LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing (original) (raw)
Abstract
Little is known about the protein constituents of autophagosome membranes in mammalian cells. Here we demonstrate that the rat microtubule-associated protein 1 light chain 3 (LC3), a homologue of Apg8p essential for autophagy in yeast, is associated to the autophagosome membranes after processing. Two forms of LC3, called LC3-I and -II, were produced post-translationally in various cells. LC3-I is cytosolic, whereas LC3-II is membrane bound. The autophagic vacuole fraction prepared from starved rat liver was enriched with LC3-II. Immunoelectron microscopy on LC3 revealed specific labelling of autophagosome membranes in addition to the cytoplasmic labelling. LC3-II was present both inside and outside of autophagosomes. Mutational analyses suggest that LC3-I is formed by the removal of the C-terminal 22 amino acids from newly synthesized LC3, followed by the conversion of a fraction of LC3-I into LC3-II. The amount of LC3-II is correlated with the extent of autophagosome formation. LC3-II is the first mammalian protein identified that specifically associates with autophagosome membranes.
Keywords: APG/autophagosomes/autophagy/mammalian homologue/protein cleavage
Introduction
Protein degradation, as well as synthesis, is essential to normal activity of the cell. To degrade intracellular proteins, all eukaryotic cells have two major mechanisms, namely the ubiquitin–proteasome system and autophagy. Autophagy is a pivotal physiological process for survival during starvation, differentiation and normal growth control, and may play a number of roles in various other cellular functions, via the turnover of cellular macromolecules and organelles. It is defined as the process of sequestrating cytoplasmic proteins or even entire organelles into the lytic compartment, i.e. the lysosome/vacuole (for reviews see Seglen and Bohley, 1992; Dunn, 1994; Blommaart et al., 1997; Klionsky and Ohsumi, 1999). The initial step of autophagy is the surrounding of cytoplasmic and organelle portions of the cell by a single isolation membrane. The fusion of the edges of the membrane sac with each other forms a closed double-membrane structure, so-called autophagosome or immature autophagic vacuole (AVi). Finally, the autophagosome fuses with a lysosome to become the autolysosome or the degradative autophagic vacuole (AVd). Within the AVd compartment, the sequestered content is degraded by lysosomal hydrolases. A convergence between autophagic and endosomal transport has been observed in the early stage of both pathways (Tooze et al., 1990; Liou et al., 1997). Although autophagy is a constitutive cellular event, it is enhanced in certain situations such as nutrient or serum starvation, hormonal stimulation and drug treatments.
While the morphology and regulation of autophagy have been investigated extensively for animals, including human, the molecular machinery underlying its process is poorly understood. To identify the proteins involved in autophagy, we isolated yeast mutants defective in autophagy (Tsukada and Ohsumi, 1993). We identified 13 APG genes essential for autophagy and characterized their products (Klionsky and Ohsumi, 1999). We also found by searching the expressed sequence tag (EST) database that most Apg proteins have related proteins in mammals, implying that the molecular basis of autophagy may well be conserved between yeast and human. As a matter of fact, we found a unique covalent modification of the human homologues of Apg12p (hApg12) with Apg5p (hApg5) (Mizushima et al., 1998b), which is equivalent to the yeast Apg12p–Apg5p conjugation system essential for autophagy that we reported previously (Mizushima et al., 1998a). Moreover, Liang et al. (1999) showed recently that the human homologue of Apg6p, beclin 1, promoted autophagy in human breast carcinoma MCF7 cells. To elucidate the molecular mechanism of animal autophagy, we initiated the systematic characterization of the Apg homologues.
Here we analyse the intracellular localization and processing of the rat microtubule-associated protein 1 light chain 3 (LC3). LC3 was identified originally as a protein that co-purified with microtubule-associated protein 1A and 1B from rat brain (Mann and Hammarback, 1994). It shows 28% amino acid identity with Apg8/Aut7p, which is essential for yeast autophagy (Liang et al., 1999). Our recent investigation on Apg8/Aut7p in yeast suggested that this protein plays a critical role in the formation of autophagosomes (Kirisako et al., 1999). We found two forms of the LC3 molecules in cells. We suggest that one is in the cytoplasmic form and is processed into another form associated with the autophagosome membrane. To our knowledge, LC3 is the first mammalian protein localized in the autophagosome membrane to be identified.
Results
Presence of two forms of LC3 showing distinct subcellular distributions
In the immunoblot analysis using antibodies raised against a synthetic peptide corresponding to the N-terminal region of rat LC3, we detected LC3 as two bands at 18 and 16 kDa in the lysates of various cells, including rat brain (Figure 1A, left lane), PC12 cells (Figure 1A, right lane) and HeLa cells (Figure 1B). The amounts of the 16 kDa molecule varied from cell to cell and were at a low level in brain when compared with the abundant 18 kDa molecule (Figure 1A, left lane). This may account for the fact that the authors of the first report on LC3 described only the 18 kDa band in rat brain extract (Mann and Hammarback, 1994). Both the 18 and the 16 kDa band were not observed when the antigen peptide was added in excess together with the antibody. The same bands were also recognized by antibodies raised against the full-length recombinant LC3 molecule (data not shown). Therefore, we conclude that the 16 kDa band is an LC3-related protein to which the antibodies bind specifically. Furthermore, expression via tagged LC3 cDNA revealed two bands similar to the two endogenous LC3s (Figure 6, lane 3), implying that both the 18 and 16 kDa bands derived from the same mRNA. The 18 and 16 kDa forms of LC3 were called LC3-I and LC3-II, respectively.
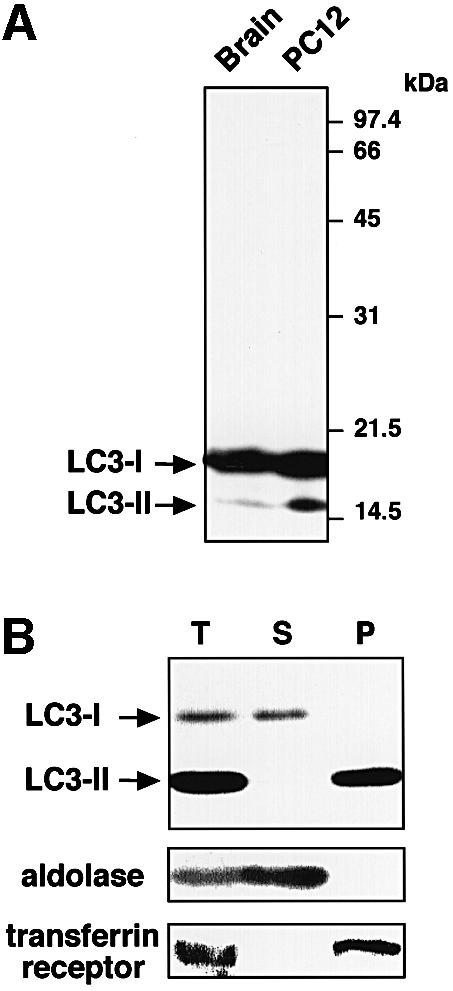
Fig. 1. Two forms of LC3 are produced and found in different locations inside the cells. (A) The lysates of rat brain (left lane) and PC12 cells (right lane) were subjected to immunoblot analysis with the anti-LC3 peptide antibody. (B) A cell homogenate prepared from starved HeLa cells (T) was fractionated into supernatant (S) and pellet (P) by centrifugation at 100 000 g. These fractions were analysed by immunoblot using antibodies against LC3, aldolase (a cytosolic marker) and transferrin receptor (a membrane protein marker). 15 and 12% SDS–polyacrylamide gels were used in (A) and (B), respectively.

Fig. 6. Post-translational processing of LC3. (A) Amino acid sequence alignment of the C-terminal segments of LC3 and its homologues. Residues identical to LC3 are shaded in black. Rn, Rattus norvegicus; Hs, Homo sapiens; Sc, Saccharomyces cerevisiae; At, Arabidopsis thaliana; Ce, Caenorhabditis elegans; Lb, Laccaria bicolor. The sequence of HsLC3 was deduced from an EST clone (au96a10.y1). (B) Constructions of proteins used in (C). Myc epitope tags at the N-terminus, HA epitope tags at the C-terminus and a hypothetical cleavage site Gly120 residue are indicated. To construct LC3G120A, a single point mutation was introduced into LC3, which leads to an amino acid substitution from glycine to alanine at position 120. To construct LC3ΔC22, the 22 C-terminal residues were deleted by PCR. LC3ΔC22,G120A was also produced by site-directed mutagenesis of LC3ΔC22. The ΔC22 mutants were tagged only with the Myc epitopes at the N-teminus. (C) HeLa cells were transiently transfected with Myc-LC3-HA (lanes 1–3 and 13–15), Myc-LC3G120A-HA (lanes 4–6 and 16–18), Myc-LC3ΔC22 (lanes 7–9) or Myc-LC3ΔC22, G120A (lanes 10–12). The cells were labelled with [35S]methionine/cysteine for 4 min and chased for 0, 6 and 90 min at 37°C. The cell lysates were immunoprecipitated with anti-Myc epitope antibody (lanes 1–12) or anti-HA epitope antibody (lanes 13–18) and the immunoprecipitates were analysed by SDS–PAGE and a bioimage analyser.
Next, we investigated the intracellular distribution of LC3-I and -II by subcellular fractionation. HeLa cell homogenate was ultracentrifuged and the resulting supernatant and pellet were examined by immunoblot analysis. Figure 1B shows a clear difference in distribution between LC3-I and -II. The former was recovered in the supernatant, while the latter was exclusively in the pellet fraction. LC3-II of the pellet could be solubilized with 1% Triton X-100 but not with 3 M urea, 2 M NaCl or 0.1 M carbonate pH 11.5 (data not shown), suggesting that it is tightly bound to the membranes. Myc-tagged LC3-I and -II expressed in cells were also cytosolic and pelletable, respectively (data not shown). These results indicate that LC3-II is associated with membrane compartments, whereas LC3-I is in the cytoplasm. For cells cultured for 1.5 h under a serum- and amino acid-depleted condition that induces autophagy, we found an apparent increase in the amount of LC3-II (Figure 1B).
LC3-II is localized in autophagosome membranes
To elucidate further the localization of LC3-II, we prepared various organelle fractions from liver homogenate of starved rat. Immunoblot analysis of the organelles for LC3 revealed that LC3-II was enriched specifically in the autophagic vacuole fraction (Figure 2, lane 4). The dense lysosomal fraction also contained LC3-II to some extent (Figure 2, lane 5). Although the quantity of LC3-I is too poor to be seen in Figure 2, it was detected exclusively in the cytosolic fraction by prolonged exposure (data not shown). These results suggest that LC3-II is localized in the membranes involved in the autophagic pathway.
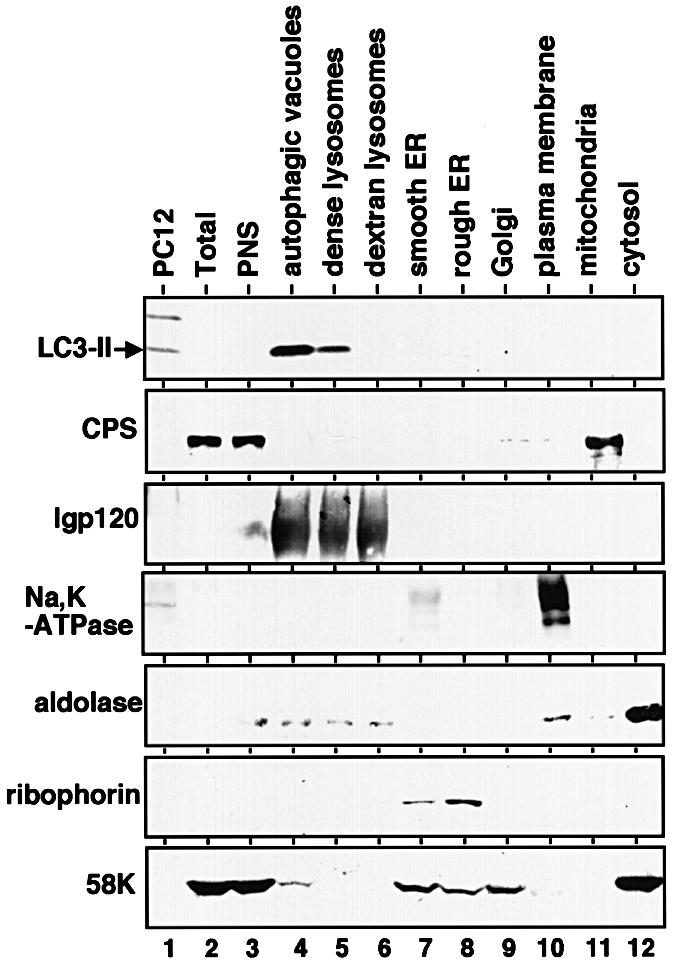
Fig. 2. LC3-II is enriched in autophagic vacuole fractions. Subcellular organelle fractions were obtained from starved rat liver as described in Materials and methods. Each fraction was subjected to immunoblot analysis using antibody against LC3 (top panel) and antibodies against each organelle marker (lower six panels). PC12, total lysate of PC12 cells as a control for the LC3-I and LC3-II positions; Total, total lysate of starved rat liver; PNS, its post-nuclear supernatant. The markers are CPS (carbamoyl phosphate synthetase 1) for the mitochondria; lgp 120 for the endosome/lysosome; Na/K-ATPase for the plasma membrane; aldolase for the cytosol; ribophorin for the ER; and 58 K protein for the Golgi complex.
We then undertook to establish the precise subcellular distribution of LC3 by immunogold labelling of cryosections. HeLa cells were transfected with LC3 fused to green fluorescent protein (GFP–LC3) for 18 h and cultured for 8 h in the presence of 10 mg/ml horseradish peroxidase (HRP) in order to fill the endocytic pathway with it. The procedure allows easy distinction between autophagosomes and autolysosomes, since the latter are accessible to endocytosed HRP while the former are not. The cells were then cultured for 1 h under the starvation conditions. We confirmed the generation of forms I and II of GFP–LC3 in the transfected cells by immunoblot analysis (data not shown). The cells were subjected to the immunogold and silver enhancement method using anti-GFP antibody. The silver-enhanced gold particles showing the presence of GFP–LC3 were associated specifically with autophagosome membranes (Figure 3A–E, arrowheads) in addition to the cytoplasmic distribution. Together with the cell fractionation data, these results lead us to believe that the molecules associated with the membranes are LC3-II, while those in the cytoplasm are LC3-I. Autolysosomes, which were discerned from autophagosomes by HRP staining (Figure 3, arrows), were also labelled; however, fewer gold particles were associated in comparison with autophagosomes. The densities of the gold particles were calculated to be 36.4 ± 20.5/µm of membranes for autophagosomes and 2.5 ± 2.0 for autolysosomes. Little binding of the particles was observed in the section of the untransfected cells (data not shown). We also observed localization of endogenous LC3 in autophagosomes as well as the cytoplasm by using anti-LC3 antibody in HeLa cells (data not shown) and in mouse embryonic stem (ES) cells (Figure 3F and G), which were cultured under the starvation conditions. It was clearly shown that the gold particles representing endogenous LC3 were associated specifically with membranes of typical autophagosomes in ES cells. Hence, it is most likely that LC3-II is localized mainly on the membranes of autophagosomes and some in autolysosomes, in contrast to the cytoplasmic distribution of LC3-I.

Fig. 3. LC3 is found associated with autophagosome membranes in addition to the cytoplasm. (A–E) HeLa cells transfected with GFP–LC3 were incubated at 37°C for 8 h in the presence of 10 mg/ml HRP and were cultured in Hanks’ solution at 37°C for 60 min. The cells were fixed and stained with DAB to detect the endocytosed HRP. Then, the localization of GFP–LC3 in the cells was examined by silver-enhanced immunogold electron microscopy using antibody against GFP. Arrowheads and arrows in (A–E), respectively, indicate autophagosomes and autolysosomes containing HRP. Bar, 1 µm. (F and G) ES cells were cultured in Hanks’ solution at 37°C for 1 h and fixed. The localization of endogenous LC3 was examined by silver-enhanced immunogold electron microscopy using antibody against LC3. The open and closed arrowheads indicate LC3 associated with inner and outer membranes of the autophagosome, respectively. Bar, 1 µm.
That the LC3-II target compartments are not lysosomes but autophagosomes is supported by comparing the distribution of LC3 with that of a lysosomal marker by fluorescence microscopy. HeLa cells transfected with GFP–LC3 were treated with bafilomycin A1 under the starvation conditions. The drug is an inhibitor of vacuolar-type proton ATPase, which was reported to accumulate autophagosomes under starvation conditions by inhibiting the fusion between autophagosomes and lysosomes (Yamamoto et al., 1998). Thus, treatment with bafilomycin A1 ensures the separation of autophagosomes and lysosomes. The cells were processed for immunofluorescence microscopy using antibody against lamp1, a lysosomal membrane protein. As expected, GFP–LC3 showed punctate staining (Figure 4A) in addition to the diffuse pattern (not clear in Figure 4, due to a high contrast photograph). The punctate staining may represent LC3-II and showed no co-localization with lamp1 (Figure 4B), excluding the possibility that LC3-II is a lysosomal protein and that its appearance in autophagic vacuoles is due to a fusion between autophagosomes and lysosomes.

Fig. 4. GFP–LC3 does not co-localize with a lysosomal protein lamp1 in bafilomycin A1-treated cells under starvation conditions. HeLa cells transiently transfected with GFP–LC3 were cultured at 37°C for 90 min in Hanks’ solution containing 0.1 µM bafilomycin A1 and analysed by immunofluorescence confocal microscopy using antibody against lamp1 and rhodamine-conjugated second antibody. (A) GFP–LC3 labelling, (B) lamp1 staining and (C) a merged image of the same field are shown. Bar, 10 µm.
To establish the topology of LC3-II localization, we performed a protease protection assay. Autophagic vacuole fractions prepared from starved rat liver were treated with pronase E to digest peripheral proteins and immunoblotted with antibody against LC3 or against betaine homocysteine methyltransferase (BHMT), a cytosolic enzyme known to be a cargo of autophagosomes (Ueno et al., 1999). All of a p44 subunit of BHMT was intact after the pronase treatment, whereas over half of LC3-II was degraded (Figure 5, lane 2). In the presence of Triton X-100, both proteins were almost completely degraded (Figure 5, lane 3). This underscores that at least part of LC3-II exists on the surface of autophagosomes; in other words, the protein is not a merely sequestered component of the compartments. Consistent with this, immunoelectron micrographs in Figure 3F and G revealed that LC3-II is attached to both the outer and inner membranes of autophagosomes (filled and open arrowheads, respectively).
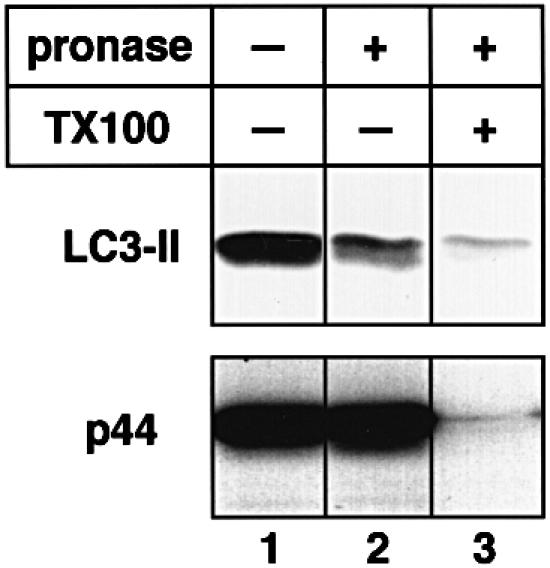
Fig. 5. LC3-II is distributed both outside and inside isolated autophagic vacuoles. Autophagic vacuole fractions were obtained from starved rat liver and were incubated at 0°C for 40 min in the absence (lane 1) or presence (lanes 2 and 3) of 0.8 mg/ml pronase E. In lane 3, 0.2% Triton X-100 was also added. The samples were subjected to immunoblotting with antibodies against LC3 or against BHMT to detect LC3-II and the p44 subunit of BHMT, an autophagic cargo marker.
LC3-II is formed through multistep processing including C-terminal cleavage
Having identified the two forms of LC3, we addressed the question of their formation. For this purpose, we constructed an LC3 tagged at both ends: with the Myc epitope at the N-terminus and with the haemagglutinin (HA) epitope at the C-terminus (Myc-LC3-HA; Figure 6B). After transfection with Myc-LC3-HA, HeLa cells were pulse-labelled with [35S]methionine/cysteine for 4 min and then chased for the times indicated. The cell lysates were immunoprecipitated with antibodies against the Myc epitope or the HA epitope, and analysed by SDS–PAGE (Figure 6C). Although two bands at 30 and 25 kDa were detected immediately after pulse labelling (Figure 6C, lane 1), the 30 kDa band disappeared during the short chase period of 6 min (lane 2) and the 25 kDa band remained. At 90 min chase, a new 23 kDa band emerged in addition to the 25 kDa band (lane 3). Since these 25 and 23 kDa bands were also observed in immunoblots (data not shown), they are thought to correspond to LC3-I and -II, respectively. Intriguingly, although the 30 kDa band was recognized by both antibodies against the Myc and the HA epitopes, the 25 and 23 kDa bands only reacted with antibody against the Myc epitope (lanes 1–3 and 13–15). Therefore, it is likely that the 30 kDa band represents the full-length molecule, while LC3-I and -II lack the C-terminal region.
The order of appearance of the bands hinted that LC3 is translated as a full-length precursor (designated proLC3: the 30 kDa band in this experiment) and processed sequentially into LC3-I and -II. This is consistent with the fact that LC3-I could be formed in vitro by mixing the HeLa cell lysate and recombinant LC3 synthesized in Escherichia coli, which was the same size as proLC3 (data not shown). To investigate this possibility further and characterize the processing of LC3, we constructed several mutant LC3s. When comparing the C-terminal sequence of LC3 with those of Apg8/Aut7p homologues found in human and other organisms, we noticed that there is a highly conserved region corresponding to Tyr113–Gly120 of LC3 (Figure 6A). In addition, the sequences downstream of the conserved glycine completely diverge from each other. Thus, we suspected that LC3-I and LC3-II were produced by the cleavage between Gly120 and Thr121 of proLC3. Based on this idea, we introduced a mutation causing the change of Gly120 to Ala (Myc-LC3G120A-HA) (Figure 6B). The major form of the mutant LC3 was proLC3 for each chase period (Figure 6C, lanes 4–6 and 16–18). The molecule was not very stable and decreased during the 90 min chase (lane 6). Although a small amount of LC3-I was detected, LC3-II was not formed at all. The result strongly suggests that the change of Gly120 to alanine affects the cleavage at the C-terminal region of proLC3, which is required for the formation of LC3-I and -II. Next, we examined Myc-LC3ΔC22, a deletion mutant lacking the sequence downstream of Gly120 (Figure 6C, lanes 7–9). No proLC3 was detected in the pulse–chase of the mutant. However, LC3-I and LC3-II formed under normal kinetic conditions, corroborating that the cleavage occurs between Gly120 and Thr121. The behaviour of LC3ΔC22 is also in agreement with the idea that LC3-II derives from LC3-I, rather than directly from proLC3. In addition, if Gly120 is changed to alanine in Myc-LC3ΔC22 (Myc-LC3ΔC22, G120A), LC3-I is formed but not LC3-II. Thus, we conclude that Gly120 is important for the formation of LC3-I from proLC3 and subsequent conversion of LC3-I into LC3-II.
The amount of LC3-II is correlated with the extent of autophagosome formation
As mentioned above, we discovered that the amount of LC3-II increased significantly in HeLa cells cultured for 90 min under the starvation conditions, which induce autophagy (Figure 7A, compare lane 2 with lane 1). The starvation-dependent increase in LC3-II was also observed in ES, HEK293 and Chinese hamster ovary cells (data not shown). This effect could be reversed by bringing the cells back into the complete medium (Figure 7A, lanes 8 and 9). To explore the relationship between this phenomenon and the autophagosome formation, we compared the rate of LC3-II increase with the rate of autophagosome formation induced by starvation. Both were comparable to each other (Figure 7B). The effect of starvation was also observed by indirect immunofluorescence microscopy for Myc-LC3 expressed in HeLa cells. Starvation for 90 min remarkably increased the dots of the punctate pattern, probably representing the LC3-II distribution, both in number and fluorescence intensity (Figure 7C).
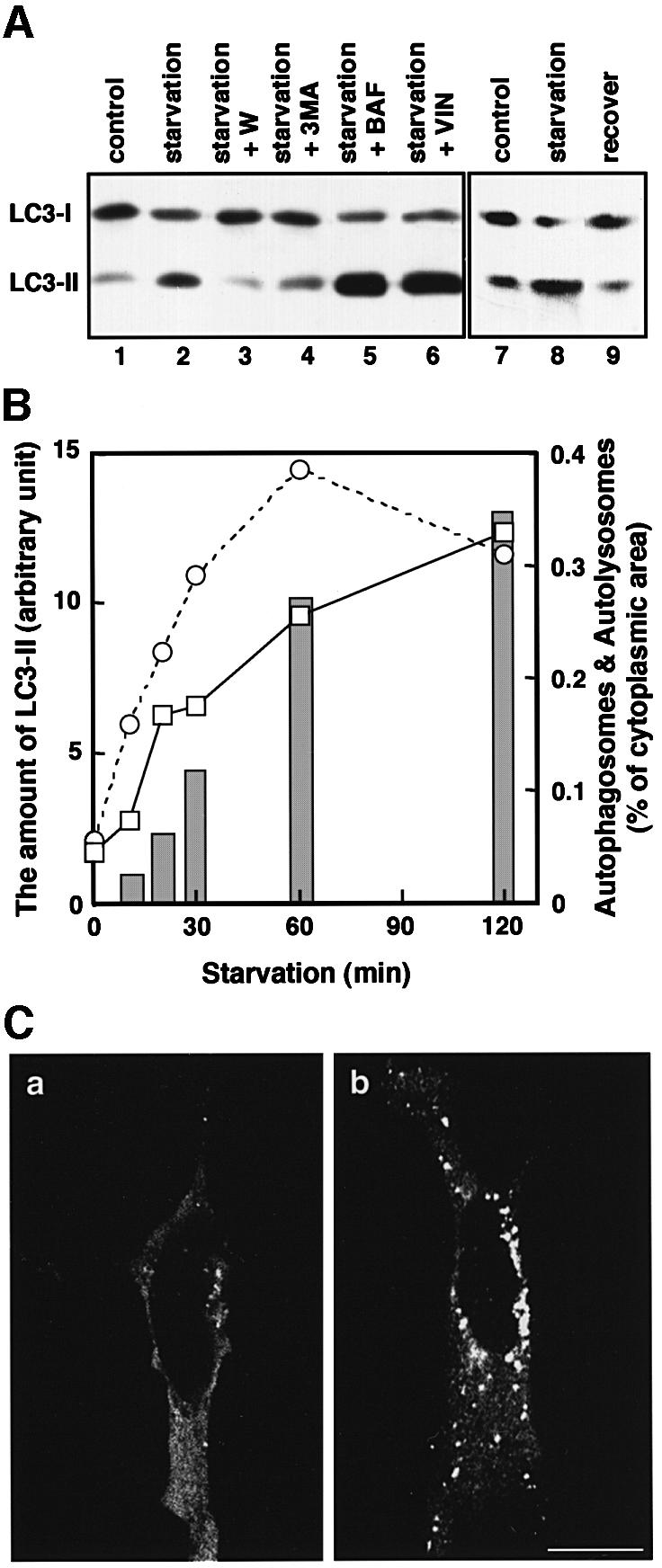
Fig. 7. The change in the amount of LC3-II corresponds to autophagosome formation. (A) HeLa cells were cultured at 37°C for 90 min in 10% FCS/DMEM (lane 1) or Hanks’ solution (lanes 2–6) containing the following reagents: 1% dimethylsulfoxide (control, lanes 1 and 2); 0.1 µM wortmannin (lane 3); 10 mM 3-methyladenine (lane 4); 0.1 µM bafilomycin A1 (lane 5); and 50 µM vinblastine (lane 6). In a separate experiment, HeLa cells were cultured at 37°C for 120 min in 10% FCS/DMEM (lane 7) or in Hanks’ solution (lane 8), and the latter was followed by re-incubation in 10% FCS/DMEM for 120 min (lane 9). After incubation, the cells were lysed and analysed by immunoblotting using antibody against LC3. A representative experiment repeated twice with duplicated dishes is shown. (B) HeLa cells (open squares with solid lines) and ES cells (open circles with dashed lines) were incubated in Hanks’ solution for the indicated time and a portion of the cells was subjected to immunoblotting using antibody against LC3. The amount of LC3-II was quantified by densitometry (left vertical axis). The remaining HeLa cells were fixed with 2.5% glutaraldehyde for conventional electron microscopy. The area of sections of autophagosomes/autolysosomes was measured on the electron micrographs and is indicated as bars (right vertical axis). (C) HeLa cells transiently transfected with Myc-LC3 were cultured in 10% FCS/DMEM (a) or in Hanks’ solution (b) at 37°C for 90 min. The cells were fixed and permeabilized for immunofluorescence confocal microscopy using anti-Myc epitope antibody and rhodamine-conjugated secondary antibody. Bar, 20 µm.
The results prompted us to test drug treatments known to affect autophagy. The starvation-dependent increment of LC3-II was blocked completely by the treatment with wortmannin or 3-methyladenine (Figure 7A, lanes 2, 3 and 4). It was shown that wortmannin prevents the formation of autophagosomes by inhibiting the activity of phosphatidylinositol 3-kinase (Blommaart et al., 1997). 3-Methyladenine is used commonly as a specific inhibitor of autophagosome formation (Seglen and Bohley, 1992) and also inhibits phosphatidylinositol 3-kinase (Blommaart et al., 1997). In contrast to these reagents, treatment with bafilomycin A1, which accumulates autophagosomes, under starvation conditions resulted in an additional large increase in LC3-II (lane 5). Vinblastine, which is a microtubule-depolymerizing reagent and inhibits fusion between autophagosomes and lysosomes, as does bafilomycin A1 (Hoyvik et al., 1986), showed a similar effect (lane 6). Taken together, it is conceivable that there is a close relationship between the amount of LC3-II and autophagosome formation.
Discussion
Autophagy is a dynamic process consisting of the formation and fusion of membrane compartments. Therefore, it is essential to identify protein components of autophagic membranes in order to unravel the mechanism of the phenomenon. We describe here that LC3-II, a processed form of LC3, is localized in autophagosomes and autolysosomes by subcellular fractionation and immunogold electron microscopy. This is in agreement with the distribution of Apg8/Aut7p, a yeast homologue of LC3. We located Apg8/Aut7p by immunogold electron microscopy in autophagosomes, autophagic bodies and intermediate structures during autophagosome formation (Kirisako et al., 1999).
Based on the results of this study, we propose that post-translational modifications generate LC3-I and LC3-II. The initial step must be a cleavage at the C-terminal region. Gly120 is important for the latter reaction. The amino acid segment Tyr121–Leu142, downstream of Gly120, should be cleaved off, although this has to be confirmed. Interestingly, it was reported previously that the genome of a cytopathogenic mutant of bovine viral diarrhoea virus contained an insertion sequence equivalent to LC3ΔC22, which lacks Tyr121–Leu142 (Meyers et al., 1998). The fact that a polyprotein translated from the virus was cleaved immediately after Gly120 of the inserted LC3ΔC22 by some cellular protease is very compatible with our hypothesis. Recently, we found that Apg8/Aut7p was also cleaved immediately after Gly116, which corresponds to Gly120 in LC3, and this proteolytic processing was required for autophagosome formation (Kirisako et al., 2000). In addition, we confirmed that a similar cleavage occurs in another Apg8/Aut7p homologue, Golgi-associated ATPase enhancer of 16 kDa (GATE16: DDBJ/EMBL/GenBank accession No. AF20262) (Sagiv et al., 2000)/ganglioside expression factor-2 (GEF2: DDBJ/EMBL/GenBank accession No. AB003515). Thus, the processing at the C-terminal region may be common in the Apg8/Aut7p family for their function. Furthermore, we could generate LC3-I in vitro from the recombinant LC3 in the presence of cell extracts (data not shown). The reaction was sensitive to _N_-ethylmaleimide, implying involvement of a cytosolic cysteine protease. We identified the yeast Apg4p as the cellular cysteine protease cleaving Apg8/Aut7p (Kirisako et al., 2000). We have also identified human homologues of Apg4p and are now in the process of their characterization. It is not clear at present that the cleavage is sufficient to form LC3-I. Although LC3-II might be produced directly from proLC3, we favour the model that LC3-I is converted to LC3-II, based on the behaviour of two mutants, LC3ΔC22 and LC3ΔC22, G120A. In either case, the nature of the modification involved in the formation of LC3-II remains cryptic, although Gly120 is evidently important. The modification should give LC3-II a membrane protein-like property. It is unclear at present whether LC3-II is formed prior to its binding to the membranes or after targeting of LC3-I to the membranes.
The cell culture in the serum- and amino acid-free medium caused the increase in the amount of LC3-II. Autophagy was induced under these conditions and we found a correlation between the rate of LC3-II increase and the rate of autophagosome formation. Wortmannin and 3-methyladenine, two inhibitors of autophagosome formation, suppressed the starvation-induced increase of LC3-II, whereas the drugs that accumulate autophagosomes, such as vinblastine and bafilomycin A1, had a strong LC3-II-increasing effect, which was synergistic with the starvation effect. Consequently, it is likely that the amount of LC3-II reflects the number of autophagosomes. If so, LC3-II may define the number of autophagosomes, i.e. it may regulate the formation of the compartments. In the later stage of autophagy, LC3-II may degrade or recycle back to cytosolic LC3-I, since autolysosomes were less labelled than autophagosomes, with gold particles representing LC3-II in immunoelectron microscopy (the labelling density was 2.5 ± 2.0/µm and 36.4 ± 20.5/µm of membranes, respectively).
When we initiated the analysis of LC3, only one mammalian protein homologous to the yeast Apg8/Aut7p was known. However, two other homologues, GATE16 and γ-aminobutyric acid A receptor-associated protein (GABARAP; Wang et al., 1999), were identified later in mammals. GATE16 was reported to be a modulator of intra-Golgi membrane transport (Sagiv et al., 2000) and GABARAP is known to bind to GABAA receptors (Wang et al., 1999). Therefore, LC3 seems to be the only one to play a role in autophagy. Since there is no homologue of Apg8/Aut7p in yeast, it may be an ancestor of an Apg8/Aut7p family including LC3, GABARAP and GATE16. The diversified members of the LC3 family in animals may have evolved to have a specialized function in different places inside the cell.
It was shown previously that purified recombinant LC3 binds to microtubules assembled from purified tubulin in vitro (Mann and Hammarback, 1994). This feature inspired us to consider a possible involvement of microtubules in the function of LC3 in autophagy. We also suspected that the increment of LC3-II induced by vinblastine is attributable to the breakdown of microtubules. However, other microtubule-depolymerizing reagents such as nocodazole and colchicine did not affect the amount of LC3-II (data not shown), ruling out this possibility. Lang et al. (1998) previously proposed that Apg8/Aut7p functions in the transport of autophagosomes by attaching to microtubules via another protein Apg4/Aut2p. Conversely, we provided evidence for a normal autophagy in yeast treated with nocodazole (Kirisako et al., 1999). Although microtubules are not required for the Apg8/Aut7p function or autophagy in yeast, the situation in mammalian cells may be different: microtubules may assist efficient transport of autophagosomes in mammalian cells, which are much larger than yeast cells. To substantiate the association of LC3-II with microtubules in vivo, we counterstained cells expressing GFP–LC3 with antibody against tubulin. Most of the dots stained did not co-localize with the microtubule meshes, although some seemed to have done so (our unpublished observation). The hypothesis that LC3-II functions by linking autophagosomes to microtubule is still a possibility to be examined.
LC3-II would be a good marker for autophagosomes, which so far have been defined by morphology but not by molecular composition. Detailed characterization of LC3 would provide us with clues to the many questions about the autophagy in mammals. We are now at a starting point for the molecular dissection of this mysterious organelle, the autophagosome.
Materials and methods
DNA constructions
cDNA encoding rat LC3 was obtained by RT–PCR from rat brain total cDNA with the LC3-s5 primer (5′-CCGGAATTCATGCCGTCCGAGAAGACCTT-3′) and LC3rc3 primer (5′-TTCGAATTCGCACCATAGTTATAAACCAG-3′). It was then subcloned into the _Eco_RI site of the eukaryotic expression vector pCI-neo (Promega, Madison, WI). The C-terminal deletion mutant of LC3 named LC3ΔC22, encoding amino acids 1–120, was generated by PCR. A point mutation for the glycine at position 120 to alanine of LC3 and LC3ΔC22 (LC3G120A and LC3ΔC22, G120A, respectively) was also created by PCR-based site-directed mutagenesis. The mutation was confirmed by DNA sequencing. The 3×Myc and 3×HA epitope-tagged constructs were obtained by insertion of the tags immediately after the first methionine codon and before the stop codon, respectively. To obtain pGFP-LC3, LC3 cDNA was inserted into the _Bgl_II and _Eco_RI sites of pEGFP-C1, a GFP fusion protein expression vector (Clontech Laboratories). For the expression of GST fusion protein, pGST-LC3 was obtained by subcloning LC3 cDNA into the _Bam_HI and _Eco_RI sites of pGEX-2T (Pharmacia).
Antibodies
Anti-LC3 antibody against a synthetic peptide corresponding to the N-terminal 14 amino acids of LC3 and an additional cysteine (PSDRPFKQRRSFADC) was prepared by immunization of rabbits according to the method previously described (Yoshimori et al., 1990) and affinity purified on an immobilized peptide–Sepharose column. Antibody against the recombinant LC3 was also produced in rabbits as previously described (Smith and Johnson, 1988) and purified on an immobilized GST–LC3–glutathione–Sepharose column. The following antibodies were also used: mouse monoclonal anti-human transferrin receptor antibody N-2 (Yoshimori et al., 1988), rabbit anti-rat aldolase serum (Kominami et al., 1983), rabbit anti-rat carbamoyl phosphate synthetase 1 serum (Lusty, 1978), rabbit anti-rat ribophorin 1 serum (Crimaudo et al., 1987), rabbit anti-rat lgp120 serum (donated by Dr H. Tsuji, Fukuyama University, Fukuyama, Japan), rabbit polyclonal anti-rat Na/K-ATPase antibody (donated by Dr K.Omori, Kansai Medical University, Osaka, Japan), mouse monoclonal anti-Golgi 58k antibody (Sigma), rabbit polyclonal anti-GFP antibody (Clontech Laboratories, Palo Alto, CA), mouse monoclonal anti-human lamp-1 antibody (donated by Dr T.August, Johns Hopkins University, Baltimore, MD), rabbit polyclonal anti-rat BHMT antibody (Ueno et al., 1999), mouse monoclonal anti-Myc epitope antibody 9E10 (BabCo), mouse monoclonal anti-HA epitope antibody 16B12 (BabCo), Fluorolink rhodamine-labelled goat anti-rabbit antibody (Amersham Pharmacia Biotech) and HRP-conjugated goat antibody against mouse and rabbit IgG (Jackson Immuno Research Laboratories, West Grove, PA).
Cell culture, transfection and immunoblotting
Media and reagents for cell culture were from Life Technologies (Grand Island, NY). HeLa cells and neuroendocrine PC12 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS), 5 U/ml penicillin and 50 µg/ml streptomycin. ES cells were grown in DMEM containing 20% FCS, 0.1 mM non-essential amino acids solution, 1 µM 2-mercaptoethanol, 2 mM glutamine, 1000 U/ml leukaemia inhibitory factor (LIF), 5 U/ml penicillin and 50 µg/ml streptomycin. HeLa cells at subconfluency were transfected with the indicated cDNAs using FuGENE 6 reagent (Roche Molecular Biochemicals). Cells were analysed at 18 h after transfection. Mock transfection was performed using the empty pCI-neo expression vector. For immunoblotting, proteins were separated by SDS–PAGE (12 or 15% gel) and transferred to a polyvinylidene difluoride membrane. The membrane was treated with antibodies according to a previously described method (Yoshimori et al., 2000).
Starvation and drug treatments of cells
To obtain starvation conditions, HeLa cells were washed three times with Hanks’ solution and incubated in the same solution for 1–1.5 h at 37°C. Drug treatment was achieved by incubation of the cells in Hanks’ solution containing 50 µM wortmannin (Sigma), 10 mM 3-methyladenine (Sigma), 100 nM bafilomycin A1 (Wako Pure Chemical Industries, Ltd, Osaka, Japan) or 50 µM vinblastine (Sigma) for 90 min at 37°C. After the treatment, the cells were lysed with 1% Triton X-100 in Tris-buffered saline for 20 min on ice and were subjected to immunoblot analysis using anti-LC3 antibody. To examine reversibility of the starvation effect, after starvation for 120 min, HeLa cells were cultured in 10% FCS/DMEM for 120 min at 37°C.
Subcellular fractionation
Subcellular fractionation was performed at high speed in order to obtain a cytosolic supernatant and a pellet containing total membrane as previously described (Yoshimori et al., 2000). For detailed analysis, various subcellular organelle fractions were prepared from starved rat liver. Smooth and rough endoplasmic reticulum membranes, plasma membranes and Golgi-enriched membranes were prepared according to the procedures of Hortsch and Meyer (1985), Hino et al. (1978) and Hubbard et al. (1983), respectively. Autophagic vacuolar membranes were prepared from leupeptin/E64c-administered rat liver as previously described (Ueno et al., 1991). As for lysosomes, two different preparations were used in this study. Lysosomes prepared from dextran-injected rat liver as described by Ueno et al. (1991) are designated as dextran-lysosomes in this study. The other preparation was obtained from normal rat liver: mitochondrial/lysosomal fraction (4 ml) isolated from starved rat liver (Ueno et al., 1991) was loaded onto 50 ml of 40% Percoll in 5 mM Tes–NaOH pH 7.5 and 0.3 M sucrose, and centrifuged at 55 000 g for 45 min. Lysosomes isolated from other membranes in the lower half of centrifugation tubes were pooled, mixed with an equal volume of 40% Percoll in 5 mM Tes–NaOH pH 7.5 and 0.3 M sucrose, and recentrifuged at 55 000 g for 45 min. The denser lysosomal layer was pooled, diluted with 4 vols of 5 mM Tes–NaOH pH 7.5 and 0.3 M sucrose, and centrifuged at 12 000 g for 20 min to remove the Percoll. This step was repeated two to three times. The lysosomal pellets were suspended in a minimal volume of 5 mM Tes–NaOH pH 7.5 and 0.3 M sucrose. These fractions are designated as dense lysosomes. Membranes of autophagic vacuoles, dextran-lysosomes and dense lysosomes were isolated according to a previous publication (Ueno et al., 1991), except that the final carbonate treatment of the membranes was omitted.
Immunoelectron and immunofluorescence microscopy
GFP–LC3-transfected HeLa cells were cultured in 10% FCS/DMEM containing 10 mg/ml HRP for 8 h prior to culture in Hanks’ solution for 1 h. The cells were fixed and stained with DAB to detect endocytosed HRP as previously described (Yoshimori et al., 2000). The cells were then subjected to the pre-embedding silver enhancement immunogold method for immunoelectron microscopy using antibody against GFP as previously described (Yoshimori et al., 2000). The number of gold particles and the length on membranes of autophagosomes and autolysosomes were measured on the electron micrographs by NIH image software. ES cells were also subjected to immunoelectron microscopy using antibodies against LC3 to detect endogenous LC3.
For immunofluorescence microscopy, HeLa cells grown on coverslips and transfected with Myc-tagged LC3 or GFP–LC3 were cultured under the conditions indicated, fixed and stained with antibody against Myc epitope or against lamp1 as previously described (Yoshimori et al., 2000).
Protease protection assay
Autophagic vacuole fractions, freshly prepared as described above, were incubated at 0°C for 40 min in a medium containing 5 mM Tes pH 7.5, 0.3 M sucrose and 0.8 mg/ml pronase E (Sigma) in the presence or absence of 0.2% Triton X-100 as previously described (Ueno et al., 1999). The samples were then precipitated with trichloroacetic acid and analysed by immunoblotting using antibody against LC3 or BHMT.
Pulse–chase experiment
The HeLa cells grown in 35 mm culture dishes were transfected with Myc-LC3-HA, Myc-LC3G120A-HA, Myc- LC3ΔC22 or Myc- LC3ΔC22, G120A. The cells were washed with methionine-free DMEM, pulse-labelled with 0.2 mCi of [35S]methionine/cysteine (NEN Life Science Products) for 4 min at 37°C and chased for the times indicated at 37°C. The cells were lysed with 1% Triton X-100 in phosphate-buffered saline on ice and immunoprecipitated with antibodies against the Myc epitope or HA epitope as previously described (Yoshimori et al., 2000). Immunoprecipitated materials were analysed on 12% SDS–polyacrylamide gels and visualized by a bioimage analyser BAS2000 (Fuji Film, Tokyo, Japan).
Measurement of the increased rate of LC3-II and autophagosome formation
HeLa cells and ES cells were incubated at 37°C in Hanks’ solution for the times indicated. To determine the amount of LC3-II, a portion of the cells was subjected to immunoblot analysis using antibody against LC3, and the density of the LC3-II bands was measured by the NIH image software. To measure autophagosome formation, the remaining HeLa cells were fixed with 2.5% glutaraldehyde and processed for conventional electron microscopy as previously described (Yoshimori et al., 2000). The area of sections of autophagosomes/autolysosomes was measured on the electron micrographs by the NIH image software.
Acknowledgments
Acknowledgements
We are grateful to Dr K.Ogura for providing the GEF2 cDNA, and Drs H.Tsuji, K.Omori and T.August for providing the antibodies. We thank Dr N.Ishihara in our laboratory and the NIBB Center for Analytical Instruments for the excellent technical assistance with subcellular fractionation and for synthesizing peptides, respectively. We also thank Drs I.Miwako and M.Ohashi for helpful discussions. This work was supported by a grant-in-aid for Scientific Research from the Ministry of Education, Science, Culture and Sports of Japan.
References
- Blommaart E.F., Luiken,J.J. and Meijer,A.J. (1997) Autophagic proteolysis: control and specificity. Histochem. J., 29, 365–385. [DOI] [PubMed] [Google Scholar]
- Crimaudo C., Hortsch,M., Gausepohl,H. and Meyer,D.I. (1987) Human ribophorins I and II: the primary structure and membrane topology of two highly conserved rough endoplasmic reticulum-specific glycoproteins. EMBO J., 6, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W.A. (1994) Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol., 4, 139–143. [DOI] [PubMed] [Google Scholar]
- Hino Y., Asano,A., Sato,R. and Shimizu,S. (1978) Biochemical studies of rat liver Golgi apparatus. I. Isolation and preliminary characterization. J. Biochem. (Tokyo), 83, 909–923. [DOI] [PubMed] [Google Scholar]
- Hortsch M. and Meyer,D.I. (1985) Immunochemical analysis of rough and smooth microsomes from rat liver. Segregation of docking protein in rough membranes. Eur. J. Biochem., 150, 559–564. [DOI] [PubMed] [Google Scholar]
- Hoyvik H., Gordon,P.B. and Seglen,P.O. (1986) Use of a hydrolysable probe, [14C]lactose, to distinguish between pre-lysosomal and lysosomal steps in the autophagic pathway. Exp. Cell Res., 166, 1–14. [DOI] [PubMed] [Google Scholar]
- Hubbard A.L., Wall,D.A. and Ma,A. (1983) Isolation of rat hepatocyte plasma membranes. I. Presence of the three major domains. J. Cell Biol., 96, 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T., Baba,M., Ishihara,N., Miyazawa,K., Ohsumi,M., Yoshimori,T., Noda,T. and Ohsumi,Y. (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol., 147, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T. et al. (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J. and Ohsumi,Y. (1999) Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol., 15, 1–32. [DOI] [PubMed] [Google Scholar]
- Kominami E., Hashida,S., Khairallah,E.A. and Katunuma,N. (1983) Sequestration of cytoplasmic enzymes in an autophagic vacuole–lysosomal system induced by injection of leupeptin. J. Biol. Chem., 258, 6093–6100. [PubMed] [Google Scholar]
- Lang T., Schaeffeler,E., Bernreuther,D., Bredschneider,M., Wolf,D.H. and Thumm,M. (1998) Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J., 17, 3597–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.H., Jackson,S., Seaman,M., Brown,K., Kempkes,B., Hibshoosh,H. and Levine,B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402, 672–676. [DOI] [PubMed] [Google Scholar]
- Liou W., Geuze,H.J., Geelen,M.J. and Slot,J.W. (1997) The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol., 136, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusty C.J. (1978) Carbamoylphosphate synthetase I of rat-liver mitochondria. Purification, properties and polypeptide molecular weight. Eur. J. Biochem., 85, 373–383. [DOI] [PubMed] [Google Scholar]
- Mann S.S. and Hammarback,J.A. (1994) Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem., 269, 11492–11497. [PubMed] [Google Scholar]
- Meyers G., Stoll,D. and Gunn,M. (1998) Insertion of a sequence encoding light chain 3 of microtubule-associated proteins 1A and 1B in a pestivirus genome: connection with virus cytopathogenicity and induction of lethal disease in cattle. J. Virol., 72, 4139–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Noda,T., Yoshimori,T., Tanaka,Y., Ishii,T., George,M.D., Klionsky,D.J., Ohsumi,M. and Ohsumi,Y. (1998a) A protein conjugation system essential for autophagy. Nature, 395, 395–398. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Sugita,H., Yoshimori,T. and Ohsumi,Y. (1998b) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem., 273, 33889–33892. [DOI] [PubMed] [Google Scholar]
- Sagiv Y., Legesse-Miller,A., Porat,A. and Elazar,Z. (2000) GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J., 19, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen P.O. and Bohley,P. (1992) Autophagy and other vacuolar protein degradation mechanisms. Experientia, 48, 158–172. [DOI] [PubMed] [Google Scholar]
- Smith D.B. and Johnson,K.S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione _S_-transferase. Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Tooze J., Hollinshead,M., Ludwig,T., Howell,K., Hoflack,B. and Kern,H. (1990) In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. J. Cell Biol., 111, 329–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M. and Ohsumi,Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett., 333, 169–174. [DOI] [PubMed] [Google Scholar]
- Ueno T., Muno,D. and Kominami,E. (1991) Membrane markers of endoplasmic reticulum preserved in autophagic vacuolar membranes isolated from leupeptin-administered rat liver. J. Biol. Chem., 266, 18995–18999. [PubMed] [Google Scholar]
- Ueno T., Ishidoh,K., Mineki,R., Tanida,I., Murayama,K., Kadowaki,M. and Kominami,E. (1999) Autolysosomal membrane-associated betaine homocysteine methyltransferase. Limited degradation fragment of a sequestered cytosolic enzyme monitoring autophagy. J. Biol. Chem., 274, 15222–15229. [DOI] [PubMed] [Google Scholar]
- Wang H., Bedford,F.K., Brandon,N.J., Moss,S.J. and Olsen,R.W. (1999) GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature, 397, 69–72. [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Tagawa,Y., Yoshimori,T., Moriyama,Y., Masaki,R. and Tashiro,Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4–II-E cells. Cell Struct. Funct., 23, 33–42. [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Shimonishi,Y. and Uchida,T. (1988) Binding properties of monoclonal antibody to the cytoplasmic domain of transferrin receptor. Cell Struct. Funct., 13, 311–324. [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Semba,T., Takemoto,H., Akagi,S., Yamamoto,A. and Tashiro,Y. (1990) Protein disulfide-isomerase in rat exocrine pancreatic cells is exported from the endoplasmic reticulum despite possessing the retention signal. J. Biol. Chem., 265, 15984–15990. [PubMed] [Google Scholar]
- Yoshimori T. et al. (2000) The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol. Biol. Cell, 11, 747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]