Identification of an erythroid-enriched endoribonuclease activity involved in specific mRNA cleavage (original) (raw)
Abstract
Stability of the human α–globin mRNA is conferred by a ribonucleoprotein complex termed the α–complex, which acts by impeding deadenylation. Using our recently devised in vitro decay assay, we demonstrate that the α–complex also functions by protecting the 3′–untranslated region (3′-UTR) from an erythroid-enriched, sequence-specific endoribonuclease activity. The cleavage site was mapped to a region protected by the α–complex and is regulated by the presence of the α–complex. Similar endoribonuclease cleavage products were also detected in erythroid cells expressing an exogenous α–globin gene. Nucleotide substitution of the target sequence renders the RNA refractory to the endoribonuclease activity. Insertion of the target sequence onto a heterologous RNA confers sequence-specific cleavage on the chimeric RNA, demonstrating the sequence specificity of this activity. We conclude that the α–complex stabilizes the α–globin mRNA in erythroid cells by a multifaceted approach, one aspect of which is to protect the 3′–UTR from specific endoribonuclease cleavage.
Keywords: α–complex/endoribonuclease/ErEN/in vitro mRNA decay assay/mRNA stability
Introduction
The steady-state accumulation of cytoplasmic mRNA is the consequence of the rates of both mRNA synthesis and degradation. Following transcription, the mRNA undergoes various processing events and is transported into the cytoplasm where it is utilized as a substrate for translation. An inherent property of each mRNA is the rate of turnover throughout the individual steps (Jacobson and Peltz, 1996). Eukaryotic mRNAs have a wide range of half-lives and their turnover is an important step in the regulation of eukaryotic gene expression. Short-lived proto-oncogene and cytokine mRNAs have half-lives of 5–30 min (Chen and Shyu, 1995), while the half-lives of some stable mRNAs are several days (Lodish and Small, 1976; Ross and Sullivan, 1985). The general stabilizing elements, which include the 5′ cap and 3′–polyadenylated tail, are found on almost all mRNAs and provide a basal level of stability by protecting the mRNA from exonuclease degradation. Differential stability of mRNAs is determined selectively by the interaction of RNA-binding proteins with _cis_-elements that commonly localize within the 3′–untranslated region (3′–UTR) (Jackson, 1993; Decker and Parker, 1995).
Much of our current understanding of mRNA turnover comes from studies in yeast, where deadenylation followed by decapping and subsequent 5′–3′ exoribonuclease decay appears to be the major mechanism (Larimer et al., 1992; Decker and Parker, 1994; Muhlrad et al., 1995; Beelman et al., 1996; LaGrandeur and Parker, 1998). Another pathway is the 3′–5′ decay pathway, in which the deadenylated mRNA is digested directly in a 3′→5′ direction by a complex of exonucleases termed the exosome (Mitchell et al., 1997; Jacobs et al., 1998). Deadenylation also appears to be the predominant initial step in vertebrate mRNA decay as well, although this pathway is less clear (Shyu et al., 1991; Couttet et al., 1997).
Distinct ribonuclease activities have been identified in higher eukaryotic cells (Schoenberg and Chernokalskaya, 1997). A poly(A)-specific 3′–5′ exoribonuclease that was cloned recently encodes a 74 kDa protein termed deadenylating nuclease (DAN) (Korner et al., 1998). The deadenylating activity of DAN could be the major activity involved in the initial phase of vertebrate mRNA turnover. A polysome-associated 33 kDa protein with 3′–5′ exonuclease activity has been identified and purified (Ross et al., 1987), yet the gene encoding this protein still remains elusive. Several endoribonuclease activities have also been identified. mRNAs targeted by endonuclease activity include those for: c–myc, insulin-like growth factor 2 (IGF–II), transferrin receptor (TfR), ApoII, Xlhbox 2B, 9E3 and albumin (Ross, 1995). Among the vertebrate endonucleases characterized, only one, a 120 kDa protein that cleaves the Xlhbox 2B mRNA, has been demonstrated to contain a sequence-specific cleavage activity (Brown et al., 1993), yet the gene remains to be identified. Recently, the gene encoding the endonuclease that cleaves Xenopus albumin mRNA was cloned. This protein, termed PMR-1, is a member of the peroxidase gene family but does not contain peroxidase activity (Chernokalskaya et al., 1998).
The stability of α–globin mRNA is conferred by a cytosine-rich element (CRE) in the 3′–UTR that forms an mRNP complex (α–complex) (Wang et al., 1995; Weiss and Liebhaber, 1995). Depending on the in vitro binding conditions, the α–complex consists of either a multiprotein complex that includes the poly(C)-binding αCP1 and αCP2 proteins (Wang et al., 1995; Kiledjian et al., 1997) or the αCP1 and αCP2 proteins alone (Chkheidze et al., 1999). However, it is still unclear whether the αCP proteins alone or the multiprotein complex constitute the functional unit involved in stabilizing mRNA. In addition to a role in stabilizing α–globin mRNA, the αCP proteins have been implicated in the stability of other mRNAs including those for collagen α1(I) and tyrosine hydroxylase (Stefanovic et al., 1995; Holcik and Liebhaber, 1997; Paulding and Czyzyk-Krzeska, 1999). The αCPs (also referred to as PCBP or hnRNP E) have also been implicated in the translational regulation of 15-lipoxygenase, poliovirus, hepatitis A virus and human papillomavirus mRNAs (Blyn et al., 1997; Gamarnik and Andino, 1997; Ostareck et al., 1997; Collier et al., 1998; Graff et al., 1998). They have recently been demonstrated specifically to bind a number of mRNAs that have cytosine-rich regions including the TAPA–1 and cox II mRNAs, although the functional significance of this binding is still unknown (Trifillis et al., 1999).
In vitro mRNA decay systems have greatly increased our ability to address the mechanism of vertebrate mRNA turnover (Brewer and Ross, 1990; Ross, 1993; Brewer, 1998; Ford et al., 1999; Wang et al., 1999). Reconstituted systems overcome limitations confronted in vivo that include difficulty in the detection of decay intermediates and the lack of genetic manipulations that are possible in yeast mRNA turnover analysis. We have recently devised an in vitro mRNA decay assay using the stable α–globin mRNA with post-polysomal S130 extract. Using this assay system, we demonstrated that one mechanism by which the α–complex stabilizes mRNA is by an interaction with the poly(A)-binding protein (PABP) to slow the rate of deadenylation (Wang et al., 1999). We now report that the α–complex also contributes to the stabilization of the α–globin mRNA by binding to and protecting the 3′–UTR from cleavage by a sequence-specific, erythroid-enriched endoribonuclease activity.
Results
Identification of an endonuclease activity in the α–globin 3′–UTR
Our previous work using an in vitro decay assay (IVDA) had demonstrated that the α–complex contributes to the stabilization of the α–globin mRNA by an interaction with PABP to slow down deadenylation (Wang et al., 1999). Throughout these studies, we had occasionally detected decay intermediates that corresponded to products within the 3′–UTR. To determine the nature of these intermediates, we undertook an in vitro analysis with unadenylated RNA probe and murine erythroleukemia (MEL) cytosolic S130 extract. MEL cells maintain the α–complex-mediated stabilization of the human α–globin mRNA in vivo (Weiss and Liebhaber, 1995) as well as in vitro (data not shown). Our initial studies where decay intermediates were observed occasionally were carried out at 37°C. We reasoned that potential decay intermediates might be less stable and difficult to detect at this temperature. As shown in Figure 1, a reaction carried out at 25°C using a uniformly labeled, unadenylated α–globin wild-type 3′–UTR (αwt) probe enabled the detection of two prominent intermediate bands (Int–I and Int–II) within 5 min of incubation (lane 2). Int–II appears to be less stable and disappears quickly, while Int–1 persists for longer. Interestingly, addition of oligo(dC), which competes for and removes the α–complex (Wang et al., 1999), accentuates the appearance and persistence of both intermediates (Figure 1, compare lanes 2–5 with lanes 6–9). Therefore, the removal of the α–complex enhances the generation of the intermediates, suggesting that the α–complex might be involved in protecting the RNA from ribonuclease digestion. Interestingly, the sum of the two prominent decay intermediate sizes equals the size of the full-length 3′–UTR input RNA. This is an indication that the intermediates are the product of an endoribonuclease activity and not exoribonuclease pause sites.
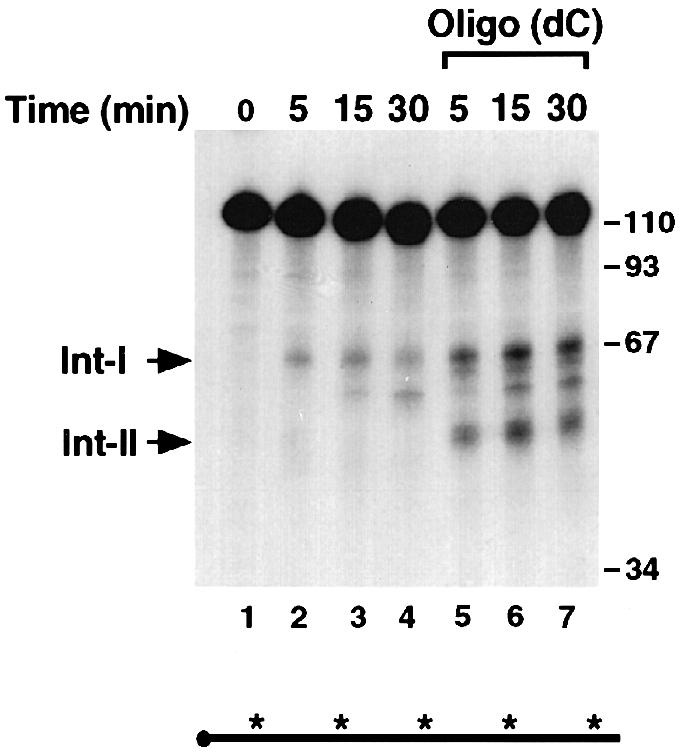
Fig. 1. Two major decay intermediates are detected in the α–globin 3′–UTR. IVDA reactions were carried out with uniformly labeled, capped αwt 3′–UTR in MEL S130 extract at 25°C. The input RNA is shown in lane 1. Lanes 2–4 and 5–7 show the reactions in the absence or presence of oligo(dC), respectively, for the times indicated. Reactions were stopped with ULB buffer, and the resulting RNAs were isolated, resolved on an 8% polyacrylamide–7 M urea gel and visualized by autoradiography. The two major decay intermediates are denoted on the left as intermediate I (Int–I) and intermediate II (Int–II). The DNA size markers are indicated on the right of the figure in nucleotides. The horizontal line below the gel represents the RNA probe used, and the closed circle denotes the 5′ cap. The asterisks represent 32P labeling, and its presence throughout the RNA indicates that it is uniformly labeled.
To determine directly whether the intermediates detected in Figure 1 are the product of an endoribonuclease activity, IVDAs were carried out with αwt RNA either uniformly labeled, 5′–end-labeled or 3′–end-labeled with 32P. Oligo(dC) was included in these reactions to detect the intermediates more readily. The RNA products were resolved on a sequencing gel to distinguish single nucleotide differences. As shown in the left panel of Figure 2, the two decay intermediates (Int–I and Int–II) are detected using a uniformly labeled αwt RNA at the initial 5 min time point, and these intermediates persist throughout the duration of the 60 min experiment (Figure 2, lanes 2–7). With increasing time, the decay intermediates accumulate and are chased into smaller products. When an αwt RNA labeled at the 5′ cap was used, only Int–I was detected (Figure 2, lanes 10–15). The use of 3′–end-labeled αwt RNA results in the detection of only Int–II (Figure 2, lanes 18–23). We therefore conclude that Int–I and II are the products of an endoribonuclease cleavage where Int–I corresponds to the 5′ fragment and Int–II to the 3′ fragment. These data demonstrate that a specific endoribonuclease cleaves the αwt RNA.

Fig. 2. An endoribonuclease activity targets the α–globin 3′–UTR in vitro. IVDAs were carried out in MEL S130 extract in the presence of 20 pmol of oligo(dC) at 25°C using uniformly labeled (lanes 1–7), 5′–end-labeled (lanes 9–15) or 3′–end-labeled (lanes 17–23) αwt probe. The input RNAs are shown in lanes 1, 9 and 17. Reaction times ranged from 5 to 60 min in each panel. An alkaline RNA ladder is shown in lanes 8, 16 and 24. The intermediates are indicated on the left of each panel. The labeling of the probes is indicated schematically by the asterisks under each panel. Uniformly labeled probe is as described in the legend to Figure 1. An asterisk at the left or right end of the horizontal line denotes that the probe is 5′ or 3′ end labeled, respectively.
Following cleavage by the endoribonuclease, the two intermediates appear to be degraded by a 3′–5′ exoribonuclease activity. The smaller decay fragments for Int–I are identical for both uniformly labeled and 5′–end-labeled RNA (Figure 2, compare lanes 2–7 with lanes 10–15). This demonstrated that the 5′ end is intact and the decay is occurring from the 3′ end most likely by a 3′–5′ exoribonuclease. This premise is substantiated further by the decay pattern of the 3′–end-labeled Int–II. Unlike the uniformly labeled Int–II in lanes 2–7, which generates smaller fragments with increasing time, only the initial cleavage product is detected with the 3′–end-labeled Int–II (lanes 18–23). This further underscores a 3′–5′ exoribonuclease being involved in clearing the endoribonuclease cleavage products. It should also be noted that the Int–II fragment is not degraded efficiently in the 5′→3′ direction even though it does not contain a 5′ cap. These data demonstrate that both endonuclease cleavage products are cleared predominantly by a 3′–5′ exoribonuclease activity in this assay system.
The endoribonuclease cleavage site was mapped using a 5′–end-labeled probe that would only detect the Int–I fragment (Figure 3A). An alkaline RNA ladder was included to identify each nucleotide, and a partial digestion of the 5′–end-labeled αwt probe with RNases U2 and T1, which cleave after adenosine and guanine residues, respectively, was also included to enable sequence identification. In agreement with the data presented in Figure 1, incubation of the 3′–UTR with S130 extract results in the accumulation of Int–I, which becomes more intense with the addition of oligo(dC) (Figure 3A, compare lanes 2 and 3 with lanes 4 and 5). These data also indicate that the cleavage product was not altered by the addition of oligo(dC). Interestingly, addition of EDTA to the reaction did not significantly affect the activity of the endoribonuclease but minimized formation of the subsequent exoribonuclease products (Figure 3A, lanes 6 and 7), which demonstrates that the endoribonuclease is less sensitive to EDTA relative to the exoribonuclease activity. A comparison of the major initial cleavage product with the alkaline RNA ladder reveals the prominent cleavage site to be 63 nucleotides downstream of the stop codon (Figure 3C). The cleavage site on the Int-II 3′ fragment was mapped using a 3′–end-labeled RNA probe. As shown in Figure 3B, the corresponding 47 nucleotide 3′ fragment is detected (lanes 2 and 3). Consistent with a role for the α–complex in protecting the target sequence, the site is contained within the region previously mapped by Holick and Liebhaber (1997) as a region protected by the α–complex (Figure 3C).
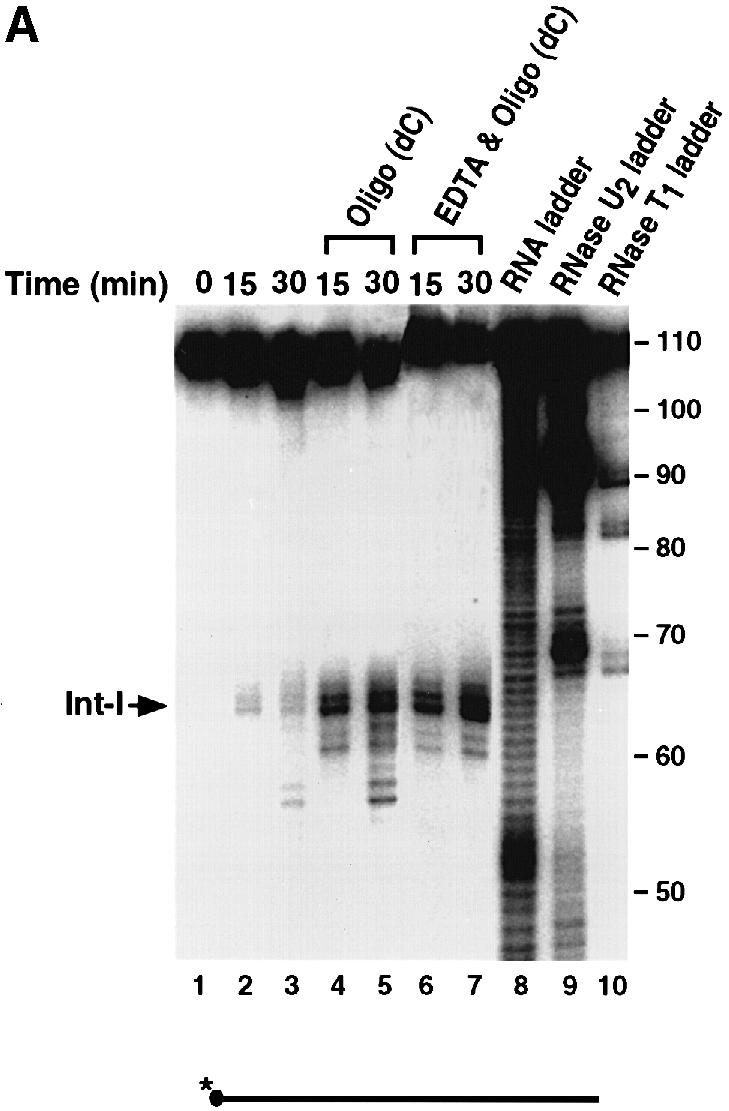
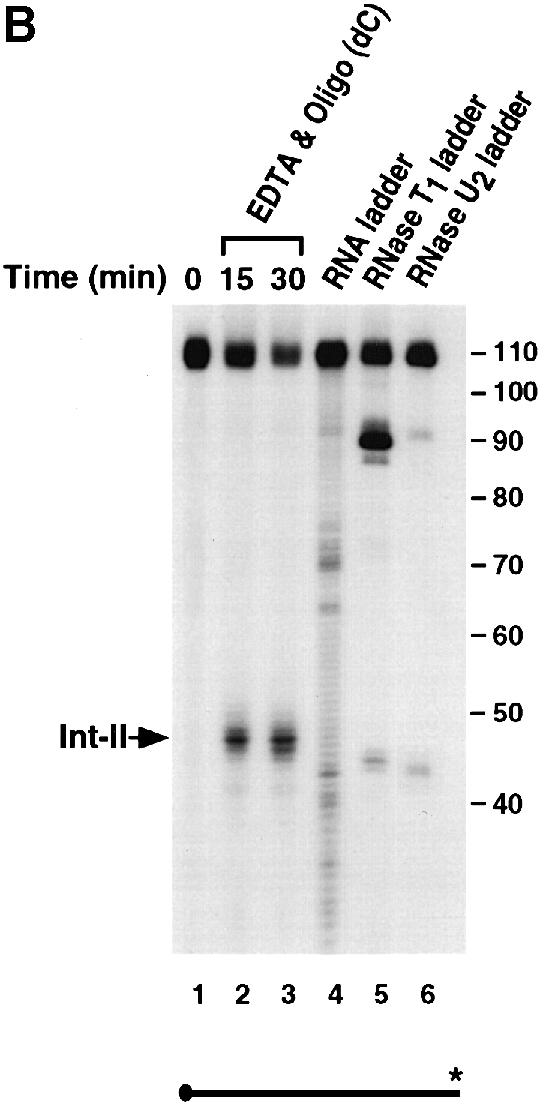

Fig. 3. The endoribonucleolytic cleavage site is within a region protected by the α–complex. (A) An IVDA of 5′–end-labeled RNA in MEL S130 extract is shown. Oligo(dC) was added in lanes 4 and 5, and both oligo(dC) and EDTA were added in lanes 6 and 7. The alkaline RNA, RNase U2 and RNase T1 ladders are shown in lanes 8, 9 and 10, respectively, and the corresponding RNA sizes are denoted on the right. The 5′ intermediate (Int–I) is indicated on the left. The 5′–end-labeled RNA schematic is shown at the bottom. (B) In vitro decay of 3′–end-labeled RNA in MEL S130 extract. The 3′ intermediate (Int–II) is indicated on the left and labeling is as described above. (C) The α–globin 3′–UTR sequence. The endoribonuclease cleavage site at nucleotide 63 is represented by the arrow. The shaded box represents the region protected by the α–complex as reported by Holcik and Liebhaber (1997).
An endoribonuclease activity is involved in the turnover of α–globin mRNA
Having identified the in vitro endoribonuclease cleavage site, we next determined whether these products could also be detected in vivo, which would indicate that the endoribonuclease can target α–globin mRNA in cells. If the in vitro pathway of α–globin mRNA turnover is representative of the in vivo pathway, fragments of 63 and 47 nucleotides, as illustrated at the bottom of Figure 4, should be produced when using an RNase protection assay. MEL cells were transfected with a plasmid expressing the human α2–globin gene and stably transformed cells (MEL–α2) were isolated. Expression of the α–globin gene in the MEL-α2 cells was shown by the presence of the protected full-length 3′–UTR in lanes 3 and 4 of Figure 4 but not in untransfected MEL cells (lane 2). In addition to the full-length intact 3′–UTR, two prominent bands corresponding to the expected sizes were also detected in MEL cells expressing the α–globin transgene (Figure 4, lanes 3 and 4). No bands were detected when RNA from untransfected MEL cells was used (lane 2). Consistent with the in vitro results, the intermediates were more stable when the cells were grown at 25°C compared with 37°C (Figure 4, compare lanes 3 and 4; see Materials and methods). The identity of the isolated Int–I and II fragments was confirmed by sequencing the reverse-transcribed and PCR-amplified RNA isolated from the gel (data not shown). These data demonstrate that the endoribonuclease activity detected in vitro is also involved in the natural turnover of α–globin mRNA in erythroid cells.

Fig. 4. An endoribonuclease cleaves α–globin mRNA in cells. Total RNA from MEL cells (lane 2), MEL-α2 cells grown at 37°C (lane 3) or at 25°C (lane 4) was analyzed by an RNase protection assay. A schematic diagram of the riboprobe and expected intermediate bands is shown at the bottom. The probe is antisense to the 3′–UTR (filled region) and extends 60 nucleotides downstream of the poly(A) addition site (unfilled region). The resulting RNase-resistant RNAs corresponding to the full-length 3′–UTR (110 nucleotides), Int–I (63 nucleotides) and Int–II (47 nucleotides) are indicated on the right.
Sequence specificity of the endoribonuclease
The sequence requirement at the cleavage site was tested by mutational analysis. Three nucleotides on either side of the cleavage site were substituted with alternating guanine and adenine dinucleotides (αmt-GA). The αmt-GA substitution prevents formation of the α–complex on the 3′–UTR (data not shown) and renders the RNA refractory to the endoribonuclease activity (Figure 5, lanes 5–8). Under the same assay conditions, the wild-type RNA is an efficient substrate (Figure 5, lanes 1–4) for this activity.

Fig. 5. Sequences at the endoribonuclease site are necessary. IVDA reactions were carried out with αwt RNA and αmt-GA RNA in which the six nucleotides around the endoribonucleolytic cleavage site were substituted with GAGAGA. αwt (lanes 1–4) and αmt-GA RNA (lanes 5–8) were incubated with MEL S130 extract containing oligo(dC) and EDTA for the times indicated. The arrow within the RNA schematic represents the site of cleavage and the ‘X’ depicts a block of the cleavage activity. The filled box denotes the six nucleotide substitution mutation. The intermediates and DNA size markers are as described in the legend to Figure 1.
To determine whether the endoribonuclease cleavage could be conferred on a heterologous RNA, a segment of the α–globin 3′–UTR (nucleotides 37–81) that includes the endoribonuclease cleavage site was inserted into a random sequence (pGEM4 polylinker). The ability of the endoribonuclease to recognize and cleave the chimeric RNA was determined. As shown in Figure 6A, a 45 nucleotide region of the α–globin 3′–UTR was recognized and cleaved efficiently by the endoribonuclease activity (lane 2). The two resulting prominent intermediate products (Int-A and Int-B) correspond to the expected 73 and 51 nucleotide fragments. Additional background bands larger than Int-A and one smaller than Int-B are also detected. These appear to be unrelated to the α–globin sequences since comparable bands are also detected in the pGEM4 polylinker alone (Figure 6A, lane 4). Further confirmation that the intermediates are endoribonuclease products is presented in Figure 6B where 5′–end-labeled RNA was used in the assays and only the 5′ fragment (Int-A) was detected (lanes 2 and 3). The cleavage site was mapped in the chimeric RNA to the same site as that in the wild-type α–globin 3′–UTR shown in Figure 3A (data not shown). These data demonstrate that the 45 nucleotide fragment that we will refer to as the endoribonuclease recognition sequence (ERS) is an autonomous element that can be recognized and cleaved by a sequence-specific endoribonuclease activity.
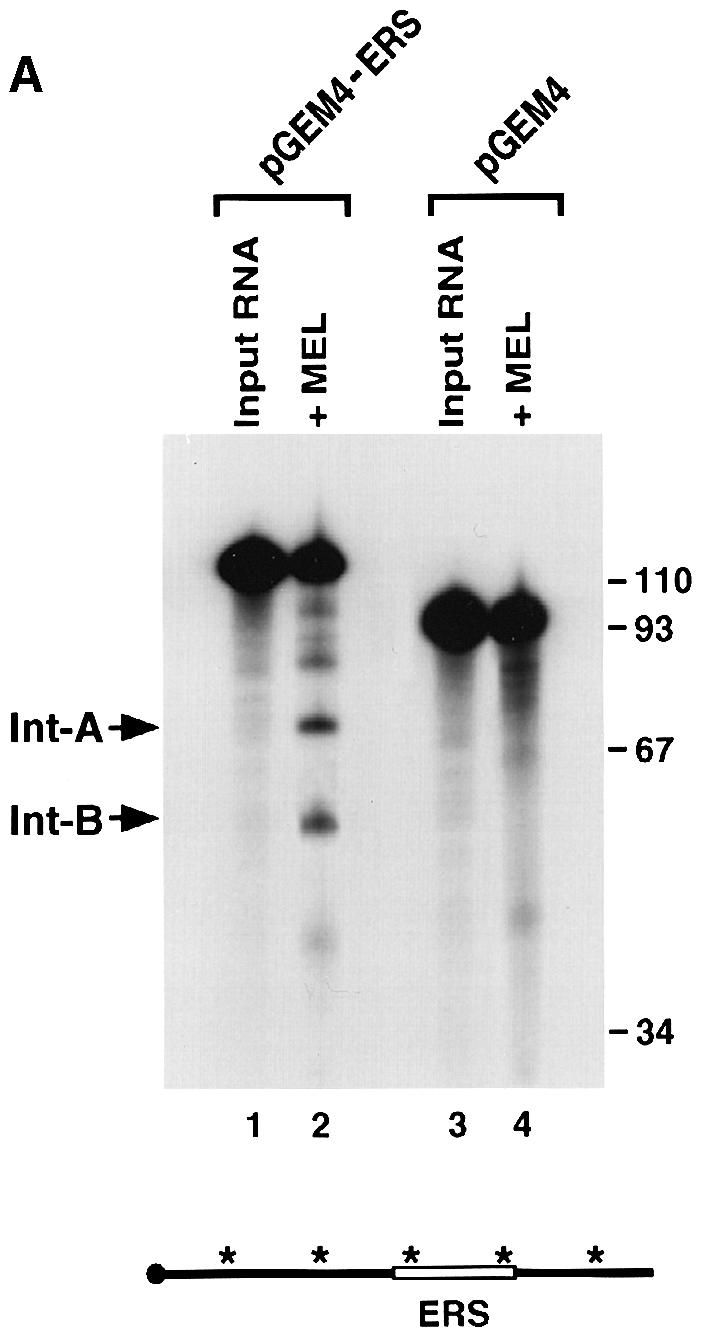
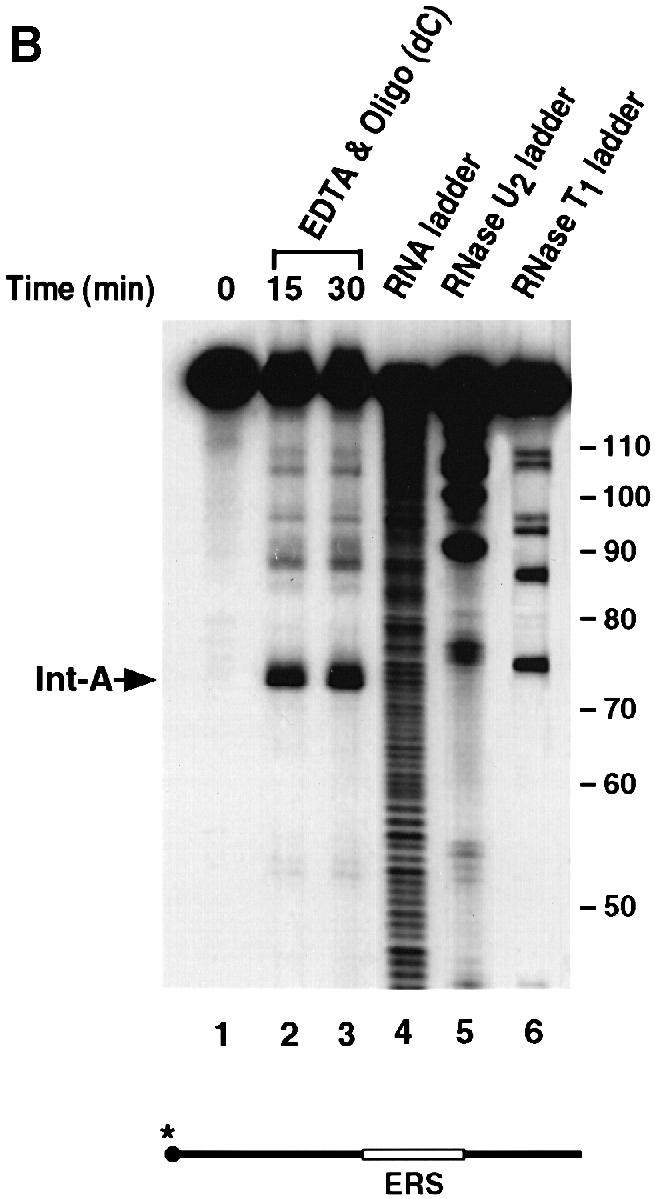
Fig. 6. The ERS can function as an autonomous element on a heterologous RNA. (A) IVDAs were carried out in MEL S130 extract containing EDTA with uniformly labeled pGEM4 polylinker RNA (lanes 3 and 4) or uniformly labeled pGEM4 polylinker containing the 45 nucleotide ERS (lanes 1 and 2). The two endoribonuclease decay intermediates are indicated as Int–A and Int–B. A schematic of the RNA is shown at the bottom as described in the legend to Figure 1, with the open box denoting the ERS. (B) An IVDA of 5′–end-labeled pGEM4-ERS chimeric RNA (shown on the bottom) with MEL S130 extract and EDTA is shown in lanes 2 and 3. The alkaline RNA, RNase U2 and RNase T1 ladders are shown in lanes 4, 5 and 6, respectively, and the corresponding RNA size marker on the right. The 5′ endoribonuclease cleavage product is indicated as Int–A on the left.
The endoribonuclease activity is ATP independent
To determine whether the endoribonuclease activity requires an energy source, the decay reactions were carried out under conditions lacking ATP. As shown in Figure 7A, ATP was not required for the endoribonuclease activity since decay intermediates were detected in the absence of ATP (compare lanes 2 and 3). To ensure that the activity was not a result of residual ATP present in the extract, apyrase was also added to remove endogenous ATP (Frassetto et al., 1993). Removal of endogenous ATP also had a minimal effect on the endoribonuclease activity (Figure 7A, lane 4), although a 93 nucleotide band was detected periodically when apyrase was included. The nature and significance of this band is unknown. Furthermore, the endoribonuclease activity is present mostly in the soluble S130 extract (Figure 7B, lanes 2–4) and only residual amounts are detected in the polysomal fraction (lanes 5–7) or the ribosomal high salt wash (RSW) fraction (lanes 8–10). Therefore, the endoribonuclease activity is ATP independent and localized predominantly in the soluble cytosolic fraction, as determined by the in vitro decay conditions employed.
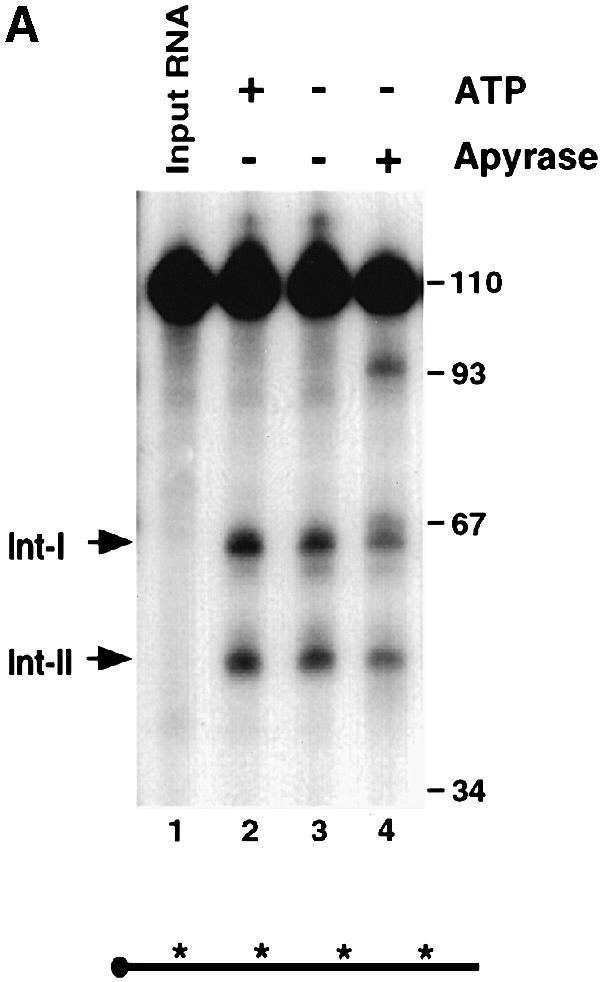

Fig. 7. The endoribonuclease activity is ATP independent and not polysome associated. (A) Uniformly labeled αwt probe (shown schematically at the bottom) was incubated with MEL S130 extract for 15 min in the presence of EDTA. ATP was included in lane 2 and excluded in lanes 3 and 4. Apyrase was also included in lane 4 to sequester endogenous ATP in the extract. The intermediates are indicated on the left. (B) IVDAs with 5′–end-labeled αwt RNA were carried out with MEL S130 extract (lanes 2–4), MEL polysomal fraction (lanes 5–7) or MEL RSW fraction (lanes 8–10) for the times indicated. The 5′ intermediate is denoted on the left, and the DNA size markers are in nucleotides.
The endoribonuclease activity is enriched in erythroid cells
To address the tissue specificity of the endoribonuclease activity, the IVDAs were carried out with extracts from non-erythroid cells. Extract from MEL cells (Figure 8, lanes 2 and 3) was compared with extracts from mouse hybridoma cells (SP2/0; lanes 4 and 5), macrophage cells (Raw246.7; lanes 6 and 7) and fibroblast cells (NIH 3T3; lanes 8 and 9). Under identical assay conditions, the activity of the erythroid cell extract was dramatically higher than that of the non-erythroid cell extracts. Similar results were obtained from human K562 erythroid cell extract (Figure 8B, lane 2) and non-erythroid cell extracts from B cells (Ramos; lanes 4 and 5), T cells (Jurkat; lanes 6 and 7) and fibroblasts (HeLa; lanes 8 and 9). The cleavage products are the result of a protein activity as indicated by the sensitivity of this activity to proteinase K treatment (compare lanes 2 and 3 in Figure 8A and B). Although the activity is enriched in erythroid cell extract, it is not exclusively erythroid specific since the appropriate size intermediate products are detected in the non-erythroid cells upon overexposure of the gel (data not shown). We will refer to this activity as the erythroid-enriched endoribonuclease, ErEN. These data are consistent with ErEN being involved in the regulation of α–globin mRNA turnover, since expression of α–globin is erythroid specific.
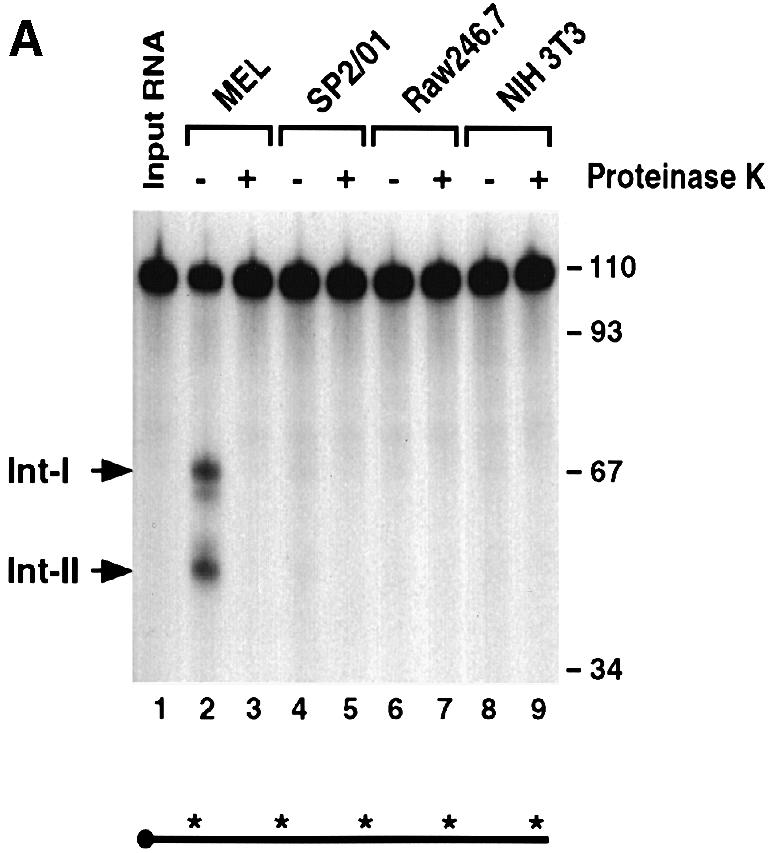
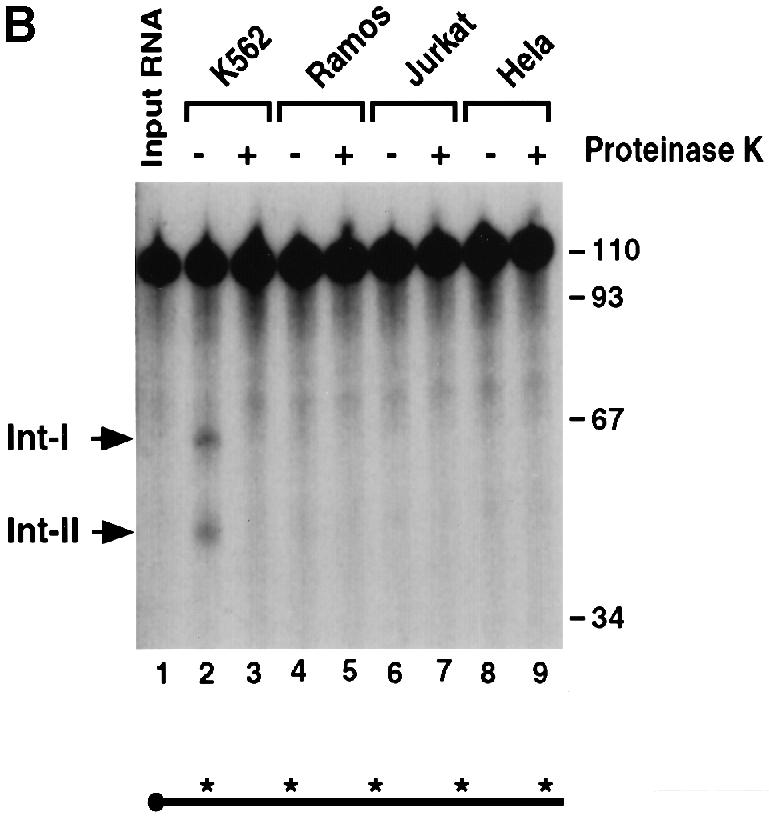
Fig. 8. The endoribonuclease activity is enriched in erythroid cells. (A) An IVDA was carried out with mouse erythroid S130 extract (lanes 2–3) and non-erythroid S130 extract (lanes 4–9) with uniformly labeled αwt probe in the absence or presence of proteinase K as indicated. Reactions were carried out for 15 min in the presence of oligo(dC). Labeling is as described in the legend to Figure 1. (B) IVDAs were carried out with human erythroid S130 extract (lanes 2–3) and non-erythroid S130 extract (lanes 4–9) as described above.
We determined whether the coordinately expressed β–globin mRNA is also regulated by ErEN. The 3′–UTR of β–globin contains the major stability determinant of this RNA (Russell and Liebhaber, 1996). Use of the β–globin 3′–UTR in an IVDA with MEL extract revealed several low abundance intermediate bands (Figure 9, lane 5). The same intermediates are also detected with HeLa S130 extract (Figure 9, lane 6). These data demonstrate that the intermediates are not the products of ErEN, otherwise extract from the erythroid and non-erythroid cells would yield distinct products as is observed with the α–globin 3′–UTR (Figure 9, lanes 2 and 3). Therefore, ErEN can target α–globin mRNA specifically but not β–globin mRNA.
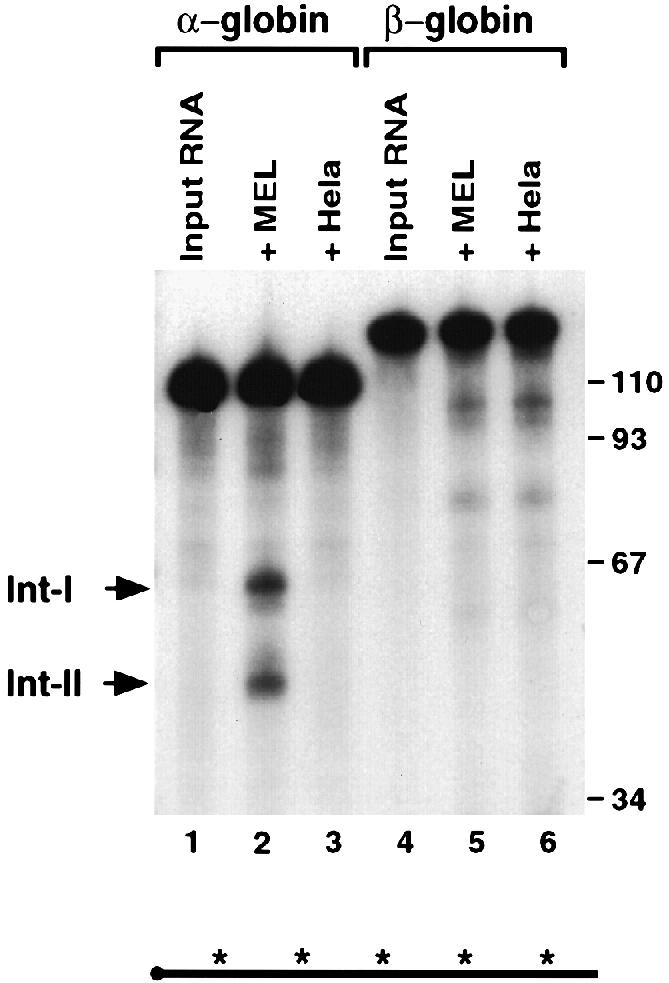
Fig. 9. The endoribonuclease activity is α–globin mRNA specific. IVDA reactions using uniformly labeled α–globin (lanes 1–3) or β–globin (lanes 4–6) 3′–UTRs were carried out in MEL S130 extract (lanes 2 and 5) or HeLa S130 extract (lanes 3 and 6) in the presence of oligo(dC) and EDTA for 15 min. Labeling is as described in the legend to Figure 1.
The murine α–globin mRNA is cleaved by an endoribonuclease
Comparison of the human α–globin (hα) ERS sequence with the corresponding region in the murine α–globin (mα) reveals an overall 60% identity, with the highest degree of conservation at the cleavage site. To test whether endoribonuclease cleavage occurs within the α–globin mRNA of other species, we determined whether the mα–3′–UTR is also targeted by an endoribonuclease. An IVDA using the 96 nucleotide mα–3′–UTR probe was carried out with MEL S130 extract. The input mα–3′–UTR probe should be cleaved in half to produce two 48 nucleotide intermediates if the position of cleavage is coincident with the hα–3′–UTR. As shown in Figure 10A, use of the mα–3′–UTR in this assay produces several bands within this size range (compare lanes 1 and 2). Wang and Liebhaber (1996) have demonstrated that the mα–3′–UTR forms an mRNA stability complex. Surprisingly, this complex is distinct from the hα–complex and contains a prominent poly(CU)-binding activity rather than a poly(C)-binding activity (Wang and Liebhaber, 1996). As shown in lane 4, addition of oligo(dCT), which competes for the mα–complex, accentuated the prevalence of the putative 48 nucleotide endoribonuclease cleavage products, while addition of a control oligonucleotide had no effect (Figure 10A, compare lanes 3 and 4). Furthermore, this activity was enriched in erythroid cells relative to non-erythroid HeLa cells even in the presence of oligo(dCT) competitor (Figure 10A, compare lanes 4 and 5). As a further confirmation that the mα–3′–UTR is targeted by an endoribonuclease activity, a second probe that contains an additional 48 nucleotides of polylinker sequence added to the 3′ terminus of the RNA was used. Cleavage of this RNA would produce the 48 nucleotide 5′ fragment as above and a 96 nucleotide 3′ fragment. As shown in Figure 10B, both the 5′ and 3′ products were detected when uniformly labeled RNA was incubated with MEL S130 extract (lane 2). Only the 5′ fragment was detected with the 5′–end-labeled RNA (Figure 10B, lane 4), and only the 3′ fragment was detected with the 3′–end-labeled RNA (lane 6). These data collectively suggest that an ErEN activity also cleaves the mα–3′–UTR in a region analogous to the hα–3′–UTR. They also suggest that the murine stability complex might be involved in regulating the access of the nuclease to the 3′–UTR.
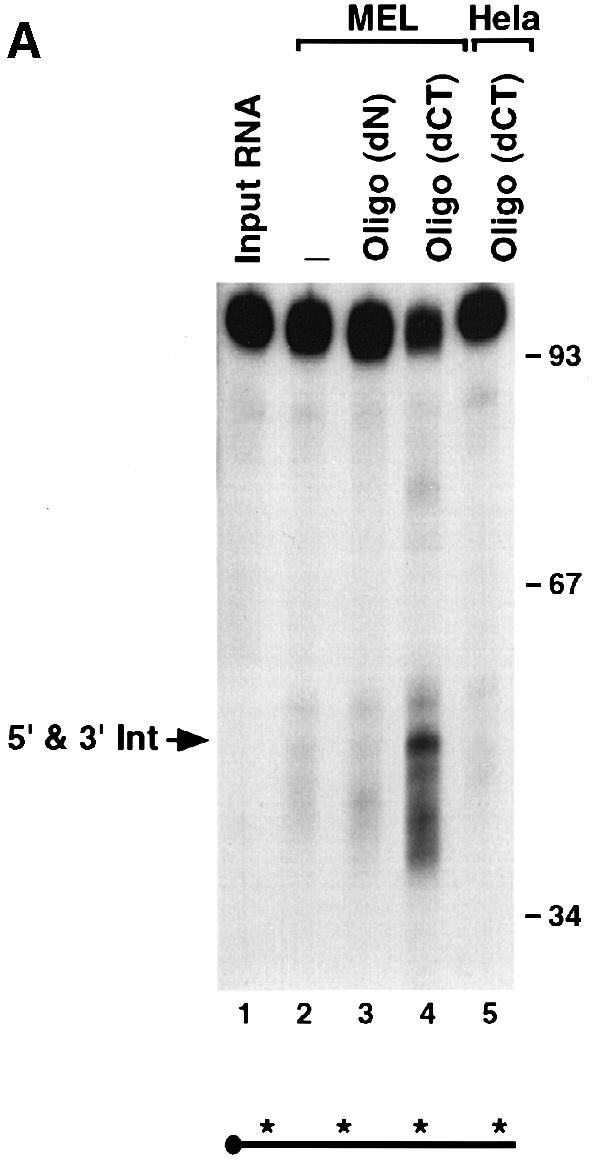
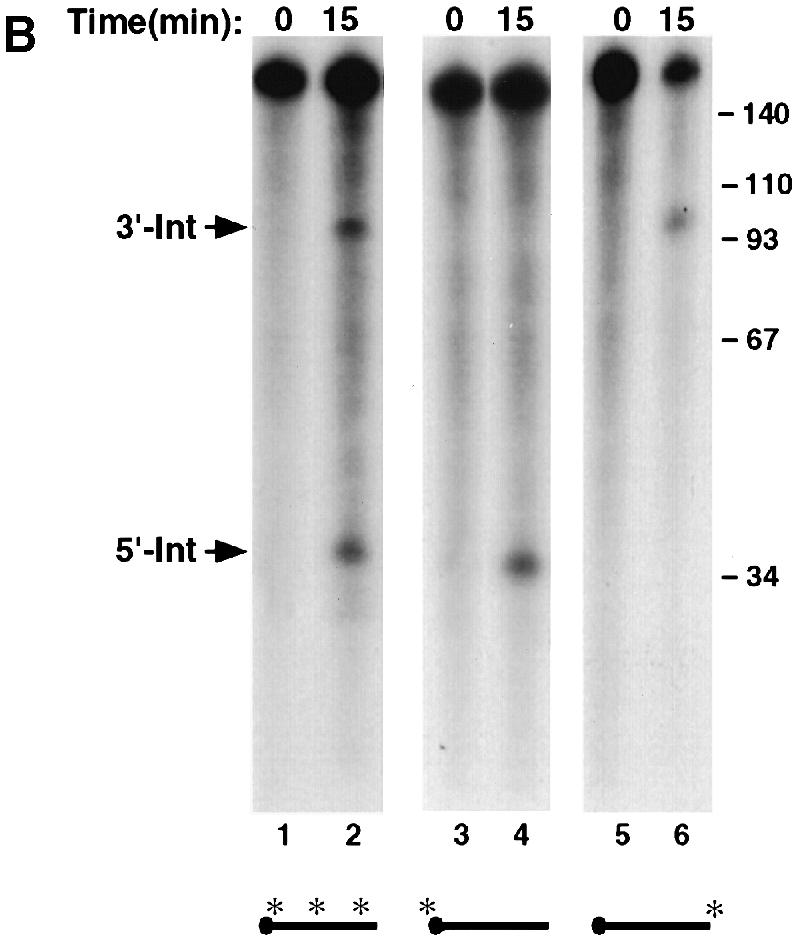
Fig. 10. The mα-3′–UTR is cleaved by an erythroid-enriched endoribonuclease. (A) IVDA reactions with mα-3′–UTR were carried out using uniformly labeled RNA and either MEL (lanes 2–4) or HeLa (lane 5) S130 extract. Reactions included either non-specific competitor (lane 3) or specific competitor for the murine stability complex [oligo(dC); lanes 4 and 5]. The co-migrating 48 nucleotide 5′ and 3′ fragments are indicated on the left and the DNA size markers on the right. (B) An IVDA with the mα-3′–UTR containing a polylinker sequence on the 3′ end of the RNA was used to resolve the resulting intermediate fragments. The 5′ and 3′ intermediates are indicated on the left. Lanes 1 and 2 contain uniformly labeled RNA (both intermediates are detected). Lanes 3 and 4 contain a 5′–end-labeled RNA where only the 5′ intermediate is detected, and lanes 5 and 6 contain 3′–end-labeled RNA where the 3′ intermediate results. The DNA size markers are shown on the left and a schematic of the RNA probes is shown at the bottom.
Discussion
In this report, we have presented evidence that a step in the turnover pathway of α–globin mRNA involves the cleavage of the 3′–UTR by a specific ErEN. The endoribonuclease products were detected in vivo as well as in vitro, further validating the in vitro turnover observations. The cleavage site was mapped to a region previously demonstrated by Holcik and Liebhaber (1997) to be protected by the α–complex, implying that access of ErEN to the 3′–UTR is regulated by the α–complex. Additional support for the involvement of the α–complex hindering access of ErEN to the RNA is provided by the use of oligo(dC) competition that removes the α–complex and enables more efficient access of the RNA to the nuclease (Figures 1 and 3). We had demonstrated previously that the α–complex influences α–globin mRNA deadenylation by interacting with PABP to slow the rate of deadenylation (Wang et al., 1999). Collectively, these data indicate that in addition to its effect on deadenylation, the α–complex also functions to stabilize mRNA by protecting the RNA from an endoribonuclease activity.
Our previous work focused on the interaction of the α–complex with PABP and its effect on deadenylation using polyadenylated RNA substrates (Wang et al., 1999). In those studies, stable decay intermediates within the RNA were not detected reproducibly. Detection of decay intermediates in the current study can be attributed to two factors. First, a shift of the reaction temperature to 25°C attenuates the exoribonucleolytic clearing of the intermediate products and enables their detection. Our original studies were carried out at 37°C. At that temperature, the endoribonuclease intermediate products were cleared rapidly and not detected efficiently. Secondly, the use of an unadenylated RNA probe removes any potential influence of the poly(A) tail and permits a clearer determination of the decay intermediates. In fact, the endoribonuclease cleavage activity is influenced by the poly(A) tail and functions subsequently to poly(A) tail removal in vitro (Z.Wang and M.Kiledjian, in preparation).
Sequence substitution of the endoribonuclease target site disrupts cleavage of the mutant RNA by the nuclease. This is an indication that the sequence around the cleavage site is necessary for proper activity. However, a 24 nucleotide sequence centered around the cleavage site at nucleotide 63 was unable to act as an autonomous element when placed onto a heterologous RNA (our unpublished observation). Therefore, unlike DNA restriction enzymes, the recognition sequence for ErEN is not contained totally within the target cleavage site. A longer segment of the 3′–UTR that extends from nucleotides 37 to 81 contains all the sequence elements necessary to be recognized and cleaved by ErEN (Figure 6). The studies presented in this report use the major α2–globin mRNA. The α1–globin gene that shares considerable homology in the 3′–UTR with the α2–globin mRNA can also be recognized and cleaved by ErEN in a sequence-specific manner (our unpublished observation). However, β–globin mRNA, which does not share sequence homology with α–globin, is not targeted by this activity (Figure 9). A detailed analysis of the recognition sequence will determine the minimal sequence requirements more precisely.
The stability determinant of both α- and β–globin mRNA resides within their respective 3′–UTRs, although the turnover of each mRNA appears to be distinct (Wang et al., 1995, 1999; Weiss and Liebhaber, 1995; Russell and Liebhaber, 1996). The β–globin 3′–UTR does not contain a CRE and consequently does not form the α–complex (Wang et al., 1995). However, low molecular weight intermediates that correspond to regions within the rabbit β–globin 3′–UTR have been detected in rabbit reticulocyte lysate. The nuclease attributed to this activity is polysome associated (Bandyopadhyay et al., 1990) unlike ErEN, which is non-polysome associated (Figure 8). It appears that distinct nucleases are involved in the regulation of α- and β–globin mRNA turnover. Additional mRNAs regulated by the various nucleases remain to be determined.
It is interesting that ErEN activity is highly enriched in erythroid cells while the α–complex is present both in erythroid and non-erythroid cells. The α–complex appears to be dispensable for the stability of ectopically expressed α–globin in non-erythroid cells. Substitution mutations that are unable to form the α–complex are less stable than the wild-type α–globin in erythroid cells and are as stable as the wild type in non-erythroid cells (Weiss and Liebhaber, 1995). This inability of non-erythroid cells to distinguish between the αwt and the substitution mutants can be explained partially by the activity of ErEN. In erythroid cells, the substitution mutants, which do not form the α–complex, are subjected to ErEN degradation, while αwt RNA is protected by the α–complex. On the other hand, in non-erythroid cells, substitution mutants show higher stability because the ErEN activity is missing. This explanation can account for most of the substitution mutations reported by Weiss and Liebhaber (1995) that can still be recognized by the endoribonuclease activity unless the substitution is at the cleavage site (our unpublished observations). Collectively, these observations suggest an involvement of tissue-specific components in selective erythroid mRNA stability.
Several examples have been documented where an endoribonuclease activity is important in the regulation of mRNA stability. The abundance of the TfR mRNA is regulated by intracellular iron levels (Kuhn, 1991; Klausner et al., 1993). Under conditions where the iron levels are reduced, the TfR mRNA is stabilized. This stability results from the binding of the iron response protein (IRP) to the iron response element (IRE) within the 3′–UTR, which protects the mRNA from cleavage by an endoribonuclease (Harford, 1993). An increase in intracellular iron concentration results in disassociation of IRP from the IRE and exposes the endoribonuclease site. Consequently, the mRNA is destabilized (Koeller et al., 1991; Binder et al., 1994). In the case of the maternal homeobox mRNA, Xhlbox2B, its mRNA levels are high in early previtellogenic oocytes but decrease at late stages of oogenesis. The 3′–UTR of this mRNA also contains a region that is bound by a protein factor which can protect the mRNA from cleavage by an endoribonuclease. A direct correlation between a decrease in the amount of the protective factor and a decrease in Xhlbox2B mRNA exists in late stages of oogenesis, suggesting that the protective factor plays an important role in regulating the mRNA turnover (Brown et al., 1993). Our observations with α–globin mRNA are analogous to these examples. The α–complex can stabilize the α–globin mRNA by protecting an endoribonuclease site within the 3′–UTR. These results suggest a general regulatory mechanism for mRNA turnover where mRNA containing endoribonuclease sites can be protected by a protein factor. Altering the binding of the protective factor to the RNA can subsequently alter the stability of this mRNA.
The fact that an endoribonuclease specifically cleaves the α–globin mRNA appears somewhat paradoxical. Why would a stable mRNA contain a target sequence for a specific endoribonuclease? We propose that this provides a safety mechanism to ensure that an erythroid cell does not produce excess α–globin relative to β–globin. A functional hemoglobin tetramer consists of two α–globin and two β–globin subunits. Excess free α–globin chains, but not free β–globin, precipitate onto and damage the cell membrane, leading to ineffective erythropoiesis and peripheral hemolysis (Nathan and Gunn, 1966; Weatherall and Clegg, 1972; Schwartz and Benz, 1990). The significance of ErEN might be to ensure that excess toxic α–globin does not accumulate in the cell. The observation that β–globin is not recognized by ErEN is consistent with this premise. The presence of an endoribonuclease activity in the mα-3′–UTR suggests that this might be a common regulatory mechanism of α–globin mRNA in different species. Despite the fact that distinct protein complexes appear to regulate the stability of the human and murine α–globin mRNAs (Wang and Liebhaber, 1996), they appear to retain the same function of protecting the mRNA from an endoribonuclease. However, it is currently unknown whether both the human and murine α–globin mRNAs are cleaved by the same endoribonuclease. A role for a specific endoribonulcease could also be envisaged during terminal erythroid differentiation, when all mRNAs, including globin mRNAs, are cleared from the cell. During this stage of development, the level of αCPs, and hence the ability of the α–complex to form and protect the α–globin mRNA, decreases (Morales et al., 1997). This should allow ErEN access to the mRNA and ensure that toxic α–globin chains do not accumulate at the terminal differentiation stage. Alternatively, the activity of ErEN might be altered by a modification or by other proteins that enable a more pleiotropic activity to target a wider range of mRNAs for endonucleolytic digestion. This could explain how α–globin is stabilized selectively relative to non–globin mRNAs. Identification and analysis of the endoribonuclease could begin addressing these issues.
Materials and methods
Plasmid constructs
The human α2–globin expression plasmid, pSV2Aneo-α2, has been described previously (Weiss and Liebhaber, 1995). The α–globin 3′–UTR substitution mutation, αmt-GA, replaces the wild-type sequence at nucleotides 61–66 downstream of the stop codon with GAGAGA. The mutation was generated by overlap PCR and inserted into the PCR-trap vector (GenHunter). The mutation was confirmed by sequence analysis. The plasmid pGEM4-ERS contains 45 bases from the α–globin 3′–UTR (nucleotides 37–81) inserted into the polylinker region of pGEM4 (Promega). The plasmid was constructed by synthesizing both strands of DNA such that _Eco_RI and _Bam_HI overhangs were generated upon annealing and inserted into the same sites in pGEM4.
Extract preparation
MEL cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum containing 100 U/ml penicillin and 100 μg/ml streptomycin. Isolation of the S130 extract was carried out as previously described (Kiledjian et al., 1999). Briefly, cells were washed twice in phosphate-buffered saline (PBS) and resuspended in 1.5 ml per 108 cells of buffer A [10 mM Tris pH 7.5, 1 mM potassium acetate, 1.5 mM magnesium acetate, 2 mM dithiothreitol (DTT)]. Cells were lysed with 25 strokes of a type–B Dounce homogenizer and nuclei removed with a 10 min 2000 g centrifugation. The supernatant was layered over buffer A containing 30% (w/v) sucrose and centrifuged at 130 000 g for 2 h. The high speed supernatant (S130 extract) was removed without disturbing the S130–sucrose interface, supplemented with glycerol to a final concentration of 5% (v/v) and frozen in aliquots at –70°C. The remaining sucrose cushion was aspirated and the polysome pellet was rinsed twice with buffer A, resuspended in buffer A with 5% glycerol and stored in aliquots at –70°C. RSW was prepared from the polysome fraction as described in Brewer and Ross (1990). Polysomes from 6 × 108 cells were resuspended in 2 ml of buffer A at 4°C, and 2 M KCl was added dropwise with stirring to bring the final concentration to 0.3 M KCl. After 15 min of stirring at 4°C, polysomes were pelleted through a cushion of buffer A containing 30% (w/v) sucrose at 130 000 g. The resulting RSW supernatant was dialyzed against buffer A and concentrated. The RSW was stored at –70°C with 5% glycerol. S130 extract from Raw246.7 (kindly provided by D.Denhardt, Rutgers University), Ramos and Jurkat (kindly provided by L.Covey, Rutgers University), SP2/0 and NIH 3T3 cells was made as described above.
MEL–α2 cells, which are MEL cells stably transformed with the human α2–globin gene, were generated by transfecting MEL cells with the pSV2Aneo–α2 plasmid. The plasmid was linearized at the unique _Kpn_I site 500 bp upstream of the α2–globin promoter. Transfections were carried out using the TransFast (Promega) system with 107 MEL cells and 1 μg of linearized plasmid as described by the manufacturer. After 2 days, cells were collected and grown in selective DMEM containing 400 μg of G418 per ml. The selective medium was replaced every 3 days. Stably transformed cells were expanded and used in the RNA isolation.
RNA probe and marker production
The α–globin 3′–UTR was PCR amplified from the pSV2Aneo–α2 plasmid with T7 bacteriophage promoter containing the 5′ primer (CGTAATACGACTCACTATAGGGCTGGAGCCTCGGTAGCCGT) and the 3′ primer (GCCGCCCACTCAGACTTT). The human β–globin 3′–UTR was PCR amplified from the plasmid pSPK_B/c_ (Ross et al., 1987) with the 5′ primer containing the T7 polymerase promoter (CGTAATACGACTCACTATAGGGGCTCGCTTTCTTGCTGTCC) and the 3′ primer (CAATGAAAATAAATGTTTTTTATTAGGCAGAATCC). The PCR products were phenol/chloroform extracted once, chloroform extracted twice, ethanol precipitated and washed with 70% ethanol. RNA transcripts were generated with T7 polymerase (Promega) according to the manufacturer's conditions using 200 ng of template. mα–globin mRNA was reverse transcribed using oligo(dT) from MEL RNA and PCR amplified with the following primers: 5′ primer, CGTAATACGACTCACTATAGGGCTGCCTTCTGCGGGGCTTGC, which includes the T7 promoter; and 3′ primer, CTTCTTCCTACTCAGGCTTTATTC. The fragment was inserted into pGEM4 (Promega) to generate pGem4–mα and confirmed by sequencing. RNA probe was generated with T7 RNA polymerase using either a PCR template amplified with the mouse 5′ and 3′ primers for the 96 nucleotide probe or from a PCR template amplified with the mouse 5′ primer and the SP6 promoter primer to yield the 144 nucleotide probe. When generating uniformly labeled riboprobes, the m7G(5′)ppp(5′)G cap analog and [α-32P]UTP were included. To generate 5′–end-labeled RNA, unlabeled RNA was synthesized without a cap and vaccinia virus capping enzyme was used to cap the 5′ end with [α–32P]GTP as described in Wang et al. (1999). 3′–end-labeled RNA was produced as described by England and Uhlenbeck (1978) with slight modification. Briefly, 20 pmol of capped unlabeled RNA was incubated with 60 U of T4 RNA ligase overnight at 4°C in T4 RNA ligase buffer with 10% dimethylsulfoxide (DMSO), 10% glycerol and 50 μCi of [5′–32P]pCp. The reactions were terminated with ULB (7 M urea, 2% SDS, 0.35 M NaCl, 10 mM EDTA and 10 mM Tris pH 7.5), and ethanol precipitated with 20 μg of glycogen carrier. All RNA probes used in the IVDAs were gel purified on an 8% denaturing polyacrylamide gel as described in Wang et al. (1999).
An alkaline RNA ladder was generated by incubating 104 c.p.m. of 5′–end-labeled probe in 50 mM NaOH for 20 s at 95°C. Reactions were stopped immediately with 3 M NaOAc pH 4.8, 7 M urea. To make the RNase T1 ladder, 104 c.p.m. of 5′–end-labeled probe and 0.5 μg of carrier tRNA were incubated with 10 U of RNase T1 at 37°C for 5 min in 10 mM Tris pH 7.5, 100 mM KCl, 10 mM MgCl2. The reactions were stopped with 9 M urea, 20 mM EDTA. The RNase U2 ladder was produced by incubating 1 U of RNase U2 with 104 c.p.m. of 5′–end-labeled probe at 55°C for 12 min in 16 mM NaOAc pH 3.5, 0.8 mM EDTA, 0.5 mg/ml carrier tRNA and 3.5 M urea. The RNA markers were used directly on the sequencing gels without further manipulation.
In vitro mRNA decay assays
IVDA reactions were carried out as described in Wang et al. (1999) using 0.1 pmol (103 c.p.m.) of 5′–capped RNA probe. Where indicated, 5 mM EDTA was included in the reactions. The competitor oligonucleotides used were thioated to minimize potential degradation. Oligo(dN) is a random 20mer, oligo(dC) is 16 cytosine residues and oligo(dCT) is CCTTCTTCCTTCCTCCTTCTTCCCTTCCTTTCCTTCCTTC as reported in Wang and Liebhaber (1996).
RNase protection assay
Total RNA was isolated from MEL–α2 cells either grown continuously at 37°C or shifted to 25°C 2 h prior to harvesting. RNA was isolated using Trizol reagent according to the manufacturer's conditions (Gibco-BRL). The RNA was subsequently treated with RNase-free DNase (1 U/μg RNA) for 1 h at 37°C and used in RNase protection assays (RPAs). The riboprobe used was antisense to the entire α–globin 3′–UTR and extended 60 nucleotides downstream of the poly(A) addition site. RPAs were carried out as described by Kiledjian and Kadesch (1990), resolved on an 8% polyacrylamide urea gel and visualized by autoradiography. The intermediate bands were identified and excised from the gel. The RNA was eluted from the gel slice with elution buffer as described in Wang et al. (1999). The Int–I fragment was reverse transcribed with a primer complementary to the mRNA at nucleotides 43–63 of the 3′–UTR (GGAGGGGAGGAGGGCCCGTTG) and PCR amplified with the 5′ primer (GCTGGAGCCTCGGTAGCCGT). The Int–II fragment was reverse transcribed with a primer complementary to the 3′ end of the 3′–UTR (GCCGCCCACTCAGACTTT) and PCR amplified with a 5′ primer corresponding to the endoribonuclease cleavage site (TTGCACCGGCCCTTCCTGGT). Both RT–PCR products were cloned into the PCR Trap vector (GenHunter) and confirmed by DNA sequencing.
Acknowledgments
Acknowledgements
We thank N.D.Rodgers for helpful discussions throughout the course of this work and A.R.Fuentes for generating the αmt-GA construct. We also thank S.Gunderson, N.D.Rodgers, P.Trifillis and C.Williams for critical reading of the manuscript. This work was supported by the National Institutes of Health grant DK51611 to M.K.
References
- Bandyopadhyay R., Coutts, M., Krowczynska, A. and Brawerman, G. (1990) Nuclease activity associated with mammalian mRNA in its native state: possible basis for selectivity in mRNA decay. Mol. Cell. Biol., 10, 2060–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelman C.A., Stevens, A., Caponigro, G., LaGrandeur, T.E., Hatfield, L., Fortner, D.M. and Parker, R. (1996) An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature, 382, 642–646. [DOI] [PubMed] [Google Scholar]
- Binder R., Horowitz, J.A., Basilion, J.P., Koeller, D.M., Klausner, R.D. and Harford, J.B. (1994) Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3′ UTR and does not involve poly(A) tail shortening. EMBO J., 13, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn L.B., Towner, J.S., Semler, B.L. and Ehrenfeld, E. (1997) Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol., 71, 6243–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. (1998) Characterization of c-myc 3′ to 5′ mRNA decay activities in an in vitro system. J. Biol. Chem., 273, 34770–34774. [DOI] [PubMed] [Google Scholar]
- Brewer G. and Ross, J. (1990) Messenger RNA turnover in cell-free extracts. Methods Enzymol., 181, 202–209. [DOI] [PubMed] [Google Scholar]
- Brown B.D., Zipkin, I.D. and Harland, R.M. (1993) Sequence-specific endonucleolytic cleavage and protection of mRNA in Xenopus and Drosophila. Genes Dev., 7, 1620–1631. [DOI] [PubMed] [Google Scholar]
- Chen C.Y. and Shyu, A.B. (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- Chernokalskaya E., Dubell, A.N., Cunningham, K.S., Hanson, M.N., Dompenciel, R.E. and Schoenberg, D.R. (1998) A polysomal ribonuclease involved in the destabilization of albumin mRNA is a novel member of the peroxidase gene family. RNA, 4, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chkheidze A.N., Lyakhov, D.L., Makeyev, A.V., Morales, J., Kong, J. and Liebhaber, S.A. (1999) Assembly of the α–globin mRNA stability complex reflects binary interaction between the pyrimidine-rich 3′ untranslated region determinant and poly(C) binding protein αCP. Mol. Cell. Biol., 19, 4572–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier B., Goobar-Larsson, L., Sokolowski, M. and Schwartz, S. (1998) Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogenous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J. Biol. Chem., 273, 22648–22656. [DOI] [PubMed] [Google Scholar]
- Couttet P., Fromont–Racine, M., Steel, D., Pictet, R. and Grange, T. (1997) Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc. Natl Acad. Sci. USA, 94, 5628–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker C.J. and Parker, R. (1994) Mechanisms of mRNA degradation in eukaryotes. Trends Biochem. Sci., 19, 336–340. [DOI] [PubMed] [Google Scholar]
- Decker C.J. and Parker, R. (1995) Diversity of cytoplasmic functions for the 3′ untranslated region of eukaryotic transcripts. Curr. Opin. Cell Biol., 7, 386–392. [DOI] [PubMed] [Google Scholar]
- England T.E. and Uhlenbeck, O.C. (1978) 3′–terminal labelling of RNA with T4 RNA ligase. Nature, 275, 560–561. [DOI] [PubMed] [Google Scholar]
- Ford L.P., Watson, J., Keene, J.D. and Wilusz, J. (1999) ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev., 13, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassetto S.S., Dias, R.D. and Sarkis, J.J. (1993) Characterization of an ATP diphosphohydrolase activity (apyrase, EC 3.6.1.5) in rat blood platelets. Mol. Cell. Biochem., 129, 47–55. [DOI] [PubMed] [Google Scholar]
- Gamarnik A.V. and Andino, R. (1997) Two functional complexes formed by KH domain containing proteins with the 5′ noncoding region of poliovirus RNA. RNA, 3, 882–892. [PMC free article] [PubMed] [Google Scholar]
- Harford J.B. (1993) Iron regulation of transferrin receptor mRNA stability. In Blasco,J. and Brawerman,G. (eds), Control of mRNA Stability. Academic Press, San Diego, CA, pp. 239–266. [Google Scholar]
- Holcik M. and Liebhaber, S.A. (1997) Four highly stable eukaryotic mRNAs assemble 3′ untranslated region RNA–protein complexes sharing cis and trans components. Proc. Natl Acad. Sci. USA, 94, 2410–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R.J. (1993) Cytoplasmic regulation of mRNA function: the importance of the 3′ untranslated region. Cell, 74, 9–14. [DOI] [PubMed] [Google Scholar]
- Jacobs J.S., Anderson, A.R. and Parker, R.P. (1998) The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J., 17, 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A. and Peltz, S.W. (1996) Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem., 65, 693–739. [DOI] [PubMed] [Google Scholar]
- Kiledjian M., DeMaria, C.T., Brewer, G. and Novick, K. (1997) Identification of AUF1 (heterogeneous nuclear ribonucleoprotein D) as a component of the α–globin mRNA stability complex [published erratum appears in Mol. Cell. Biol., 1997, 17, 6202]. Mol. Cell. Biol., 17, 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiledjian M., Day, N. and Trifillis, P. (1999) Purification and RNA binding properties of the polycytidylate-binding proteins αCP1 and αCP2. Methods, 17, 84–91. [DOI] [PubMed] [Google Scholar]
- Klausner R.D., Rouault, T.A. and Harford, J.B. (1993) Regulating the fate of mRNA: the control of cellular iron metabolism. Cell, 72, 19–28. [DOI] [PubMed] [Google Scholar]
- Koeller D.M., Horowitz, J.A., Casey, J.L., Klausner, R.D. and Harford, J.B. (1991) Translation and the stability of mRNAs encoding the transferrin receptor and c-fos. Proc. Natl Acad. Sci. USA, 88, 7778–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner C.G., Wormington, M., Muckenthaler, M., Schneider, S., Dehlin, E. and Wahle, E. (1998) The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J., 17, 5427–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn L.C. (1991) mRNA–protein interactions regulate critical pathways in cellular iron metabolism. Br. J. Haematol., 79, 1–5. [DOI] [PubMed] [Google Scholar]
- LaGrandeur T.E. and Parker, R. (1998) Isolation and characterization of Dcp1p, the yeast mRNA decapping enzyme. EMBO J., 17, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer F.W., Hsu, C.L., Maupin, M.K. and Stevens, A. (1992) Characterization of the XRN1 gene encoding a 5′→3′ exoribonuclease: sequence data and analysis of disparate protein and mRNA levels of gene-disrupted yeast cells. Gene, 120, 51–57. [DOI] [PubMed] [Google Scholar]
- Lodish H.F. and Small, B. (1976) Different lifetimes of reticulocyte messenger RNA. Cell, 7, 59–65. [DOI] [PubMed] [Google Scholar]
- Mitchell P., Petfalski, E., Shevchenko, A., Mann, M. and Tollervey, D. (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell, 91, 457–466. [DOI] [PubMed] [Google Scholar]
- Morales J., Russell, J.E. and Liebhaber, S.A. (1997) Destabilization of human α–globin mRNA by translation anti-termination is controlled during erythroid differentiation and is paralleled by phased shortening of the poly(A) tail. J. Biol. Chem., 272, 6607–6613. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Decker, C.J. and Parker, R. (1995) Turnover mechanisms of the stable yeast PGK1 mRNA. Mol. Cell. Biol., 15, 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.G. and Gunn, R.B. (1966) Thalassemia: the consequences of unbalanced hemoglobin synthesis. Am. J. Med., 41, 815–830. [DOI] [PubMed] [Google Scholar]
- Ostareck D.H., Ostareck–Lederer, A., Wilm, M., Thiele, B.J., Mann, M. and Hentze, M.W. (1997) mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell, 89, 597–606. [DOI] [PubMed] [Google Scholar]
- Paulding W.R. and Czyzyk–Krzeska, M.F. (1999) Regulation of tyrosine hydroxylase mRNA stability by protein-binding, pyrimidine-rich sequence in the 3′–untranslated region. J. Biol. Chem., 274, 2532–2538. [DOI] [PubMed] [Google Scholar]
- Ross J. (1993) mRNA decay in cell-free systems. In Blasco,J. and Brawerman,G. (eds), Control of mRNA Stability. Academic Press, San Diego, CA, pp. 417–448. [Google Scholar]
- Ross J. (1995) mRNA stability in mammalian cells. Microbiol. Rev., 59, 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. and Sullivan, T.D. (1985) Half-lives of β and γ globin messenger RNAs and of protein synthetic capacity in cultured human reticulocytes. Blood, 66, 1149–1154. [PubMed] [Google Scholar]
- Ross J., Kobs, G., Brewer, G. and Peltz, S.W. (1987) Properties of the exonuclease activity that degrades H4 histone mRNA. J. Biol. Chem., 262, 9374–9381. [PubMed] [Google Scholar]
- Russell J.E. and Liebhaber, S.A. (1996) The stability of human β–globin mRNA is dependent on structural determinants positioned within its 3′ untranslated region. Blood, 87, 5314–5323. [PubMed] [Google Scholar]
- Schoenberg D.R. and Chernokalskaya,E. (1997) Ribonucleases involved in eukaryotic mRNA turnover. In Harford,J.B. and Morris,D.R. (eds), mRNA Metabolism and Post-Transcriptional Gene Regulation. Wiley-Liss, New York, NY, pp. 217–240. [Google Scholar]
- Schwartz E. and Benz,E.J. (1990) The thalassemia syndromes. In Hoffman,R., Benz,E.J., Shatill,S., Furie,B. and Cohen,H.J. (eds), Hematology: Basic Principles and Practice. Churchill Livingstone, New York, NY, pp. 368–392. [Google Scholar]
- Shyu A.B., Belasco, J.G. and Greenberg, M.E. (1991) Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev., 5, 221–231. [DOI] [PubMed] [Google Scholar]
- Stefanovic B., Hellerbrand, C. and Brenner, D.A. (1995) Post-transcriptional regulation of collagen α 1 (I) mRNA in hepatic stellate cells. Nucleic Acids Res. Symp. Ser., 33, 212–214. [PubMed] [Google Scholar]
- Trifillis P., Day, N. and Kiledjian, M. (1999) Finding the right RNA: identification of cellular mRNA substrates for RNA-binding proteins. RNA, 5, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. and Liebhaber, S.A. (1996) Complementary change in cis determinants and trans factors in the evolution of an mRNP stability complex. EMBO J., 15, 5040–5051. [PMC free article] [PubMed] [Google Scholar]
- Wang X., Kiledjian, M., Weiss, I.M. and Liebhaber, S.A. (1995) Detection and characterization of a 3′ untranslated region ribonucleoprotein complex associated with human α–globin mRNA stability [published erratum appears in Mol. Cell. Biol., 1995, 15, 2331]. Mol. Cell. Biol., 15, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Day, D., Trifillis, P. and Kiledjian, M. (1999) An mRNA stability complex functions with the poly(A)-binding protein to stabilize mRNA in vitro. Mol. Cell. Biol., 19, 4552–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall D.J. and Clegg, J.B. (1972) The Thalassemia Syndromes. Blackwell Scientific Publications, Oxford, UK, pp. 75–144. [Google Scholar]
- Weiss I.M. and Liebhaber, S.A. (1995) Erythroid cell-specific mRNA stability elements in the α 2–globin 3′ nontranslated region. Mol. Cell. Biol., 15, 2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]