GreA and GreB proteins revive backtracked RNA polymerase in vivo by promoting transcript trimming (original) (raw)
Abstract
The GreA and GreB proteins of Escherichia coli show a multitude of effects on transcription elongation in vitro, yet their physiological functions are poorly understood. Here, we investigated whether and how these factors influence lateral oscillations of RNA polymerase (RNAP) in vivo, observed at a protein readblock. When RNAP is stalled within an (ATC/TAG)n sequence, it appears to oscillate between an upstream and a downstream position on the template, 3 bp apart, with concomitant trimming of the transcript 3′ terminus and its re-synthesis. Using a set of mutant E.coli strains, we show that the presence of GreA or GreB in the cell is essential to induce this trimming. We show further that in contrast to a ternary complex that is stabilized at the downstream position, the oscillating complex relies heavily on the GreA/GreB-induced ‘cleavage-and-restart’ process to become catalytically competent. Clearly, by promoting transcript shortening and re-alignment of the catalytic register, the Gre factors function in vivo to rescue RNAP from being arrested at template positions where the lateral stability of the ternary complex is impaired.
Keywords: elongation factors/GreA/GreB/RNA polymerase/transcription elongation complex
Introduction
RNA polymerases (RNAPs) carry out transcript elongation in a discontinuous manner. Frequently, DNA sequences through which the polymerase must pass to complete an RNA chain lead the enzyme to stop RNA synthesis for varying lengths of time before resuming elongation. This process of RNAP pausing is physiologically significant for both positive and negative control of transcription elongation (Das, 1993; Henkin, 1996; Landick et al., 1996; Uptain et al., 1997; Artsimovitch and Landick, 2000). In some instances in vitro, a fraction of the paused polymerases eventually transforms into an arrested state in which the ternary complex neither elongates nor dissociates (Davenport et al., 2000 and references therein). According to current models, this loss of catalytic activity results from a backward translocation of RNAP along the DNA and RNA chains (Landick, 1997; von Hippel, 1998; Nudler, 1999). The upstream movement shifts the transcription bubble, the RNA–DNA hybrid and the catalytic center away from the 3′ end of the transcript, which becomes extruded out of the polymerase (Reeder and Hawley, 1996; Komissarova and Kashlev, 1997a,b; Nudler et al., 1997; Samkurashvili and Luse, 1998; Sidorenkov et al., 1998). Thus, unless the sliding process is reversed, resumption of RNA synthesis requires trimming of the transcript to generate a new 3′ hydroxyl group in register with the catalytic center of the enzyme.
A special class of elongation factors, which includes GreA and GreB for the bacterial RNAP and SII for the eukaryotic RNAP II, stimulates transcript cleavage within ternary complexes that are artificially halted by NTP starvation (Borukhov et al., 1992, 1993; Reines, 1992, 1994; Reines et al., 1992; Kassavetis and Geiduschek, 1993). These in vitro observations raised the attractive possibility that the Gre/SII family of elongation factors may play a key role in vivo (i) to mitigate pausing and suppress elongation arrest and (ii) to enhance transcription fidelity (Erie et al., 1993; Jeon and Agarwal, 1996; Thomas et al., 1998). To date, however, evidence that GreA and GreB as well as SII act as cleavage factors inside the cells is still lacking. Ternary transcription complexes made with a variety of purified RNAPs have been shown to possess substantial endonucleolytic cleavage activity in the absence of the known stimulatory factors (Surratt et al., 1991; Hagler and Schuman, 1992; Wang and Hawley, 1993; Rudd et al., 1994; Tschochner, 1996; Sastry and Ross, 1997; Chedin et al., 1998). In particular, experiments with Escherichia coli RNAP purified from a _greA_– _greB_– double mutant indicated that the cleavage reaction is inherent to the polymerase and that Gre factors simply enhance this intrinsic property (Orlova et al., 1995). Nevertheless, the biological function of the transcript trimming activity of any RNAP has yet to be revealed.
We have recently shown that a transcription elongation complex, halted within an (ATC/TAG)n sequence due to a readblock imposed by the lac repressor in E.coli, oscillates between an upstream and a downstream position on the template, with accompanying transcript cleavage and re-synthesis (Toulmé et al., 1999). We have now investigated the influence of GreA and GreB on RNAP oscillations in this system. Results reported below provide direct evidence for the transcript cleavage reaction mediated by GreA and GreB inside the cell. They also show that the Gre factor-induced transcript shortening is essential for RNAP to read through template sequences that impair the lateral stability of the ternary complex.
Results
GreA and GreB induce transcript cleavage within an oscillating ternary complex in vivo
To investigate the cellular function of the Gre factors, we analyzed in vivo the structural behavior of an RNAP elongation complex readblocked on a reporter plasmid (Figure 1). On this template, transcription is initiated from a constitutive promoter and the elongation complex is stalled by a physical readblock, imposed by the lac repressor bound to its operator. By a combination of in situ DNA footprinting and RNA 3′ end mapping experiments, we have shown previously (Toulmé et al., 1999) that the ternary complex halted within an (ATC/TAG)n sequence, which potentially generates an unstable RNA–DNA hybrid, is distributed between a downstream (–6) and an upstream (–9) translocated position (Figure 1, plasmid pATC6a). In contrast, a template sequence that should yield a stable RNA–DNA hybrid holds the ternary complex fixed in the downstream location (plasmid pATC6b).

Fig. 1. Schematic models of a stable and an oscillating elongation complex halted in vivo by the lac repressor bound to its operator. The middle section of the figure shows the relevant part of plasmids pATC6a and pATC6b with the non-template strand sequence of the repeats. These plasmids, which have been described previously (Toulmé et al., 1999), are pKK232-8 derivatives that carry the β-lactamase (Amp) and chloramphenicol acetyltransferase (CAT) genes. The transcription of the cat gene initiated at the constitutive hisR promoter goes through the repeats and the operator sequences that are inserted at position +40 relative to the transcription start site. Positions –6 and –9 relative to the upstream edge of the operator motif are also indicated, as the mRNA start site (right-angled arrow). See text for details.
We envisaged that the multiple forms of the ternary complex observed within the (ATC/TAG)n stretch are in a dynamic equilibrium, where the readblocked polymerase oscillates between two positions on the template with accompanying transcript cleavage and re-synthesis. According to this scenario, the polymerase first elongates the transcript to the point where the operator-bound lac repressor becomes a physical barrier. Within such a readblocked complex, the catalytic center of the enzyme is in register with position –6 (the location of the 3′ terminal RNA nucleotide) with respect to the upstream edge of the operator motif. When the RNA–DNA hybrid within the complex is relatively strong (5 rC–dG bp in the putative 8 bp heteroduplex), the polymerase remains fixed at this single site on the template. However, when the complex contains a weak hybrid (3 rC–dG bp in the heteroduplex), the polymerase becomes prone to slide backward. The backsliding of RNAP relocates the catalytic center in register with position –9, where transcript cleavage occurs. This generates the upstream translocated complex. Re-elongation of the transcript from the new 3′ end brings RNAP back to the downstream, readblocked location.
Conceivably, transcript cleavage within the backslided ternary complex is an intrinsic activity of polymerase, or the RNA trimming is induced by Gre factors. To obtain evidence to support these possibilities, we extended the footprinting analyses with E.coli cells that lack functional GreA, GreB or both. The pATC6a plasmid that harbors the oscillation-inducing template sequence was moved into a set of four isogenic strains: wild type, greA::KanR, Δ_greB::CamR_ and the _greA_– _greB_– double mutant. We first mapped the 3′ ends of the truncated transcripts generated by the readblocked ternary complex.
Cellular RNAs were extracted from the transformed cells and the RNA 3′ ends were determined by S1 nuclease protection experiments (see Materials and methods). In agreement with previous results (Toulmé et al., 1999), the RNAs extracted from wild-type cells grown in the absence of the inducer isopropyl-β-d-thiogalactopyranoside (IPTG) contained short transcripts with 3′ ends distributed primarily between positions –6 and –9 with respect to the lac operator (Figure 2A). This signature of the apparent ‘cleavage-and-restart’ process within the oscillating complex was also observed for the RNAs extracted from either the _greA_– or the _greB_– mutants. In striking contrast, a major band corresponding to the 3′ end at position –6 was detected for the RNAs produced in the double-mutant strain. The intensity of this band is approximately equal to the sum of the two signals (positions –6 and –9) observed for either the wild type or single mutants (Figure 2B). These results are consistent with the hypothesis that the RNAP readblocked within the (ATC/TAG)n tract backslides, and that it resumes elongation upon transcript shortening promoted by GreA and GreB.

Fig. 2. Nuclease S1 mapping of the 3′ ends of RNAs produced from pATC6a in wild-type (Wt), _greA_–, _greB_– or _greA_– _greB_– (Dble) mutant strains. (A) Autoradiogram showing the distribution of the RNA 3′ ends. In this and subsequent figures, the + or – signs at the top of the lanes denote the addition or omission, respectively, of the inducer (IPTG). The arrowheads indicate the –6 and –9 positions of the 3′ ends of the transcript relative to the upstream edge of the operator motif. (B) Densitometric scans of the S1-protected bands obtained in the absence of IPTG, quantified and processed on a PhosphorImager (Molecular Dynamics) with Imagequant software Version 3.3 for data processing. The data reported in the graph point to variations in the intensity of the –6 and –9 signals in the _greA_–, _greB_– and _greA_– _greB_– mutant strains compared with the wild-type strain.
The apparent transcript cleavage activity in the readblocked ternary complex is more efficient with GreA than with GreB. A quantitative analysis of the S1 nuclease protection patterns obtained with the single mutants revealed a reproducible, albeit modest, difference between the distributions of the RNA 3′ ends (see densitometric scans in Figure 2B). Whereas the 3′ ends in the _greB_– mutant are divided in nearly equal amounts between positions –6 and –9, as was observed with the wild-type strain, the RNAs produced in the _greA_– mutant have more 3′ ends at position –6.
Another diagnostic for the cleavage and re-synthesis process within the oscillating complex is obtained by analyzing the methylation status of the G residue at position –6 on the template strand. The in situ dimethylsulfate (DMS) probing data, shown in Figure 3, reveal the readblock-dependent hypermethylation of the G-6 residue in wild-type cells and single mutants, but clearly not in the double mutant. The extent of methylation of the G-6 residue in the double mutant is only slightly greater than that of the surrounding G residues. These results strongly support our previous suggestion that the hyper-reactivity of the G-6 residue with DMS is associated with the dynamic equilibrium, the template base residue being repeatedly blocked in a highly exposed configuration during the re-incorporation of the rC residue into the transcript (Toulmé et al., 1999). Thus, the absence of both GreA and GreB renders the stalled elongation complex transcriptionally quiescent by impairing the dynamic process of cleavage and re-synthesis between the –6 and –9 positions.

Fig. 3. In situ DMS modification patterns of the template strand of pATC6a in wild-type (Wt), _greA_–, _greB_– and _greA_– _greB_– (Dble) mutant strains. The position of the G-6 residue is indicated by an arrow.
Interestingly, these in situ DMS probing experiments (Figure 3) also show a slightly lower hypermethylation of the G-6 residue in the _greA_– cells compared with the _greB_– or wild-type strains (densitometric scans not shown). In agreement with the S1 mapping data described above, these results emphasize a lower frequency of rC-6 re-incorporation in the _greA_– strain due to the reduced efficiency of transcript cleavage.
The catalytic competence of the backslided RNA polymerase requires functional GreA or GreB
The molecular properties of the ternary complex generated in the _greA_– _greB_– double mutant with plasmid pATC6a could be readily explained if the readblocked RNAP is no longer sliding back and forth within the (ATC/TAG)n tract, but instead is trapped in the downstream position. Indeed, similar characteristics were found previously in wild-type cells for a ternary complex that is stably positioned at the downstream location (as in the case of the pATC6b variant). Alternatively, we imagine that in the absence of GreA and GreB, the ternary complex in pATC6a is still repeatedly switching between the upstream and downstream positions without breaking the 3′ tip of the transcript. A distinguishing feature between the oscillating and non-oscillating elongation complexes is the apparent size of the transcription bubble. Whereas in the laterally stabilized complex the DNA bubble is limited to 8 bp, the oscillating complex is characterized by an appropriately larger DNA bubble (Toulmé et al., 1999). Therefore, we extended our comparative studies by analyzing the accessibility of the non-template strand to the single-strand-specific chemical probe chloroacetaldehyde (CAA).
Escherichia coli cells bearing pATC6a were treated with CAA, and following plasmid DNA extraction the modified bases on the non-template strand were revealed by primer extension. As shown in Figure 4, the CAA reactivity patterns of pATC6a modified in the wild-type and double-mutant strains are virtually identical, although the modifications at the upstream and downstream margins of the footprint differ slightly between the two strains. The CAA reactivities are spread over a region of ∼14 bases located upstream from position –6. This apparent acces-sibility to the probe of a large region of the non-template strand must result from the superposition of the different forms of the complex that are in equilibrium. As shown above (Figure 2), the ternary complexes of pATC6a in the _greA_– _greB_– double mutant have a single 3′-ended transcript (position –6). Thus, these CAA probing results indicate that in the absence of functional Gre factors, the ternary complexes readblocked within the (ATC/TAG)n stretch do slide back and forth between the upstream and downstream locations; however, they do not shorten the 3′ terminal part of the transcript. Notably, the slight shift of the CAA footprint towards the upstream positions observed in the double mutant might reflect the fact that the oscillating ternary complexes spend more time at the backward location when GreA and GreB are absent (Figure 4). In the view where the backward sliding of the polymerase extrudes the RNA 3′ tip from the active center, the upstream form of the complex should be catalytically inactive. Evidence supporting this notion is presented below.

Fig. 4. Primer extension analysis of CAA modification on the non-template strand of pATC6a in wild-type (Wt) or _greA_– _greB_– (Dble) mutant strains. The two arrows at positions –6 and –18 delimit the apparent footprint of the complex, which is in equilibrium between the upstream and downstream conformations.
The operator-bound lac repressor is expected to cause only a transient block to the emergent elongation complex. Once the two complexes are in close apposition on the DNA, the duration of the block is a function of the dwelling time of the lac repressor on its binding site. However, the efficiency with which the polymerase will read through the operator motif, following repressor dissociation, depends upon whether the ternary complex is able to escape rapidly from this position before the lac repressor re-binds DNA. Thus, the level of transcription readthrough under repressing conditions should be related to the catalytic competence of the halted ternary complex.
Therefore, we measured the amounts of downstream cat mRNA produced in the different strains harboring pATC6a. A 32P end-labeled oligonucleotide complementary to the early part of the cat message was used to quantitate mRNA by primer extension with reverse transcriptase. A second oligonucleotide was also included to detect the plasmid-encoded β-lactamase transcript (bla) for normalization. Under repressing conditions, a fraction of RNAPs did transcribe beyond the operator motif, and this level of readthrough was clearly affected by the absence of both GreA and GreB (Figure 5A). Curiously, the gre mutations showed a defect in cat gene expression that is independent from the lac repressor impediment, since a reduction in cat mRNA was also observed under inducing conditions. We have not addressed the molecular basis of this defect. We note, however, that this could be due to a reduced frequency of RNA chain initiation in the gre mutants (Hsu et al., 1995; A.Das et al., manuscript in preparation). Alternatively, in the absence of GreA and GreB, RNA chain elongation could have been delayed at some natural pause sites within the early part of the cat gene. To evaluate specifically the readblock-dependent effects of the Gre factors, we normalized, for each strain, the level of cat mRNA produced under repressing conditions to that obtained under inducing conditions. This analysis revealed a clear defect of RNAP in overcoming the readblock in the absence of GreA and GreB (Figure 5B). Whereas the efficiency of readthrough was almost unaffected in the _greB_– mutant compared with the wild-type cells, the _greA_– strain displayed a 30% reduction of cat mRNA. By comparison, the readthrough was reduced by 60% in the double mutant.
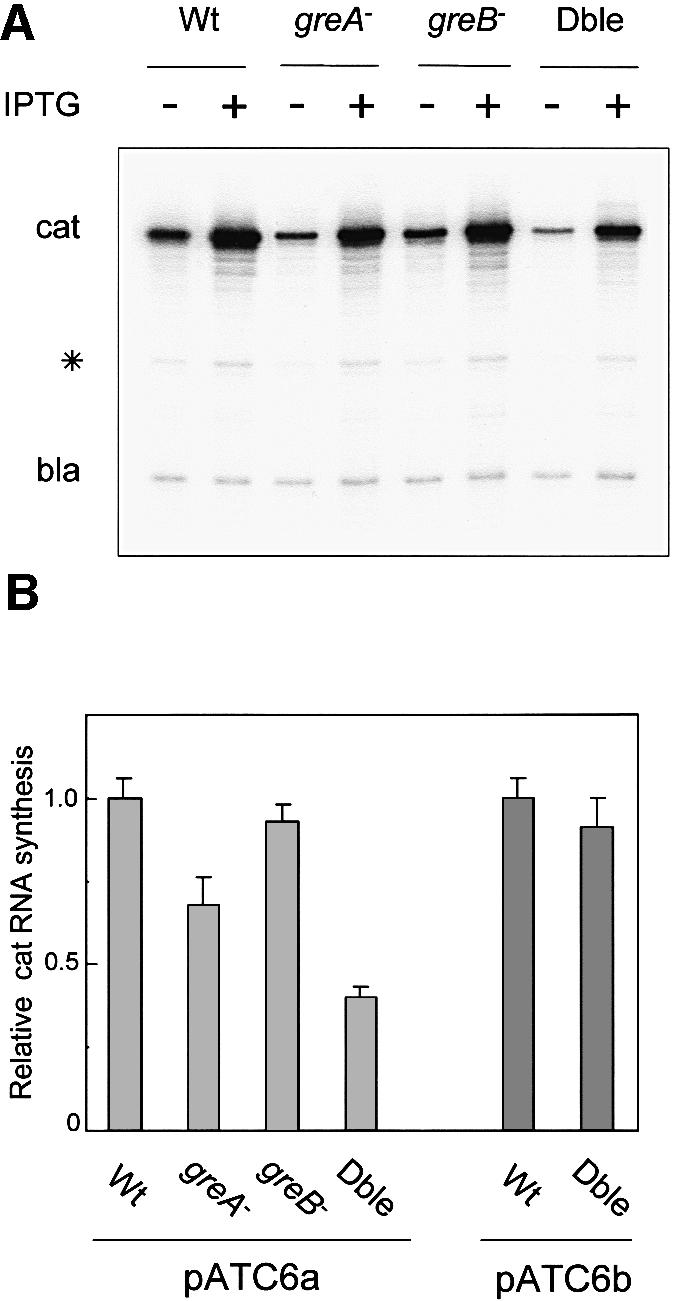
Fig. 5. cat mRNA quantitation in wild-type (Wt), _greA_–, _greB_– and _greA_– _greB_– (Dble) mutant cells transformed with either pATC6a or pATC6b. (A) Autoradiogram of the extension products obtained after in vitro reverse transcription of the RNAs with 32P primers hybridizing to either the cat or the bla transcripts. The asterisk indicates a non-specific site of arrest during reverse transcription of the cat mRNA. (B) Quantification of the extension products shown in (A) was performed and processed based on densitometric scans as described in Figure 2. For each lane, the value plotted indicates the relative level of cat mRNA synthesis, calculated as explained in the text. The error bars reflect the standard deviation from a mean of three independent experiments. The 1.0 value for the wild type is an arbitrary unit. On the right-hand side of the histogram, the data obtained with the pATC6b plasmid introduced in the wild-type and the double-mutant strains is also reported to underline the Gre factor-independent readthrough when the ternary complex is stabilized in the downstream position (experimental data not shown).
We infer that the ternary complex in pATC6a slides back and forth between a catalytically dormant (upstream) and an active form (downstream). Upon repressor dissociation, only the active form escapes before the lac repressor re-binds. By promoting transcript trimming and realignment of the catalytic register, the Gre factors reactivate the backtracked ternary complex more efficiently than do the oscillations without cleavage. Our conclusion is clearly supported by the results with the pATC6b variant (Figure 5B). In this case, where the ternary complex is fixed in the downstream location (active form), the efficiency of transcription readthrough is virtually unaffected by the absence of the two Gre factors.
Discussion
Transcription processivity, essential for gene expression and control, relies on the perplexing grips of RNAP that hold the template DNA and the nascent transcript in a stable but flexible manner. The stability of these grips prevents the random dissociation of the ternary complex, while their flexibility enables the catalytic center to rapidly reposition itself (15–50 ms) at every cycle of nucleotide addition. During typical elongation, the polymerase has little or no reason to march backward. Yet, RNAP does slide backward at certain template sites, when it is deprived of nucleotides, or when RNAP encounters some natural pause sites or a physical barrier imposed by a protein bound on the DNA. The backsliding event renders RNAP catalytically incompetent owing to the out-of-register catalytic center. As a result, transcription stalls for varying periods of time until RNAP regains catalytic activity. The experiments presented in this paper show that elongation factors GreA and GreB play a significant role in the cell to revive RNAP from its backslided, catalytically incompetent state. They do so by promoting transcript cleavage, which generates a new 3′ terminus of the transcript in register with the catalytic center (Figure 1).
We have shown that the backslided RNAP fails to trim the RNA 3′ terminus in vivo when both GreA and GreB are absent. This contrasts with previous in vitro results demonstrating that both E.coli RNAP and eukaryotic RNAP II possess intrinsic transcript cleavage activity (Rudd et al., 1994; Orlova et al., 1995). Notably, in these in vitro experiments, transcript hydrolysis at internal positions was induced by specific conditions, such as NTP starvation and high pH or the presence of pyrophosphate activity. These studies suggested that the endonucleolytic cleavage reaction is probably catalyzed by the same active center that performs RNA synthesis. Our findings do not exclude this possibility, but they clearly indicate that within the cellular environment of E.coli, the interaction of GreA or GreB with the ternary complex is essential for the cleavage reaction to occur. We suspect that this rule is likely to hold true for eukaryotes as well.
GreA and GreB are closely related proteins that share a substantial amino acid sequence homology, have a similar structural organization and are conserved in the bacterial kingdom (Koulich et al., 1997). However, the two proteins appear to act differently within ternary complexes in vitro. Whereas GreA-induced transcript cleavage produces short RNAs, two or three nucleotides long, GreB (like SII) produces 2–18 nucleotide RNA fragments (Borukhov et al., 1992, 1993; Reines, 1992; Reines et al., 1992). Indeed, our in vivo results presented here reveal a differential sensitivity of RNAP for the Gre factors, and they provide evidence that this differential specificity is biologically significant. While the two factors clearly substitute for each other in reviving backtracked RNAP in our system, GreA is more efficient than GreB. Apparently, GreA is more specialized for modulating RNAP where backtracking is limited over a short distance. The molecular mechanism underlying this specialization is still unclear. Recent experiments suggest that the functional property of the two Gre proteins is determined in part by the size of the respective basic patch residing on the N-terminal coiled-coil domain (Kulish et al., 2000).
The ‘Zipping’ model of transcript elongation (Reeder and Hawley, 1996; Komissarova and Kashlev, 1997a,b; Nudler et al., 1997; Toulmé et al., 1999) holds that the lateral stability of the ternary complex at each template position is governed by the direct competition between the DNA–DNA and RNA–DNA base pairs that can form at the upstream and downstream branching points of the transcription bubble. Accordingly, we expect that the back-and-forth oscillations of the ternary complex over short distances, such as that operational in our experimental system, are relatively common under the dynamic conditions of transcription in vivo. RNAP oscillations should be induced at template sequences where the competition between the base pairs at the two branching points of the bubble is unfavorable to elongation. Furthermore, misincorporation, which hinders further catalysis, should favor a backward slide of RNAP. Our results show clearly that the oscillating complex relies heavily on the ‘cleavage-and-restart’ mechanism mediated by Gre factors to escape from the protein readblock. In the absence of GreA and GreB, the ternary complex spends time switching between active and inactive isoforms. Thus, the Gre proteins are processivity factors that preclude RNAP from being retarded at positions where the lateral stability of the ternary complex is impaired. They probably function to prevent RNAP from extensive backtracking that would lead to an irreversible arrest. Finally, the Gre factors must also play a significant role in editing the misincorporated RNAs in the cell. It would be surprising if bacteria do not utilize GreA and GreB to control specific genes by suppression of transcription arrest and RNA editing.
The disruption of the two gre genes in E.coli is not lethal under normal growth conditions, but renders the cells thermosensitive (Orlova et al., 1995). It is tempting to speculate that the lateral oscillations of the ternary complex are aggravated by high temperature, which in turn makes the cell more dependent on the Gre factors for proper gene expression and control (A.Das et al., manuscript in preparation). We imagine that the high temperature defect might be amplified by some genetic alterations in RNAP that favor its propensity for backward sliding, either directly or indirectly. This would certainly explain why certain RNAP mutants confer high temperature lethality that is rescued by an elevated level of GreA (Sparkowski and Das, 1990, 1992).
Materials and methods
Enzymes, chemicals and oligonucleotides
Restriction enzymes and T4 polynucleotide kinase were obtained from New England Biolabs. The Klenow fragment of DNA polymerase I was purchased from Amersham. Superscript RNase H– Reverse Transcriptase from M-MLV, S1 nuclease and T4 DNA ligase were from Life Technologies. DMS and most chemicals, including antibiotics, were from Sigma. CAA was bought from Fluka Chemie and double distilled before use (boiling point 78–80°C). Unlabeled dNTPs were from Boehringer Mannheim, whereas [α-32P]dATP, [γ-32P]ATP and all the oligonucleotides were from Amersham.
Bacterial strains and plasmids
The four E.coli recipient strains used are isogenic to W3110 (E.coli Genetic Stock Center). The AD8782, AD8786 and AD8775 strains harbor the _greA_– (_greA::_KanR), the _greB_– (Δ_greB::_CamR) and the _greA_– _greB_– double mutations, respectively (A.Das et al., manuscript in preparation). In all subsequent in vivo experiments, the strains were grown with the appropriate antibiotics at the following concentrations: kanamycin, 18 µg/ml; chloramphenicol, 15 µg/ml.
The pATC6a and pATC6b plasmids described previously (Toulmé et al., 1999) are pKK232-8-based plasmids. These constructs contain a constitutive promoter that drives transcription of the cat gene through the (ATC/TAG)n repeats and the lac operator motif (Figure 1). The introduction of either of these plasmids into the strains described above is via selection on agar plates containing 100 µg/ml ampicillin in addition to the aforementioned antibiotics. A second plasmid (pAC184IQ) that overproduces lac repressor was then introduced in these transformants by selecting tetracycline (16 µg/ml) and ampicillin resistance. The pAC184IQ plasmid was created by subcloning the lacIQ gene from pAC177IQ (Toulmé et al., 1999) into the _Pvu_II (position 515) and _Sty_I (position 2863) restriction sites of pACYC184 (New England Biolabs), thus deleting the cat gene. In general, all DNA manipulations and transformations into E.coli cells were performed according to standard procedures (Sambrook et al., 1989).
In situ DNA footprinting and RNA analyses
The in situ DNA probing with DMS or CAA and the subsequent analyses of the modifications by hot piperidine cleavage (DMS) or primer extension with the Klenow fragment of DNA polymerase I (CAA) were carried out as described (Guérin et al., 1996; Toulmé et al., 1999). The RNA extraction and the 3′ end mapping with S1 nuclease protection experiments were also performed exactly as reported previously (Toulmé et al., 1999).
Quantitative analyses of the cat and bla transcripts were carried out simultaneously with two 32P end-labeled primers that anneal between positions 4850 and 4868 for the bla gene and between positions 248 and 265 for the cat gene (the positions are those of the original vector pKK232-8). Twenty micrograms of total RNAs were mixed in water with 0.1 pmol of each labeled primer in a volume of 10 µl. The samples were heat denatured at 90°C for 2 min and chilled on ice. Four microliters of 5× reverse transcription buffer (250 mM Tris–HCl pH 8.3, 375 mM KCl, 15 mM MgCl2), 2 µl of 100 mM dithiothreitol and 5 U of RNase inhibitor (RNAguard™ from Amersham) were then added prior to hybridization at 45°C for 20 min. cDNA synthesis was initiated by the addition of a mixture containing 4 µl of dNTPs (5 mM each) and 100 U of reverse transcriptase from M-MLV, and incubation was continued at 45°C for 1 h. Nucleic acids were then ethanol precipitated, dried, and finally resolved on a 6% denaturating polyacrylamide gel. Note that the cat primer also hybridizes to the chromosome-encoded cat transcript produced in the AD8786 and AD8775 strains. However, the resulting cDNAs are easily identified by their smaller size and are not present on the part of the gel shown in Figure 5A.
Acknowledgments
Acknowledgements
This article is dedicated to the memory of Marc Leng who died on May 7, 2000. We are grateful to Marc Boudvillain and Martine Guérin for helpful discussions. This work was supported in part by l’Association de la Recherche sur le Cancer, la Ligue Contre le Cancer (comité du Loiret) and by the USPHS Grant (GM28946) from the National Institutes of Health to (A.D.).
References
- Artsimovitch I. and Landick,R. (2000) Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl Acad. Sci. USA, 97, 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S., Polyakov,A., Nikiforov,V. and Goldfarb,A. (1992) GreA protein: a transcription elongation factor from Escherichia coli. Proc. Natl Acad. Sci. USA, 89, 8899–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S., Sagitov,V. and Goldfarb,A. (1993) Transcript cleavage factors from E.coli. Cell, 72, 459–466. [DOI] [PubMed] [Google Scholar]
- Chedin S., Riva,M., Schultz,P., Sentenac,A. and Carles,C. (1998) The RNA cleavage activity of RNA polymerase III is mediated by an essential TFIIS-like subunit and is important for transcription termination. Genes Dev., 12, 3857–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A. (1993) Control of transcription termination by RNA-binding proteins. Annu. Rev. Biochem., 62, 893–930. [DOI] [PubMed] [Google Scholar]
- Davenport R.J., Wuite,G.J.L., Landick,R. and Bustamante,C. (2000) Single-molecule study of transcriptional pausing and arrest by E.coli RNA polymerase. Science, 287, 2497–2500. [DOI] [PubMed] [Google Scholar]
- Erie D.A., Hajiseyedjavadi,O., Young,M.C. and von Hippel,P.H. (1993) Multiple RNA polymerase conformations and GreA: control of the fidelity of transcription. Science, 262, 867–873. [DOI] [PubMed] [Google Scholar]
- Guérin M., Leng,M. and Rahmouni,A.R. (1996) High resolution mapping of E.coli transcription elongation complex in situ reveals protein interactions with the non-transcribed strand. EMBO J., 15, 5397–5407. [PMC free article] [PubMed] [Google Scholar]
- Hagler J. and Schuman,S. (1992) Stability of ternary transcription complexes of vaccinia virus RNA polymerase at promoter-proximal positions. J. Biol. Chem., 267, 7644–7654. [PubMed] [Google Scholar]
- Henkin T.M. (1996) Control of transcription termination in prokaryotes. Annu. Rev. Genet., 30, 35–57. [DOI] [PubMed] [Google Scholar]
- Hsu L.M., Vo,N.V. and Chamberlin,M.J. (1995) Escherichia coli transcript cleavage factors GreA and GreB stimulate promoter escape and gene expression in vivo and in vitro. Proc. Natl Acad. Sci. USA, 92, 11588–11592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon C. and Agarwal,K. (1996) Fidelity of RNA polymerase II transcription controlled by elongation factor TFIIS. Proc. Natl Acad. Sci. USA, 93, 13677–13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassavetis G.A. and Geiduschek,E.P. (1993) RNA polymerase marching backward. Science, 259, 944–945. [DOI] [PubMed] [Google Scholar]
- Komissarova N. and Kashlev,M. (1997a) Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc. Natl Acad. Sci. USA, 94, 1755–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarova N. and Kashlev,M. (1997b) RNA polymerase switches between inactivated and activated states by translocating back and forth along the DNA and the RNA. J. Biol. Chem., 272, 15329–15338. [DOI] [PubMed] [Google Scholar]
- Koulich D., Orlova,M., Malhotra,A., Sali,A., Darst,S.A. and Borukhov,S. (1997) Domain organization of Escherichia coli transcript cleavage factors GreA and GreB. J. Biol. Chem., 272, 7201–7210. [DOI] [PubMed] [Google Scholar]
- Kulish D., Lee,J., Lomakin,I., Nowicka,B., Das,A., Darst,S., Normet,K. and Borukhov,S. (2000) The functional role of basic patch, a structural element of Escherichia coli transcript cleavage factors GreA and GreB. J. Biol. Chem., 275, 12789–12798. [DOI] [PubMed] [Google Scholar]
- Landick R. (1997) RNA polymerase slides home: pause and termination site recognition. Cell, 88, 741–744. [DOI] [PubMed] [Google Scholar]
- Landick R., Turnbough,C.L.,Jr and Yanofsky,C. (1996) Transcription attenuation. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella typhimurium. American Society for Microbiology, Washington, DC, pp. 1263–1286. [Google Scholar]
- Nudler E. (1999) Transcription elongation: structural basis and mechanisms. J. Mol. Biol., 288, 1–12. [DOI] [PubMed] [Google Scholar]
- Nudler E., Mustaev,A., Lukhtanov,E. and Goldfarb,A. (1997) The RNA–DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell, 89, 33–41. [DOI] [PubMed] [Google Scholar]
- Orlova M., Newlands,J., Das,A., Goldfarb,A. and Borukhov,S. (1995) Intrinsic transcript cleavage activity of RNA polymerase. Proc. Natl Acad. Sci. USA, 92, 4596–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder T.C. and Hawley,D.K. (1996) Promoter proximal sequences modulate RNA polymerase II elongation by a novel mechanism. Cell, 87, 767–777. [DOI] [PubMed] [Google Scholar]
- Reines D. (1992) Elongation factor-dependent transcript shortening by template-engaged RNA polymerase II. J. Biol. Chem., 267, 3795–3800. [PMC free article] [PubMed] [Google Scholar]
- Reines D. (1994) Nascent RNA cleavage by transcription elongation complexes. In Conaway,R.C. and Conaway,J.W. (eds), Transcription: Mechanisms and Regulation. Raven Press, New York, NY, pp. 263–278. [Google Scholar]
- Reines D., Ghanouni,P., Li,Q.Q. and Mote,J.,Jr (1992) The RNA polymerase II elongation complex factor-dependent transcription elongation involves nascent RNA cleavage. J. Biol. Chem., 267, 15516–15522. [PMC free article] [PubMed] [Google Scholar]
- Rudd M.D., Izban,M.G. and Luse,D.S. (1994) The active site of RNA polymerase II participates in transcript cleavage within arrested ternary complexes. Proc. Natl Acad. Sci. USA, 91, 8057–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Samkurashvili I. and Luse,D.S. (1998) Structural changes in the RNA polymerase II transcription complex during transition from initiation to elongation. Mol. Cell. Biol., 18, 5343–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry S.S. and Ross,B.M. (1997) Nuclease activity of T7 RNA polymerase and the heterogeneity of transcription elongation complexes. J. Biol. Chem., 272, 8644–8652. [DOI] [PubMed] [Google Scholar]
- Sidorenkov I., Komissarova,N. and Kashlev,M. (1998) Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol. Cell, 2, 55–64. [DOI] [PubMed] [Google Scholar]
- Sparkowski J. and Das,A. (1990) The nucleotide sequence of greA, a suppressor gene that restores growth of an Escherichia coli RNA polymerase mutant at high temperature. Nucleic Acids Res., 18, 6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkowski J. and Das,A. (1992) Simultaneous gain and loss of functions caused by a single amino acid substitution in the β subunit of Escherichia coli RNA polymerase: suppression of nusA and rho mutations and conditional lethality. Genetics, 130, 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surratt C.K., Milan,S.C. and Chamberlin,M.J. (1991) Spontaneous cleavage of RNA in ternary complexes of Escherichia coli RNA polymerase and its significance for the mechanism of transcription. Proc. Natl Acad. Sci. USA, 88, 7983–7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M.J., Platas,A.A. and Hawley,D.K. (1998) Transcriptional fidelity and proofreading by RNA polymerase II. Cell, 93, 627–637. [DOI] [PubMed] [Google Scholar]
- Toulmé F., Guérin,M., Robichon,N., Leng,M. and Rahmouni,A.R. (1999) In vivo evidence for back and forth oscillations of the transcription elongation complex. EMBO J., 18, 5052–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschochner H. (1996) A novel RNA polymerase I-dependent RNase activity that shortens nascent transcripts from the 3′ end. Proc. Natl Acad. Sci. USA, 93, 12914–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain S.M., Kane,C.M. and Chamberlin,M.J. (1997) Basic mechanisms of transcript elongation and its regulation. Annu. Rev. Biochem., 66, 117–172. [DOI] [PubMed] [Google Scholar]
- von Hippel P.H. (1998) An integrated model of the transcription complex in elongation, termination and editing. Science, 281, 660–665. [DOI] [PubMed] [Google Scholar]
- Wang D. and Hawley,D.K. (1993) Identification of a 3′–5′ exonuclease activity associated with human RNA polymerase II. Proc. Natl Acad. Sci. USA, 90, 843–847. [DOI] [PMC free article] [PubMed] [Google Scholar]