Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: Implications for glaucoma (original) (raw)
Abstract
Our group recently demonstrated that autoimmune T cells directed against central nervous system-associated myelin antigens protect neurons from secondary degeneration. We further showed that the synthetic peptide copolymer 1 (Cop-1), known to suppress experimental autoimmune encephalomyelitis, can be safely substituted for the natural myelin antigen in both passive and active immunization for neuroprotection of the injured optic nerve. Here we attempted to determine whether similar immunizations are protective from retinal ganglion cell loss resulting from a direct biochemical insult caused, for example, by glutamate (a major mediator of degeneration in acute and chronic optic nerve insults) and in a rat model of ocular hypertension. Passive immunization with T cells reactive to myelin basic protein or active immunization with myelin oligodendrocyte glycoprotein-derived peptide, although neuroprotective after optic nerve injury, was ineffective against glutamate toxicity in mice and rats. In contrast, the number of surviving retinal ganglion cells per square millimeter in glutamate-injected retinas was significantly larger in mice immunized 10 days previously with Cop-1 emulsified in complete Freund's adjuvant than in mice injected with PBS in the same adjuvant (2,133 ± 270 and 1,329 ± 121, respectively, mean ± SEM; P < 0.02). A similar pattern was observed when mice were immunized on the day of glutamate injection (1,777 ± 101 compared with 1,414 ± 36;P < 0.05), but not when they were immunized 48 h later. These findings suggest that protection from glutamate toxicity requires reinforcement of the immune system by antigens that are different from those associated with myelin. The use of Cop-1 apparently circumvents this antigen specificity barrier. In the rat ocular hypertension model, which simulates glaucoma, immunization with Cop-1 significantly reduced the retinal ganglion cell loss from 27.8% ± 6.8% to 4.3% ± 1.6%, without affecting the intraocular pressure. This study may point the way to a therapy for glaucoma, a neurodegenerative disease of the optic nerve often associated with increased intraocular pressure, as well as for acute and chronic degenerative disorders in which glutamate is a prominent participant.
Recent studies in our laboratory demonstrated that the death of neurons after traumatic injury to the central nervous system (CNS) can be delayed and reduced by reinforcing a T cell immune response directed against myelin-associated self-antigens (1–3). With the use of optic nerve injury of adult rats as a model, we found that, irrespective of their antigen specificity, T cells accumulate at the lesion site after injury, and their numbers increase after their passive transfer (1, 2,4). We further demonstrated that passive transfer of T cells specific to myelin basic protein (MBP), but not to non-self-antigens, reduces the posttraumatic neuronal loss in rat models of optic nerve crush and spinal cord contusion (3, 5). We also showed that active immunization with myelin or myelin-derived peptides can lead to effective neuroprotection in both of those models (5, 6). However, because of the encephalitogenic properties of myelin, immunization with myelin or myelin-derived peptides is likely to be accompanied by devastating side effects, such as induction of an autoimmune disease.
In the course of our search for a “safe” myelin peptide, we recently observed that neuroprotection in the rat optic nerve model could be achieved by immunization with copolymer 1 (Cop-1) (7), a synthetic peptide that cross-reacts with myelin antigen and is used clinically as an immunosuppressor of myelin-associated autoimmune disease.
The above findings raised intriguing questions. Does the effectiveness of autoimmune neuroprotection depend on the type and/or the site of the neuronal insult? Is Cop-1 an adequate substitute only for nerve insults involving myelin exposure, caused by mechanical disruption of the myelin sheath? Is immunization with Cop-1 beneficial in cases where the threat persists, as in chronic diseases? These questions were addressed in the present study by using an intravitreal glutamate insult, instead of axonal injury, in the mouse and rat visual systems, and by using a rat model of chronically elevated intraocular pressure (IOP), which simulates a major risk factor in glaucoma, a neurodegenerative disease of the optic nerve.
Glutamate is an amino acid that normally functions as a CNS neurotransmitter (8, 9). When its physiological concentrations are exceeded, however, it becomes cytotoxic and acts as a major mediator of toxicity in acute and chronic injuries (including optic nerve degeneration in glaucoma) (10, 11). We have recently demonstrated that glutamate toxicity has a more deleterious effect in mature rats or mice devoid of mature T cells, either as a result of thymectomy performed at birth or because of defective T cell maturation, than in normal animals (J.K., E.Y., H.S., E. Hauben, I. Shaked, and M.S., unpublished observations; H.S., E.Y., and M.S., unpublished work). These results suggested that a glutamate insult, like mechanical insults, spontaneously evokes a T cell-dependent beneficial autoimmunity, which is amenable to boosting. In the present study we showed that active or passive immunization with a peptide derived from myelin oligodendrocyte glycoprotein (MOG) or with MBP, which provides effective neuroprotection after axonal injury (1, 2, 6), did not protect the neurons from the toxicity caused by glutamate. Protection from glutamate toxicity was achieved, however, by vaccination with Cop-1. We further showed that immunization with Cop-1 provides highly effective protection from retinal ganglion cell (RGC) death induced by ocular hypertension in the rat model of glaucoma, under conditions where the pressure remains high and is not affected by the immunization.
Materials and Methods
Animals.
All animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee. Mice of the C57BL/6J and BALB/c strains, aged 8−13 weeks, and inbred adult Lewis rats, aged 8–12 weeks, were supplied by the Animal Breeding Center of the Weizmann Institute of Science and housed in light- and temperature-controlled rooms. The rats were matched for age and size in each experiment. Before their use in experiments, animals were anesthetized by i.p. administration of ketamine (80 mg/kg) and xylazine (16 mg/kg).
Antigens.
Cop-1 was purchased from Teva Pharmaceuticals (Petah Tikva, Israel). MOG peptide (pMOG) 1−22 (GQFRVIGPGHPIRALVGDEAEL) was synthesized in the laboratory of M. Fridkin at the Department of Chemistry of the Weizmann Institute of Science, by the use of the fluorenylmethoxycarbonyl technique and an automatic multiple peptide synthesizer (AMS422; Abimed Analysentechnik, Langenfeld, Germany). MBP from the spinal cords of guinea pigs was purchased from Sigma.
Immunization.
Mice were immunized with 75 μg of Cop-1 or 300 μg of pMOG emulsified with an equal volume of complete Freund's adjuvant (CFA) containing 0.5 or 5 mg/ml Mycobacterium tuberculosis. The emulsion (total volume 0.15 ml) was injected s.c. at one site in the flank. One week later, the mice immunized with pMOG were given an identical immunization in the other flank as a booster. Control mice were injected with PBS in CFA (Difco). Rats were immunized with 200 μg of Cop-1 emulsified with an equal volume of CFA containing 5 mg/ml M. tuberculosis.
Crush Injury of the Optic Nerve.
Mice or rats were anesthetized and subjected to severe crush injury in the intraorbital portion of the optic nerve, 1−2 mm from the eyeball. With the aid of a binocular operating microscope, the conjunctiva was incised and the optic nerve exposed. Taking special care not to interfere with the blood supply, we crushed the nerve with cross-action calibrated forceps for 2 s (mice) or 30 s (rats).
Glutamate and_N_-Methyl-d-aspartate (NMDA) Injections.
The right eye of the anesthetized mouse or rat was punctured with a 27-gauge needle in the upper part of the sclera, and a 10-μl Hamilton syringe with a 30-gauge needle was inserted as far as the vitreal body. Mice were injected with a total volume of 1 μl (200 nmol) ofl-glutamate or 1 μl of NMDA (75 nmol; Research Biochemicals, Natick, MA) dissolved in saline. Rats were injected with 2 μl (375 nmol) of l-glutamate.
Elevation of IOP in Rats.
Male Lewis rats were anesthetized with a mixture of ketamine (15 mg/kg), acepromazine (1.5 mg/kg), and xylazine (0.3 mg/kg). An increase in IOP was achieved by laser photocoagulation of the limbal and episcleral veins (E.W.M., M. Wijono, and G.R., unpublished work). Rats received two laser treatments, 1 week apart, with a blue-green argon laser (1 W for 0.2 s, delivering a total of 130–150 spots of 50 μm in the two treatments; Coherent, Palo Alto, CA). IOP was measured once a week by using a TONO-PEN (Mentor, Norwell, MA).
Preinjury Stereotactic Application of Dye in Mice.
The skull was exposed and kept dry and clean with 15% hydrogen peroxide. The bregma was identified and marked. A hole was drilled above the superior colliculus of each hemisphere (0.292 mm behind and 0.05 mm lateral to the midline). With the use of a stereotactic measuring device and a Hamilton injector, the mice were injected with FluoroGold (5% in saline, 1 μl; Fluorochrome, Denver) at one point in the superior colliculus of each hemisphere, at a depth of 0.16 mm or 0.175 mm (depending on the mouse strain) from the surface of the brain. After completion of the injection, the skin was sutured. Retrograde uptake of the dye provides a marker of the living cells.
Assessment of Retinal Ganglion Cell Survival in Mice.
Mice were given a lethal dose of pentobarbitone (170 mg/kg). Their eyes were enucleated, and the retinas were detached and prepared as flattened whole mounts in paraformaldehyde (4% in PBS). Labeled cells from 4–6 selected fields of identical size (0.076 mm2) were counted. The selected fields were located at approximately the same distance from the optic disk (0.3 mm) to overcome the variation in RGC density as a function of distance from the optic disk. Fields were counted under the fluorescence microscope (magnification ×800) by observers blinded to the treatment received by the mouse. The average number of RGCs per field in each retina was calculated.
Assessment of RGC Survival in Rats.
Survival of RGCs in rats was measured after postinjury application of the fluorescent lipophilic dye 4-(4-(didecylamino)styryl)-_N_-methylpyridinium iodide (Molecular Probes Europe, Leiden, The Netherlands) distally to the optic nerve head. Labeling and measurement were carried out as follows. The optic nerve was exposed without damaging the retinal blood supply. Complete axotomy was performed 1−2 mm from the optic nerve head, and solid crystals (0.2−0.4 mm diameter) of 4-(4-(didecylamino)styryl)-_N_-methylpyridinium iodide were deposited at the site of the formed axotomy. Five days after dye application the rats were killed. The retina was detached from the eye, prepared as a flattened whole mount in 4% paraformaldehyde solution, and examined for labeled RGCs by fluorescence microscopy. In the IOP experimental animals, the RGCs were retrogradely labeled with dextran tetramethylrhodamine (DTMR) (Molecular Probes), applied 2–3 mm from the globe. After 24 h the retinas were whole-mounted, and labeled RGCs in eight regions, two in each quadrant (0.66–1.103 mm from the edge of the optic disk), were counted under 400× magnification.
Generation of a Mouse Cop-1–T Cell Line.
A mouse T cell line was generated from draining lymph node cells obtained from C57BL/6J mice immunized with Cop-1 antigen. The antigen was dissolved in PBS (1 mg/ml) and emulsified with an equal volume of CFA supplemented with 5 mg/ml M. tuberculosis (Difco). Ten days after immunization into the hind foot pads, the mice were killed and their draining lymph nodes were surgically removed and dissociated. The cells were washed and activated with the antigen (10 μg/ml) in stimulation medium containing RPMI medium 1640 supplemented withl-glutamine (2 mM), 2-mercaptoethanol (5 × 10−5 M), penicillin (100 units/ml), streptomycin (100 μg/ml), and autologous serum 0.5% (vol/vol). After incubation for 72 h at 37°C, 98% relative humidity, and 10% CO2, the cells were transferred to propagation medium consisting of RPMI medium 1640 supplemented with nonessential amino acids (1 ml/100 ml), sodium pyruvate (1 mM),l-glutamine, β-mercaptoethanol, penicillin, and streptomycin, in the same concentrations as above, with the addition of 5% FCS (vol/vol) and 10% T cell growth factor derived from the supernatant of Con A-stimulated spleen cells. Cells were grown in propagation medium for 10–14 days before being restimulated with their antigen (10 μg/ml) in the presence of irradiated (2,500 rad) spleen cells (107 cells/ml) in stimulation medium. The T cell line was expanded by repeated stimulation and propagation. Basically the line has a phenotype similar to that described in ref. 12.
Generation of a Rat Cop-1–T Cell Line.
T cell lines were generated from draining lymph node cells obtained from Lewis rats immunized with the above antigens. The antigen was dissolved in PBS (1 mg/ml) and emulsified with an equal volume of incomplete Freund's adjuvant (Difco) supplemented with 4 mg/ml_M. tuberculosis_ (Difco). Ten days after the antigen was injected into the rats' hind foot pads in 0.1 ml of the emulsion, the rats were killed and their draining lymph nodes were surgically removed and dissociated. The cells were washed and activated with the antigen (10 μg/ml) in stimulation medium containing DMEM supplemented withl-glutamine (2 mM), 2-mercaptoethanol (5 × 10−5 M), sodium pyruvate (1 mM), penicillin (100 units/ml), streptomycin (100 μg/ml), nonessential amino acids (1 ml/100 ml), and autologous serum 1% (vol/vol). After incubation for 72 h at 37°C, 98% relative humidity, and 10% CO2, the cells were transferred to propagation medium consisting of DMEM, l-glutamine, 2-mercaptoethanol, sodium pyruvate, nonessential amino acids, and antibiotics in the same concentrations as above, with the addition of 10% FCS (vol/vol) and 10% T cell growth factor derived from the supernatant of Con A-stimulated spleen cells. Cells were grown in propagation medium for 4−10 days before being restimulated with their antigen (10 μg/ml) in the presence of irradiated (2,000 rad) thymus cells (107 cells per ml) in stimulation medium. The T cell lines were expanded by repeated stimulation and propagation.
Histological Analysis.
Seven days after glutamate or saline injection the mice were killed by injection of a lethal dose of pentobarbitone (170 mg/kg), and their eyes were removed and fixed in formaldehyde (4% in PBS) for 48 h at 4°C. Sections (10 μm thick) were embedded in paraffin and stained with hematoxylin and eosin.
Results
Myelin-Associated Antigens Are Not Protective Against Glutamate Toxicity.
We have previously demonstrated that passive and active immunization with myelin-associated antigens can reduce the posttraumatic degeneration associated with optic nerve crush injury in mice and rats (1, 6) and with spinal cord contusive injury in adult rats (3, 5). To determine whether such immune neuroprotection is exerted after a nonmechanical injury as well, we first attempted to determine whether active immunization with myelin-associated antigens or passive transfer of T cells reactive to these antigens provides neuroprotection against toxicity induced by intravitreal injection of glutamate. The optic nerves of rats and mice were subjected to crush injury. With the use of our established protocols for immune neuroprotection, active immunization with MOG-derived peptides (6) was performed in mice and passive transfer of anti-MBP T cells was performed in rats (1). Glutamate insult was inflicted by intravitreal injection of glutamate at a concentration previously shown to lead to RGC death that is measurable after 1 week in both mice and rats (13).
Mice were immunized with pMOG before the intravitreal injection of glutamate (200 nmol). When the RGC survival rate was assessed 1 week after glutamate injection, no evidence of a beneficial effect of the immunization with pMOG could be detected (Fig.1A). Similarly, no beneficial effect was detectable when the glutamate injection was immediately followed by passive transfer of anti-MBP T cells in rats (Fig.1B). Thus, although vaccination with pMOG(1−22) was recently shown to induce a neuroprotective response in mice after crush injury of the optic nerve (6), no such neuroprotection was seen in the present study after the pMOG-vaccinated mice were subjected to glutamate insult.
Figure 1.

Immunization with pMOG or passive transfer of anti-MBP T cells does not protect mouse RGCs from glutamate toxicity. (A) C57BL/6J mice were immunized with pMOG 14 days before their RGCs were exposed directly to glutamate toxicity by intravitreous injection ofl-glutamate (200 nmol). Four days later the RGCs were retrogradely labeled with FluoroGold, followed after 3 days by retinal excision and counting (see Materials and Methods). RGC survival is expressed as the mean ± SEM per square millimeter. No significant differences in RGC survival after glutamate injection were observed between the group treated with pMOG in CFA (n = 8) and the control group treated with PBS in CFA (n = 7). (B) Glutamate was injected intravitreally into Lewis rats. Four days later, the RGCs were labeled by application of the dye 4-(4-(didecylamino)styryl)-_N_-methylpyridinium iodide, followed after 5 days by retinal excision and counting. Note that retinal survival in the T-cell-treated group did not differ significantly from that in the control group (no. of RGCs per square millimeter, mean ± SEM; n = 6 in each group).
These findings led us to consider two possibilities: either the loss of RGCs after glutamate toxicity is not amenable to immune neuroprotection, implying that glutamate-induced RGC death does not involve the immune system, or myelin-associated antigens such as pMOG and MBP are not the right antigens for protection against glutamate toxicity. Our recent results (J.K., E.Y., H.S., E. Hauben, I. Shaked, and M.S., unpublished observations), showing that the rate of cell death caused by glutamate is higher in rats or mice that lack mature T cells than in normal animals, strongly suggest that the beneficial physiological response to glutamate insult involves T cells. Accordingly, boosting this endogenous glutamate-induced T cell response is likely to have a beneficial effect on the injured retina.
Cop-1 Immunization Protects against Glutamate Toxicity.
Although the search for a physiological antigen that might evoke a beneficial immune response to glutamate-induced toxicity is still a prime objective of our research, at this stage we were interested in finding an antigen that might be used for purposes of exogenous immune system manipulation of the immune response to glutamate. First, we attempted to determine whether glutamate injection causes the RGCs to become accessible to lymphocytes. We found that large numbers of lymphocytes invade the glutamate-injected eye within 24 h of the glutamate injection (Fig. 2), suggesting that immune manipulation might influence the survival of RGCs after their local exposure to glutamate. Taken together, these two observations led us to believe that glutamate toxicity activates a T cell-mediated protective effect and encouraged us to search for a way to boost this beneficial immune response. In seeking an appropriate antigen, we considered as a likely candidate the synthetic polymer Cop-1, an oligopeptide used as a drug in patients with multiple sclerosis and recently shown to boost neuroprotection in a model of optic nerve crush injury of the adult rat (7).
Figure 2.
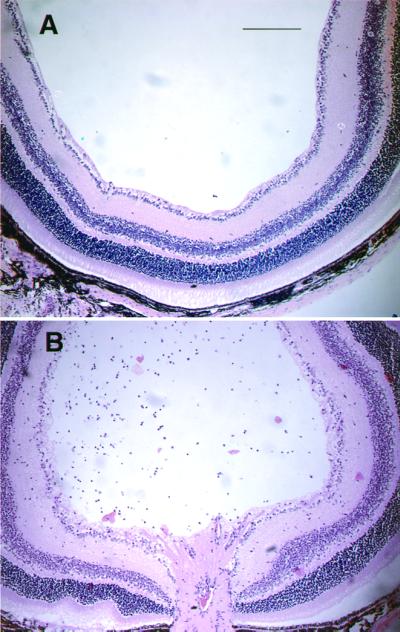
Invasion of lymphocytes after intravitreal injection of glutamate. Glutamate was injected intravitreally into C57BL/6J mice. After 24 h the eye was removed and processed for histology. Hematoxylin and eosin-stained retinal sections (10 μm thick) of both glutamate-injected and control mice are shown. (Bar = 200 μm.)
First, we attempted to determine whether immunization with Cop-1 has a beneficial effect on RGC survival after optic crush injury in mice and rats. For this study we used BALB/c mice. Immunization with Cop-1, by a protocol that was found to be beneficial after optic nerve crush injury in rats, was also beneficial after optic nerve injury in mice (Fig. 3).
Figure 3.

Survival rate of retinal ganglion cells after optic nerve injury. The RGCs of inbred adult BALB/c were retrogradely labeled with FluoroGold (see Materials and Methods) 10 days after being immunized with 50 μg of Cop-1 emulsified in CFA. Control mice were injected with PBS in CFA (n = 8−12 in each group). Three days after labeling of RGCs, mice were subjected to a severe crush injury of the intraorbital portion of the optic nerve. Two weeks after injury the retinas were excised, and their labeled RGCs were counted (see Materials and Methods). Relative to nonimmunized controls, survival rates were significantly higher (P < 0.001, Student's t test) in mice immunized with Cop-1 in CFA.
Next, we attempted to determine whether the same protocol can lead to neuroprotection from glutamate-induced toxicity. Ten days before intravitreal injection of glutamate, C57BL/6J mice were immunized with Cop-1 emulsified in adjuvant containing 5 mg/ml bacteria. This strain was selected in view of our recent finding that the loss of RGCs induced by glutamate injection in these mice is greater than in BALB/c mice because of a genetic linkage between neuronal loss and resistance to autoimmune disease (J.K., E.Y., H.S., E. Hauben, I. Shaked, and M.S., unpublished observations). Immunization with Cop-1 resulted in a significant reduction in glutamate toxicity (Fig. 4A). In an attempt to establish the therapeutic window for immunization with Cop-1 in this model, we repeated the experiment, with Cop-1 emulsified in adjuvant containing two different concentrations of bacteria (0.5 or 5 mg/ml), and immunized the mice at different times in relation to the glutamate insult. Mice immunized on the day of glutamate injection still showed significantly higher rates of RGC survival than those seen in mice injected with PBS emulsified in the corresponding adjuvant (Fig. 4 B and C). Both adjuvants yielded significant effects. The protective efficacy of Cop-1 was diminished when Cop-1 immunization was given 48 h after glutamate injection (Fig. 4D). Interestingly, Cop-1 immunization failed to protect the mice from toxicity caused by NMDA (Fig.5), recently shown by us to cause, in this in vivo model, RGC death with features different from those typical of apoptotic death (E.Y., H.S., T. Raveh, A. Kimchi, and M.S., unpublished work).
Figure 4.

Neuroprotection from glutamate toxicity by active immunization with Cop-1. (A) Ten days before glutamate injection, mice were immunized by s.c. injection with Cop-1 in CFA (5 mg/ml bacteria) or injected with PBS in CFA. The results of one experiment are shown (n = 5 in each group). The number of surviving RGCs per square millimeter (mean ± SEM) was significantly higher in the Cop-1-immunized mice than in the mice injected with PBS in CFA or in mice that received glutamate only (P < 0.02, two-tailed t test). Injection with PBS in CFA had no detectable effect on the number of RGCs. The experiment was repeated three times, with identical results. Altogether 13 animals in the Cop-1-treated group and 15 animals in the PBS-treated group were tested. (B) Immediately after intravitreal injection of glutamate, mice were immunized with Cop-1 emulsified in CFA (5 mg/ml bacteria). The number of surviving RGCs per square millimeter (mean ± SEM) was determined 1 week later. The results of one experiment are shown. The effect of immunization with Cop-1 was significant (P < 0.05; two-tailed t test; n = 12 for Cop-1 and n = 8 for the control). This experiment was repeated with 11 mice for Cop-1 immunization and eight mice for injection with PBS in CFA (5 mg/ml bacteria). (C) RGC survival after glutamate insult and immediate immunization with Cop-1 in adjuvant containing 0.5 mg/ml of bacteria. The number of surviving RGCs per square millimeter was significantly higher in the Cop-1-immunized mice (n = 15) than in the mice injected with glutamate (n = 5) (P < 0.04; two-tailed t test). (D) Survival of RGCs after immunization performed before, immediately after, or 48 h after glutamate insult. Bars show the pooled results obtained for all mice examined in each treatment, collected from repeated experiments. No effect was seen when immunization was performed 48 h after the insult.
Figure 5.

Cop-1 immunization fails to protect mice from NMDA toxicity. Ten days before injection of NMDA (75 nmol), mice (n = 5−7) were immunized by s.c. injection of Cop-1 in CFA or with PBS in CFA. Labeling of RGCs and counting of viable RGCs under fluorescence microscopy were as described for Fig. 1. RGC survival in the Cop-1-immunized mice, expressed as a percentage of survival in a normal eye, was similar to that in the PBS-injected mice (P = 0.55, Student's t test), indicating that no neuroprotection was obtained.
Adoptive Transfer of T Cells Reactive to Cop-1 Protects Against Glutamate Toxicity.
To determine whether the observed immunization with Cop-1 leads to T cell-mediated neuroprotection against glutamate toxicity, we passively transferred 5 × 106 Cop-1-reactive T cells (250 μl i.p.) into mice immediately after injection of glutamate (200 nmol). One week later, significantly more RGCs had survived in the mice injected with Cop-1-reactive T cells (1,978 ± 86, n = 6) than in control mice (1,238 ± 2, n = 3) injected i.p. with PBS (Fig.6).
Figure 6.

Cop-1-reactive T cells protect RGCs from glutamate toxicity. Immediately after injection of glutamate (200 nmol), mice were injected with Cop-1-reactive T cells or with PBS. Dye application, preparation, and counting of RGCs and calculation of RGC survival were as described for Fig. 1. Significantly more labeled RGCs were seen in the retinas of mice injected with Cop-1-reactive T cells than in the retinas of PBS-injected control mice (**, P < 0.0007, Student's t test).
Cop-1 Immunization Protects RGCs from Death Induced by Ocular Hypertension in Rats.
By 2 weeks after laser treatment in Lewis rats, their IOP had increased to 30 ± 0.4 mmHg (mean ± SEM), and it remained at approximately that level thereafter (Fig.7A). Loss of RGCs in these rats, measured 3 weeks after the initial laser treatment, can be seen in Fig. 7B. To examine the effect of Cop-1 immunization on the survival of RGCs, rats were immunized with Cop-1 emulsified in CFA on the day of the first laser treatment. Control rats were injected with PBS in the same adjuvant. After 3 weeks the percentage of RGC loss in the Cop-1-immunized rats (4.3% ± 1.6%) was significantly lower (P < 0.002, two-tailed t test) than in the PBS/CFA-injected controls (27.8% ± 6.8%) (Fig.7C). The IOP in both groups of rats remained as high as that in a group of laser-treated rats that had received no injections (Fig.7A). A similar though slightly smaller effect was seen in rats that were immunized with Cop-1 when their IOP was already high (Fig. 7D).
Figure 7.

Effect of chronically increased IOP and Cop-1 immunization on RGC survival in Lewis rats. (A) Laser cauterization causing occlusion of the episcleral and limbal veins results in an increase in IOP and subsequent death of RGCs. Three weeks after laser treatment, the mean IOP was 30.4 ± 0.42 mmHg (mean ± SEM,n = 5) in rats subjected to venous occlusion and 15.8 ± 0.2 mmHg (n = 7) in naïve rats. (B) Three weeks after venous occlusion the surviving RGCs were counted in the laser-treated and in naïve rats (bars represent mean ± SEM). (C) Rats were immunized with Cop-1 (n = 15) or injected with PBS (n = 13) in CFA immediately after laser cauterization. Bars represent RGC loss, calculated as a percentage of the number of RGCs in naïve rats (mean ± SEM). The difference in the numbers of RGCs in the two groups was significant (P < 0.002, two-tailed t test). (D) Rats were immunized, 10 days after venous occlusion, with either Cop-1 (n = 5) or PBS (n = 4). Bars represent RGC loss, calculated as a percentage of the number of RGCs (mean ± SEM) in naïve animals. A tendency toward a neuroprotective effect was observed after delayed immunization with Cop-1; the difference was significant only by one-tailed t test (P = 0.04).
Discussion
T cell-dependent immune neuroprotection, achieved by passive or active immunization with Cop-1, is shown here to be an effective therapy for glutamate-induced toxicity in mice and in a rat model of chronically high IOP. The excitatory amino acid glutamate is capable of displaying dual activity, as an essential neurotransmitter and—when its physiological concentrations are exceeded—as a devastating excitotoxic compound. In its latter role, it is one of the most common mediators of toxicity in acute and chronic degenerative disorders. Intensive research has been devoted to attenuating the cytotoxic effect of glutamate by the use of locally acting drugs, such as NMDA receptor antagonists (14). Conventional therapy of this type is often unsatisfactory, however, as in neutralizing the toxic effect it is likely to interfere with physiological function. In humans, such compounds have psychotropic and other side effects that make them unsuitable as therapeutic agents. They also have the disadvantage of interfering with the essential physiological functioning of glutamate as a ubiquitous CNS neurotransmitter. Because glutamate activity is essential for normal physiological function, yet is potentially devastating after acute injury or in chronic CNS disorders, any attempt to neutralize its harmful effect must do so without eliminating its essential activity at other sites in the body.
The results of this study show that both passive and active immunizations with Cop-1 provide effective neuroprotection from glutamate toxicity, suggesting that vaccination can be developed as a way to reduce the neuronal toxicity associated with glutamate. These observations have a number of interesting implications: (i) Cop-1, which is used as an immunosuppressive drug in patients with the autoimmune disease multiple sclerosis, is effective as a vaccination against glutamate-induced neurotoxicity; (ii) loss of CNS neurons due to a local stress signal can benefit from systemic immune manipulation; (iii) the same neurons, in this case the RCGs, can benefit from a systemic immune response regardless of the nature and site of the insult, although the antigenic specificity of the response may vary; (iv) the beneficial activity of Cop-1, although apparently not dependent on the type of insult (mechanical or biochemical), appears to critically depend on the mechanism of death that the insult activates. In this study, the induced immune activity protected the cells against death caused by glutamate but not against death caused by NMDA; (v) Cop-1, acting as an immunogen, may induce neuroprotection by a mechanism that does not necessarily require cross-recognition of myelin proteins.
Cop-1 originally was designed to mimic the activity of MBP and to induce the inflammatory demyelinating disease experimental autoimmune encephalomyelitis in rodents. It was found, however, to be nonencephalitogenic and even to suppress experimental autoimmune encephalomyelitis induced by MBP (15), proteolipid protein (16), or MOG (17). Cop-1 prevents the development of experimental autoimmune encephalomyelitis in rodents and ameliorates multiple sclerosis in humans. Studies have demonstrated partial cross-reactivity between antibody to Cop-1 and MBP or between T cells directed to these antigens (18, 19). Cop-1 can serve as an antagonist of the T cell antigen receptor for the immunodominant MBP epitope (20). It also can bind to various MHC class II molecules and prevent them from binding to T cells with specific antigen recognition properties (21). In rodents, Cop-1 induces the expression of regulatory cells that suppress the encephalitogenic T cells. Adoptive transfer of such T cells in rodents was found to prevent the development of experimental autoimmune encephalomyelitis induced by MBP (22), proteolipid protein (16), or whole spinal cord homogenate (12).
It should be stressed that there is an important difference between immune neuroprotection against secondary degeneration and immune therapy for autoimmune diseases. Whereas the former requires active involvement of beneficial T cells, the latter may benefit from either immune modulation of encephalitogenic T cells or from their suppression. Cop-1, acting as an immunogen, may serve both purposes—neuroprotection from neuronal insult and therapy for autoimmune diseases. Presumably it achieves this dual purpose by evoking a “safe” T cell response, which provides, on the one hand, the beneficial autoimmune T cell response needed for neuroprotection (1, 2, 7, 23, 24) and, on the other hand, the immune modulation required for avoidance of autoimmune disease (25).
T cells reactive to MBP were shown in our laboratory to be neuroprotective in rat models of partially crushed optic nerves (1, 24) and spinal cord contusive injury (3). Our previous findings demonstrated in vivo cross-recognition between Cop-1-reactive T cells and components of CNS myelin (7). We suggested that such recognition, by triggering T cell reactivation and thus causing the T cells to switch to a protective phenotype, might represent a possible mechanism underlying T cell neuroprotection after axonal injury. We further showed that Cop-1-reactive T cells (7), like MBP-reactive T cells (23), when activated by their specific antigen, secrete significant amounts of brain-derived neurotrophic factor, a neurotrophin that plays a major role in neuronal survival (26, 27). In a previous study we examined T-cell immunity to Cop-1 after physical trauma to the white matter, where anti-Cop-1 T cells can cross-react with exposed myelin epitopes. The present finding that immunization with Cop-1 is active against glutamate toxicity, which directly affects neuronal cell bodies under conditions where no myelin antigens are likely to be involved, may suggest that anti-Cop-1 T cells, because of their heterogeneity, respond to a variety of antigens, including those associated with the retina. The Cop-1-reactive T cells may interact directly with glutamate itself. Such interaction could convert the Cop-1-reactive T cells, or endogenous T cells, to a protective phenotype.
The possibility that Cop-1-reactive T cells might interact with the injected glutamate within the vitreous or with microglia-activated cells within the retina is supported by the large numbers of invading lymphocytes observed in the vitreous 24 h after glutamate injection. In mice injected with Cop-1, the invading lymphocytes are likely to include some that are specific to Cop-1. The finding that passive transfer of Cop-1-reactive T cells has an effect similar to that of active immunization with Cop-1 suggests that the effect of the vaccination is indeed mediated by T cells, rather than by humoral immunity or by Cop-1 itself. Because glutamate, being a mediator of secondary degeneration, appears at some distance in time from the primary insult (11), a treatment window of 24 h in the case of direct glutamate toxicity may imply that in cases of CNS trauma the window for treatment with Cop-1 is much wider. It is interesting to note that Cop-1 had no protective effect when the toxic insult was induced by NMDA. This lack of a protective effect is in line with our previous studies showing that NMDA imposes an almost immediate death signal, without clear signs of apoptosis and with no apparent opportunity for therapy other than with NMDA receptor antagonists (E.Y.et al., unpublished observations). It is therefore not surprising that immunization with Cop-1 was ineffective against NMDA-induced toxicity. It remains to be established whether the activity of Cop-1 as a neuroprotective rather than as a suppressive agent is dependent on its route of administration. It is also necessary to determine how the local accumulation of T cells specific to CNS antigens or of T cells specific to cross-reactive antigens such as Cop-1 mediates neuroprotection after CNS insults. The T cell-mediated neuroprotection demonstrated in this study might be applicable not only to acute but also to chronic injury of CNS nerves such as that seen in glaucoma in which neurons are vulnerable to degeneration and amenable to neuroprotection (28–31).
Glaucoma is a chronic condition of the optic nerve, often associated with increased IOP, and is a leading cause of blindness. It is common experience, however, that the disease may continue to progress even though the IOP remains within the normal range, suggesting that mechanical compression is probably not the sole reason for optic nerve damage and that treatment, in addition to lowering the IOP, should therefore include neuroprotective therapy (for review see refs. 29, 32, and 33). Recent studies have shown, for example, that treatment with a glutamate antagonist (34) or a nitric oxide synthase inhibitor (35) attenuates RGC death in a rat model of increased IOP. The present finding is potentially of great interest clinically because (i) it shows that neuroprotection can be achieved even when the pressure remains high [indeed, even a pressure reduced to normal is not necessarily safe for patients with glaucoma, in whom the remaining neurons are more vulnerable than normal ones (36)]; and (ii) it is based on a strategy that harnesses and augments the tissue's own defense machinery.
Acknowledgments
We thank S. Smith for editing the manuscript. M.S. holds the Maurice and Ilse Katz Professorial chair in Neuroimmunology. The work was supported by Proneuron Ltd., Industrial Park, Ness-Ziona, Israel, and in part by a grant from the Glaucoma Research Foundation awarded to M.S.
Abbreviations
CFA
complete Freund's adjuvant
CNS
central nervous system
Cop-1
copolymer-1
IOP
intraocular pressure
MBP
myelin basic protein
MOG
myelin oligodendrocyte glycoprotein
pMOG
MOG peptide
NMDA
_N_-methyl-d-aspartate
RGC
retinal ganglion cell
References
- 1.Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen I R, Schwartz M. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 2.Moalem G, Monsonego A, Shani Y, Cohen I R, Schwartz M. FASEB J. 1999;13:1207–1217. doi: 10.1096/fasebj.13.10.1207. [DOI] [PubMed] [Google Scholar]
- 3.Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Akselrod S, Neeman M, Cohen I R, Schwartz M. Lancet. 2000;355:286–287. doi: 10.1016/s0140-6736(99)05140-5. [DOI] [PubMed] [Google Scholar]
- 4.Hirschberg D L, Moalem G, He J, Mor F, Cohen I R, Schwartz M. J Neuroimmunol. 1998;89:88–96. doi: 10.1016/s0165-5728(98)00118-0. [DOI] [PubMed] [Google Scholar]
- 5.Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, et al. J Neurosci. 2000;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher J, Levkovitch-Verbin H, Schori H, Yoles E, Butovsky O, Kaye J F, Ben-Nun A, Schwartz M. J Neurosci. 2001;21:136–142. doi: 10.1523/JNEUROSCI.21-01-00136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kipnis J, Yoles E, Porat Z, Cohen A, Mor F, Sela M, Cohen I R, Schwartz M. Proc Natl Acad Sci USA. 2000;97:7446–7451. doi: 10.1073/pnas.97.13.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitt D, Werner P, Raine C S. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 9.Schoepp D D, Jane D E, Monn J A. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 10.Dreyer E B, Zurakowski D, Schumer R A, Podos S M, Lipton S A. Arch Ophthalmol. 1996;114:299–305. doi: 10.1001/archopht.1996.01100130295012. [DOI] [PubMed] [Google Scholar]
- 11.Yoles E, Schwartz M. Arch Ophthalmol. 1998;116:906–910. doi: 10.1001/archopht.116.7.906. [DOI] [PubMed] [Google Scholar]
- 12.Aharoni R, Teitelbaum D, Sela M, Arnon R. Proc Natl Acad Sci USA. 1997;94:10821–10826. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoles, E., Hauben, E., Palgi, O., Agranov, E., Kuchroo, V. K., Cohen, I. R., Weiner, H. & Schwartz, M. (2001)J. Neurosci., in press. [DOI] [PMC free article] [PubMed]
- 14.Brauner-Osborne H, Hermit M B, Nielsen B, Krogsgaard-Larsen P, Johansen T N. Eur J Pharmacol. 2000;406:41–44. doi: 10.1016/s0014-2999(00)00625-7. [DOI] [PubMed] [Google Scholar]
- 15.Teitelbaum D, Meshorer A, Hirshfeld T, Arnon R, Sela M. Eur J Immunol. 1971;1:242–248. doi: 10.1002/eji.1830010406. [DOI] [PubMed] [Google Scholar]
- 16.Teitelbaum D, Fridkis-Hareli M, Arnon R, Sela M. J Neuroimmunol. 1996;64:209–217. doi: 10.1016/0165-5728(95)00180-8. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Nun A, Mendel I, Bakimer R, Fridkis-Hareli M, Teitelbaum D, Arnon R, Sela M, Kerlero de Rosbo N. J Neurol. 1996;243:S14–S22. doi: 10.1007/BF00873697. [DOI] [PubMed] [Google Scholar]
- 18.Webb C, Teitelbaum D, Arnon R, Sela M. Eur J Immunol. 1973;3:279–286. doi: 10.1002/eji.1830030506. [DOI] [PubMed] [Google Scholar]
- 19.Teitelbaum D, Aharoni R, Arnon R, Sela M. Proc Natl Acad Sci USA. 1988;85:9724–9728. doi: 10.1073/pnas.85.24.9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aharoni R, Teitelbaum D, Sela M, Arnon R. J Neuroimmunol. 1998;91:135–146. doi: 10.1016/s0165-5728(98)00166-0. [DOI] [PubMed] [Google Scholar]
- 21.Fridkis-Hareli M, Strominger J L. J Immunol. 1998;160:4386–4397. [PubMed] [Google Scholar]
- 22.Aharoni R, Teitelbaum D, Arnon R. Eur J Immunol. 1993;23:17–25. doi: 10.1002/eji.1830230105. [DOI] [PubMed] [Google Scholar]
- 23.Moalem G, Gdalyahu A, Shani Y, Otten U, Lazarovici P, Cohen I R, Schwartz M. J Autoimmun. 2000;15:331–345. doi: 10.1006/jaut.2000.0441. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz M, Moalem G, Leibowitz-Amit R, Cohen I R. Trends Neurosci. 1999;22:295–299. doi: 10.1016/s0166-2236(99)01405-8. [DOI] [PubMed] [Google Scholar]
- 25.Neuhaus O, Farina C, Yassouridis A, Wiendl H, Then Bergh F, Dose T, Wekerle H, Hohlfeld R. Proc Natl Acad Sci USA. 2000;97:7452–7457. doi: 10.1073/pnas.97.13.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde Y A. Nature (London) 1992;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 27.Yan Q, Elliott J, Snider W D. Nature (London) 1992;360:753–755. doi: 10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz M, Yoles E. Invest Ophthalmol Visual Sci. 2000;41:349–351. [PubMed] [Google Scholar]
- 29.Schwartz M, Yoles E. Curr Opin Ophthalmol. 2000;11:107–111. doi: 10.1097/00055735-200004000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Doble A. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 31.Grunblatt E, Mandel S, Youdim M B. J Neurol. 2000;247, Suppl. 2:II95–II102. doi: 10.1007/pl00022909. [DOI] [PubMed] [Google Scholar]
- 32.Osborne N N, Chidlow G, Nash M S, Wood J P. Curr Opin Ophthalmol. 1999;10:82–92. doi: 10.1097/00055735-199904000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Weinreb R N, Levin L A. Arch Ophthalmol. 1999;117:1540–1544. doi: 10.1001/archopht.117.11.1540. [DOI] [PubMed] [Google Scholar]
- 34.Chaudhary P, Ahmed F, Sharma S C. Brain Res. 1998;792:154–158. doi: 10.1016/s0006-8993(98)00212-1. [DOI] [PubMed] [Google Scholar]
- 35.Neufeld A H, Sawada A, Becker B. Proc Natl Acad Sci USA. 1999;96:9944–9948. doi: 10.1073/pnas.96.17.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kass M A, Gordon M O. Am J Ophthalmol. 2000;130:490–491. doi: 10.1016/s0002-9394(00)00658-9. [DOI] [PubMed] [Google Scholar]