the wild-type protein (>65% of the protein remaining after 10 μg/mL of trypsin digestion). Based on these criteria, 26 of 117 of the missense mutations showed high sensitivity to tryptic digestion; 30 of 117 showed intermediate sensitivity; and 61 of 117 showed a stability that was indistinguishable from the wild-type (Supplementary Figs. S2A and S3). We also noted that the C1697R mutant showed a particularly low protein expression level.
Analysis of the phosphopeptide binding activity of BRCT missense variants
The BRCA1 BRCT domain interacts with BACH1 and several other proteins through the selective recognition of the peptide motif pSer-x-x-Phe (10–14, 44). All mutations that have been strongly linked to disease using family-based data, such as A1708E and M1775R (1, 35), fail to bind specifically to pSer-x-x-Phe–containing peptides as shown in a pilot study that analyzed the peptide binding activities of 25 BRCA1 BRCT domain variants (14). This indicates the functional importance of the BRCA1 BRCT domain as a phosphopeptide-binding module in tumor suppression.
We examined the ability of the BRCT domains containing missense variants to bind peptides derived from BACH1 using a pull-down assay in which biotinylated peptide was bound to streptavidin beads. We tested binding to a phosphorylated version of the peptide containing the BRCA1 target sequence pSer-Pro-Thr-Phe or an unphosphorylated version as a control. The results were compared with experiments with the wild-type BRCA1 BRCT as well as with the peptide binding–deficient variants A1708E and M1775R carried out in parallel. We present two analyses of the data. In the first analysis, we define “binding activity” as the fraction of the input protein that bound to the phosphorylated peptide, normalized against the binding of the wild-type protein (Supplementary Fig. S2B). In the second analysis, we define “binding specificity” as the ratio of the variant bound to the phosphopeptide compared with the nonphosphorylated counterpart, normalized to the wild-type control (Supplementary Fig. S2C). Classified in this way, we observed a range of binding activities and specificities. As observed for the protein stability assays, the variants tended to cluster near either 100% or 0% activity, with relatively few variants displaying intermediate binding activities/specificities (Supplementary Fig. S3). Thirty-eight of 117 of the variants displayed binding activities >80% of wild-type levels, whereas 35 of 117 variants displayed binding activities <20% of wild-type. The binding specificity analysis revealed 29 of 117 of the variants exhibited >80% of wild-type levels, whereas 50 of 117 of the variants exhibited <20% of wild-type levels.
Analysis of the transcriptional activation activity of BRCA1 BRCT variants
The transcriptional activation activity of the BRCA1 BRCT domain was the first identified activity of this region of the protein (15, 16). Assays for BRCA1 BRCT transcriptional activation have been established in human and yeast cell systems (45). In either method, the BRCT domain is fused to a heterologous DNA binding domain that recognizes a defined regulatory sequence within the promoter of the reporter. Pilot studies involving subsets of the BRCA1 BRCT variants showed that mutations that have been associated with cancer using family-based data abrogate, or markedly diminish, the expression of the reporter gene (31, 32, 46). Moreover, the fact that mutations that destabilize the BRCT fold are strongly associated with a loss of transactivation function indicates that this assay is a monitor of the integrity of the BRCT domain (29).
As observed for the protein stability and phosphopeptide-binding assays, a wide range of transcriptional activities were observed for the variant set, with the observed values tending to cluster either at very low or very high activity levels (Supplementary Figs. S2D and S3). Thirty-eight of 117 variants displayed transcriptional activity levels at >80% of wild-type, whereas 54 of 117 were found to be <20% of wild-type levels.
Correlation of structural and functional data with family history and clinical data
The use of functional data in cancer risk assessment is predicated on rigorous validation of the results against sets of variants for which cancer risks are well defined. Easton and colleagues (35) have assigned risks based on genetic data for a large number of BRCA1 variants. We used these data, together with available genetic information for several other variants (25, 34–40), to create a validation set of 24 variants, consisting of 14 deleterious mutations and 10 neutral variants (Table 1). We plotted the protein stability, peptide binding, and transcriptional activity data against the combined log10 likelihood ratio scores for the subset of 19 variants analyzed by Easton and colleagues (Fig. 2; likelihood ratio). In general, this analysis revealed a clear correlation between structural/functional defects and cancer risk (35). A comparison of all the genetically characterized variants with the experimental results is shown in Table 1. This comparison was used to define the sensitivities and specificities of the functional assays. Sensitivity ranged from 81.8% for phosphopeptide binding activity to 100% for phosphopeptide binding specificity and transcription assays. Specificity ranged from 75% for proteolysis assays to 87.5% for transcription assays, to 100% for binding activity assays (Supplementary Table S2).
Figure 2.
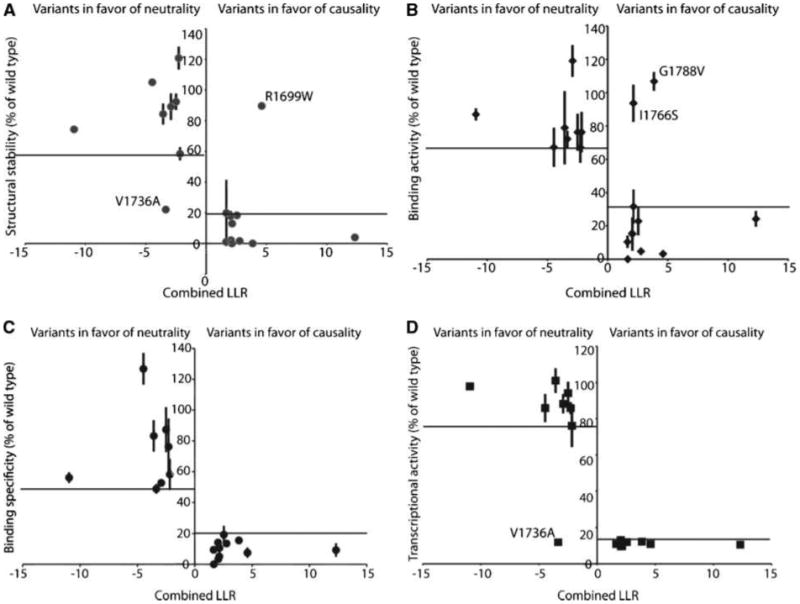
Functional analysis of 19 variants plotted against the combined log10 likelihood ratio (LLR) score as calculated. Points, mean binding activity of each of the variants as percent of wild-type activity; bars, SD. Outliers are labeled. A, structural stability. B, phosphopeptide binding activity. C, phosphopeptide binding specificity. D, transcription activation activity.
Although overall there is a good agreement between the assessed cancer risks and the functional activities of the variants, there are several variants in which cancer risk assessment and the structure/function results do not agree. For example, a significant number of cancer-associated (IARC classes 5 and 4) variants are not structurally destabilized as determined by the proteolysis assay. Each of these variants (R1699W, M1775R, and M1775K) is highly defective for specific phosphopeptide recognition and transcriptional activity. Structural studies reveal that both Arg1699 and Met1775 make critical direct contacts to the phosphopeptide (12–14), and structural investigations of the M1775R and M1775K variants reveal subtle structural rearrangements that block the +3 specificity pocket (37, 47). Two of the cancer-associated (IARC class 5) variants, I1766S and G1788V, were highly defective in the proteolysis and transcriptional assays; however, they showed significant levels of nonspecific peptide binding, independent of phosphorylation. It is likely that the unfolded state of these variants exposes hydrophobic residues that are responsible for these nonspecific interactions, as suggested previously (14). G1706E, one of the cancer-associated (IARC class 5) variants, is defective in the proteolysis and peptide-binding assays; however, it shows an intermediate level of transcriptional activity. The neutral (IARC class 1) variant V1736A exhibited the least agreement between the different assays, showing significant sensitivity to proteolysis and a strong transcriptional defect while showing intermediate/strong levels of phosphopeptide interaction.
Considering the overall high specificity and sensitivity obtained in the assays (Supplementary Table S2), we have defined limits to predict disease association for the uncharacterized variants based on the biochemical and transcriptional assay data (Table 1; Fig. 2). It is important to stress that the definition of these limits should not be used for classifying variants in terms of their pathogenicity in the absence of additional data. The cancer-associated (IARC classes 5 and 4) variants in general exhibited protein stability levels <58% of the wild-type, phosphopeptide binding activities <32% of wild-type, phosphopeptide specificity binding activities <21% of wild-type, and transcriptional activation function <35% of wild-type levels. Conversely, the neutral variants (IARC classes 1 and 2) in general had protein stability levels >59% of the wild-type, phosphopeptide binding activities >67% of wild-type, phosphopeptide specificity binding activities >49% of wild-type, and transcriptional activation function >76% of wild-type levels. Variants falling between these limits are defined as uncertain. Using these limits, we have defined the structure and functional activities of each of the variants (Fig. 3).
Figure 3.
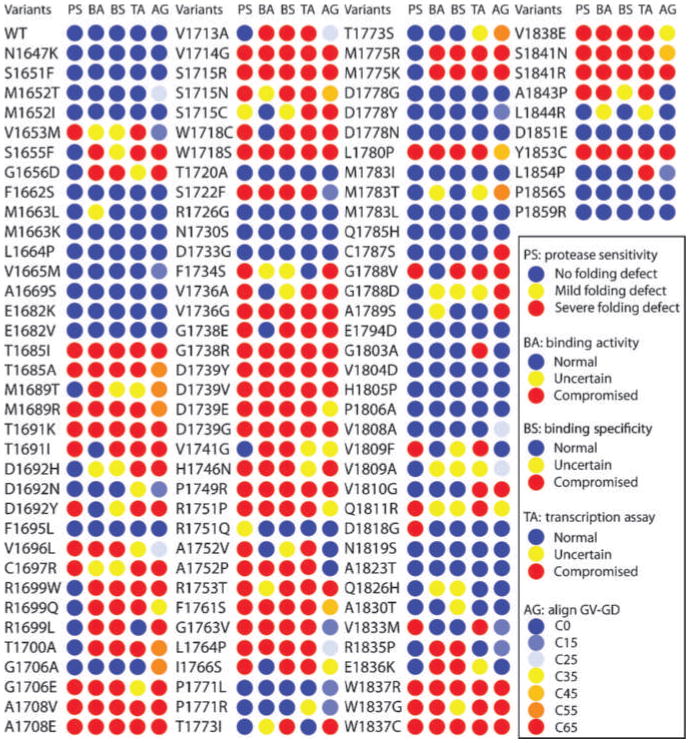
Cross-validation of structural and functional assays. The results for protease sensitivity (PS), peptide binding activity (BA), peptide binding specificity (PS), and transcriptional activity (TA) are tabulated using blue circles to indicate no defect, yellow circles to indicate an intermediate activity, and red circles to indicate a large defect. Also shown are the predicted align-GVGD scores (AG) with circles colored from blue to red to indicate no predicted effect, to a large effect, respectively.
Comparison of results from different assays and overall classification
Next, we compared the results from the different assays for the entire set of variants (Fig. 3). In 64 of 117 (54.7%) of the variants, there was complete concordance between the different assays, with 35 of 117 (29.9%) giving protein stability, peptide binding, and transcription activities within the limits that correspond to neutral variants in IARC classes 1 and 2, whereas 29 of 117 (24.8%) exhibited values in the range corresponding to cancer-associated (IARC classes 4 and 5) variants for each of the assays.
Based on these results, we have classified the variants into groups based on their functional effect (Table 2). Although classification is straightforward for those variants in which there is a complete agreement between the different assays, for the variants in which there is not a complete agreement, classification is more complex. The largest class of discordant results includes variants that exhibit defects in binding and transcription but little or no folding defect (6 of 117 variants or 5.1%). These variants likely directly affect the phosphopeptide binding surface and may only affect residues that are exposed to the solvent preserving the overall fold structure. Because phosphopeptide recognition is critical for the tumor-suppressive function of BRCA1 (14, 48), we have therefore included these variants in the strong functional effect class. Likewise, severely destabilized variants that show significant levels of phosphorylation-independent peptide binding but a strong transcriptional defect have also been categorized as strong functional effect (Table 2).
Table 2. Classifying BRCA1 BRCT domain variants based on functional effect.
| Strong functional effect* | Moderate functional effect† | Uncertain‡ | Low functional effect§ | No functional effect‖ | |||||
|---|---|---|---|---|---|---|---|---|---|
| T1685I | T1685A | G1656D | S1655F | V1653M | D1692Y | M1663L | D1692N | N1647K | S1651F |
| M1689R | T1691K | R1699L | V1696L | C1697R | A1752V | R1751Q | P1771R | M1652T | M1652I |
| T1691I | A1708V | G1706E | H1746N | T1773I | Q1811R | T1773S | A1789S | F1662S | M1663K |
| A1708E | V1714G | W1837G | A1843P | G1788D | V1833M | A1830T | L1664P | V1665M | |
| S1715R | S1715N | L1844R | M1689T | A1669S | E1682K | ||||
| W1718C | W1718S | D1692H | S1715C | E1682V | F1695L | ||||
| S1722F | V1736G | F1734S | V1736A | G1706A | T1720A | ||||
| G1738E | G1738R | V1741G | M1783T | R1726G | N1730S | ||||
| D1739Y | D1739V | G1803A | V1809A | D1733G | P1771L | ||||
| D1739E | D1739G | V1809F | V1810G | D1778G | D1778Y | ||||
| P1749R | R1751P | D1818G | Q1826H | D1778N | M1783I | ||||
| A1752P | R1753T | E1836K | L1854P | M1783L | Q1785H | ||||
| F1761S | G1763V | R1835P | C1787S | E1794D | |||||
| L1764P | I1766S | V1804D | H1805P | ||||||
| L1780P | G1788V | P1806A | V1808A | ||||||
| W1837R | W1837C | N1819S | A1823T | ||||||
| V1838E | S1841N | D1851E | P1856S | ||||||
| S1841R | Y1853C | P1859R | |||||||
| R1699W | R1699Q | ||||||||
| T1700A | V1713A | ||||||||
| M1775R | M1775K |
We have also described eight of the variants as of “moderate functional effect” in which a preponderance of the data points to a loss of function. These include variants with intermediate levels of peptide binding specificity (W1837G and A1843P) or transcriptional activity (V1696L, G1706E, and H1746N), and strong defects in all of the other assays. In addition, we have included three additional mutants in this group that show poor agreement between the different assays but are likely to directly affect the peptide-binding surface. S1655F is predicted to impede recognition of the phosphoserine in the bound peptide, whereas G1656D could perturb the structure of the phosphoserine-binding pocket, and the introduction of a negatively charged side chain in this region would be expected to further inhibit phosphopeptide recognition. R1699L would remove a critical determinant for recognition of the phenylalanine residue at the +3 position of the phosphopeptide, and indeed, the similar R1699W variant is pathogenic (IARC class 5; Table 1).
We predict a given variant to be neutral when each of the assays gives values similar to wild-type within the limits defined by comparison with the set of variants that have been characterized through family history and genetic data. In addition, we define a set of seven variants as “low functional effect” that display an intermediate activity for one of the assays and wild-type levels for the other three.
Finally, there is a group of 25 variants (21.4%) that we define as “uncertain.” In general, these variants show a poor level of agreement between the different assays, and a significant number of assay results in the intermediate range.
Correlation of mutant classification with predictions from Align-GVGD
The Align-GVGD method (http://agvgd.iarc.fr/agvgd_input.php) uses protein multiple sequence alignment (PMSA) data to provide cancer risk estimates for BRCA1 mutations (33). In this method, each missense variant is classified based on the observed amino acid fluctuation at that position in the PMSA (the Grantham variance, GV) as well as a comparison of the amino acid substitution with the range of amino acids observed at that position (Grantham difference, GD). In this way, seven classes of variants (C0, C15, C25, C35, C45, C55, and C65) that represent a graded series of GV-GD scores were defined. Variants in the C0 class correspond to variants that are well conserved in the PMSA and are therefore predicted to be neutral, whereas the C65 class contains missense variants that deviate from the PMSA and likely associated with an increased cancer risk.
To assess the accuracy of the Align GV-GD method, we compared Align GV-GD scores to the results for the structure/function assays (Fig. 4). For the variants with no functional effect, a large majority (28 of 35, 80%) falls into the C0 class, three are classified as C15, and two are classified as C25. Only one of them, G1706A, received a high score in Align GV-GD, C55. Another variant at this position (G1706E) scored as C65, but this variant was classified with moderate functional effect by structure/function assays. These results suggest substitution of a small side chain at position 1,706 is tolerated, but not substitution of the larger, charged Glu. For the variants with strong functional effect, 25 of 42 (59.5%) are in the C65 class and 16 are spread throughout classes C15 to C55 (Fig. 4B). One missense variant (A1752P) was classified C0, but this variant shows strong defect in all assays. The substitution of a proline at position 1,752 likely destabilizes the linker α-helix, which may be responsible for the strong folding defect associated with this variant. Overall, there is strong agreement between Align GV-GD predictions and the results of functional assays, indicating that sequence alignment–based predictive methods can offer a valuable measure of risk assessment for variants in which there is no direct functional data.
Figure 4.
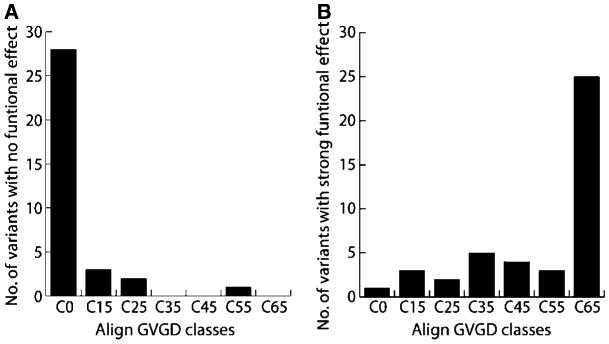
Variants classified with no functional effect (A) or strong functional effect (B) by structure/function assays (see Table 2) were analyzed with the GV-GD algorithm and sorted in one of the seven grades (C0–C65).
Discussion
Here, we present the first comprehensive assessment of the structural and functional consequences of 117 BRCA1 variants corresponding to the complete set of BRCT missense variants detected to date in the human population (100% coverage). Our protein structure assay, peptide binding, and transcriptional assays show a remarkable degree of agreement and give us confidence in the validity of these results. Correlation of this experimental data with the available clinical and family history data for a subset of these variants defines limits that can be used to tentatively predict functional effect that is likely to be reflected in the disease risk associated with these variants. In this way, we show that 42.7% of the variants fall into the “clear functional effect” or “moderate functional effect” categories (Table 2) and are likely associated with an increased cancer risk, whereas 35.9% fall into the “no functional effect” or “low functional effect” categories. The remaining 21.4% falls into the uncertain category and could perhaps represent low-penetrance variants. The fact that a large proportion of variants tested displayed some functional effect lends support to the idea of applying higher prior probabilities for causality to variants found in the BRCT domains (35).
The BRCA1 BRCT variant set provides a useful set of reagents for further detailed study of variants of particular interest. For example, variants that are moderately well folded based on proteolytic stability can often be purified and studied in more detail. Circular dicroism spectroscopy and differential scanning calorimetry have been used to quantitatively assess the fold stability of V1696L, R1699W, M1775K, M1775R, M1783T, V1808A, V1809F, and V1833M (49–51). Although our proteolytic assay indicated that the stability of two of these variants, R1699W and V1808A, were indistinguishable from wild-type, the more sensitive thermodynamic studies of the purified forms of these variants reveal a small but significant reduction in fold stability. In addition, quantitative assays of phosphopeptide binding, such as isothermal titration calorimetry, can reveal the detailed thermodynamics of peptide-BRCT interactions (50, 51). The structural rearrangements induced by three of these variants, M1775R (47), M1775K (37), and V1809F (14), have been visualized through crystallographic structural analysis. Each of the variants affect the +3 specificity pocket formed at the interface between the two BRCT repeats. For M1775R and M1775K, the substituted positively charged side chain adopts an orientation that fills the specificity pocket and blocks the binding of the Phe side chain at the +3 position of the target peptide. Intriguingly, the V1809F substitution, which occurs far from the +3 pocket, induces a series of conformational adjustments that results in the movement of Met1775 to fill the specificity pocket, explaining the loss of peptide binding specificity and transcriptional activity associated with this variant.
Nonsynonymous single nucleotide polymorphisms (SNP) that lead to amino acid substitutions are likely to have diverse phenotypic consequences and implications for human disease. Although computational methods have been proposed to assess the effect of individual SNPs (52), relatively little has been done at the experimental level to provide insight into the effects of SNPs. The database of BRCA1/BRCA2 mutations provides an important first look at the kind of genetic diversity that exists in the human population. Another example of a large mutation database is that assembled for mutations found in p53 from human tumors. Detailed studies of the stability of the p53 DNA binding domain indicate that, like the BRCA1 BRCT domain, this relatively unstable domain is sensitive to mutations that destabilize the protein fold in addition to other classes of mutations that do not significantly affect protein fold stability but instead modify DNA-binding or protein-protein interaction surfaces (53). Thus, rapid, general assays of protein structure and function such as the proteolysis assay used here could potentially prove useful for the rapid, high-throughput assessment of the effect of SNPs on protein function.
Ultimately, unambiguous classification of variants, as proposed by the IARC Unclassified Genetic variants Working Group (41), will need a comprehensive integrative model to incorporate data from several sources. Robust integrative models have been proposed but they still lack a coherent framework to incorporate functional data. A major roadblock in the development of such a model was the lack of an established series of functional assays with standardized controls, validated against available genetic data. The present study fills this gap and allows for the development of better predictive models.
Although a systematic large-scale classification of all BRCA1 variants into the IARC classes has not yet been done, it is expected that a large number will fall into class 3 (uncertain), due to the lack of genetic-based data. Conceivably, even large-scale efforts may not be able to gather sufficient genetic data on individual variants because most are extremely rare. Thus, we anticipate that the use of structural and functional information will be instrumental to move variants from the class 3 into classes that provide a basis for better risk assessment and clinical decisions. In addition, the fact that variants can now also be functionally and structurally rationalized will provide an impulse for further functional dissection of the tumor-suppressive activities of BRCA1.
Supplementary Material
Supplementary Methods, Figures, Tables
Supplementary Tables
Acknowledgments
Grant Support: Canadian Cancer Society Research Institute (J.N.M. Glover), the Howard Hughes International Scholar Program (J.N.M. Glover), an Alberta Heritage Foundation for Medical Research Studentship (M.S. Lee), a fellowship from CNPq, Brazil (S.M. Marsillac), and NIH award CA116167 (A.N.A. Monteiro).
Footnotes
Reprints and Subscriptions: To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at pubs@aacr.org.
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 2.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–82. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 3.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–32. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–76. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- 5.Varma AK, Brown RS, Birrane G, Ladias JA. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005;44:10941–6. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 6.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–86. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang B, Matsuoka S, Ballif BA, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–8. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol. 2007;14:710–5. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–92. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem. 2003;278:52914–8. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- 11.Botuyan MV, Nomine Y, Yu X, et al. Structural basis of BACH1 phosphopeptide recognition by BRCA1 tandem BRCT domains. Structure. 2004;12:1137–46. doi: 10.1016/j.str.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiozaki EN, Gu L, Yan N, Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol Cell. 2004;14:405–12. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 13.Clapperton JA, Manke IA, Lowery DM, et al. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004;11:512–8. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 14.Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11:519–25. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 15.Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci U S A. 1996;93:13595–9. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678–9. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- 17.Ouchi T, Monteiro AN, August A, Aaronson SA, Hanafusa H. BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci U S A. 1998;95:2302–6. doi: 10.1073/pnas.95.5.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan S, Wang J, Yuan R, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354–6. doi: 10.1126/science.284.5418.1354. [DOI] [PubMed] [Google Scholar]
- 19.Wu LC, Wang ZW, Tsan JT, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–40. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 20.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–7. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 21.Brzovic PS, Keeffe JR, Nishikawa H, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003;100:5646–51. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 23.Lee MS, Edwards RA, Thede GL, Glover JN. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the γ-H2AX histone tail. J Biol Chem. 2005;280:32053–6. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 24.Lovelock PK, Healey S, Au W, et al. Genetic, functional, and histopathological evaluation of two C-terminal BRCA1 missense variants. J Med Genet. 2006;43:74–83. doi: 10.1136/jmg.2005.033258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chenevix-Trench G, Healey S, Lakhani S, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–27. doi: 10.1158/0008-5472.CAN-05-3546. [DOI] [PubMed] [Google Scholar]
- 26.Abkevich V, Zharkikh A, Deffenbaugh AM, et al. Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. 2004;41:492–507. doi: 10.1136/jmg.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chasman D, Adams RM. Predicting the functional consequences of nonsynonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J Mol Biol. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- 28.Karchin R, Monteiro AN, Tavtigian SV, Carvalho MA, Sali A. Functional impact of missense variants in BRCA1 predicted by supervised learning. PLoS Comput Biol. 2007;3:e26. doi: 10.1371/journal.pcbi.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams RS, Chasman DI, Hau DD, Hui B, Lau AY, Glover JN. Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. J Biol Chem. 2003;278:53007–16. doi: 10.1074/jbc.M310182200. [DOI] [PubMed] [Google Scholar]
- 30.Hayes F, Cayanan C, Barilla D, Monteiro AN. Functional assay for BRCA1: mutagenesis of the COOH-terminal region reveals critical residues for transcription activation. Cancer Res. 2000;60:2411–8. [PMC free article] [PubMed] [Google Scholar]
- 31.Phelan CM, Dapic V, Tice B, et al. Classification of BRCA1 missense variants of unknown clinical significance. J Med Genet. 2005;42:138–46. doi: 10.1136/jmg.2004.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho MA, Marsillac SM, Karchin R, et al. Determination of cancer risk associated with germ line BRCA1 missense variants by functional analysis. Cancer Res. 2007;67:1494–501. doi: 10.1158/0008-5472.CAN-06-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29:1342–54. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tavtigian SV, Deffenbaugh AM, Yin L, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–83. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lovelock PK, Spurdle AB, Mok MT, et al. Identification of BRCA1 missense substitutions that confer partial functional activity: potential moderate risk variants? Breast Cancer Res. 2007;9:R82. doi: 10.1186/bcr1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tischkowitz M, Hamel N, Carvalho MA, et al. Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet. 2008;16:820–32. doi: 10.1038/ejhg.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spurdle AB, Lakhani SR, Healey S, et al. Clinical classification of BRCA1 and BRCA2 DNA sequence variants: the value of cytokeratin profiles and evolutionary analysis-a report from the kConFab Investigators. J Clin Oncol. 2008;26:1657–63. doi: 10.1200/JCO.2007.13.2779. [DOI] [PubMed] [Google Scholar]
- 39.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–44. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judkins T, Hendrickson BC, Deffenbaugh AM, et al. Application of embryonic lethal or other obvious phenotypes to characterize the clinical significance of genetic variants found in trans with known deleterious mutations. Cancer Res. 2005;65:10096–103. doi: 10.1158/0008-5472.CAN-05-1241. [DOI] [PubMed] [Google Scholar]
- 41.Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–91. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 43.Williams RS, Green R, Glover JN. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat Struct Biol. 2001;8:838–42. doi: 10.1038/nsb1001-838. [DOI] [PubMed] [Google Scholar]
- 44.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 45.Carvalho MA, Couch FJ, Monteiro AN. Functional assays for BRCA1 and BRCA2. Int J Biochem Cell Biol. 2007;39:298–310. doi: 10.1016/j.biocel.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallon-Christersson J, Cayanan C, Haraldsson K, et al. Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet. 2001;10:353–60. doi: 10.1093/hmg/10.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams RS, Glover JN. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J Biol Chem. 2003;278:2630–5. doi: 10.1074/jbc.M210019200. [DOI] [PubMed] [Google Scholar]
- 48.Mirkovic N, Marti-Renom MA, Weber BL, Sali A, Monteiro AN. Structure-based assessment of missense mutations in human BRCA1: implications for breast and ovarian cancer predisposition. Cancer Res. 2004;64:3790–7. doi: 10.1158/0008-5472.CAN-03-3009. [DOI] [PubMed] [Google Scholar]
- 49.Ekblad CM, Wilkinson HR, Schymkowitz JW, Rousseau F, Freund SM, Itzhaki LS. Characterisation of the BRCT domains of the breast cancer susceptibility gene product BRCA1. J Mol Biol. 2002;320:431–42. doi: 10.1016/s0022-2836(02)00478-3. [DOI] [PubMed] [Google Scholar]
- 50.Nikolopoulos G, Pyrpassopoulos S, Thanassoulas A, et al. Thermal unfolding of human BRCA1 BRCT-domain variants. Biochim Biophys Acta. 2007;1774:772–80. doi: 10.1016/j.bbapap.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 51.Drikos I, Nounesis G, Vorgias CE. Characterization of cancer-linked BRCA1-BRCT missense variants and their interaction with phosphoprotein targets. Proteins. 2009;77:464–76. doi: 10.1002/prot.22460. [DOI] [PubMed] [Google Scholar]
- 52.Sunyaev S, Lathe W, III, Bork P. Integration of genome data and protein structures: prediction of protein folds, protein interactions and “molecular phenotypes” of single nucleotide polymorphisms. Curr Opin Struct Biol. 2001;11:125–30. doi: 10.1016/s0959-440x(00)00175-5. [DOI] [PubMed] [Google Scholar]
- 53.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–82. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Figures, Tables
Supplementary Tables