VEGFR2 and Src Kinase Inhibitors Suppress Andes Virus-Induced Endothelial Cell Permeability (original) (raw)
Abstract
Hantaviruses predominantly infect human endothelial cells and, in the absence of cell lysis, cause two diseases resulting from increased vascular permeability. Andes virus (ANDV) causes a highly lethal acute pulmonary edema termed hantavirus pulmonary syndrome (HPS). ANDV infection enhances the permeability of endothelial cells in response to vascular endothelial growth factor (VEGF) by increasing signaling responses directed by the VEGFR2-Src-VE-cadherin pathway, which directs adherens junction (AJ) disassembly. Here we demonstrate that inhibiting pathway-specific VEGFR2 and Src family kinases (SFKs) blocks ANDV-induced endothelial cell permeability. Small interfering RNA (siRNA) knockdown of Src within ANDV-infected endothelial cells resulted in an ∼70% decrease in endothelial cell permeability compared to that for siRNA controls. This finding suggested that existing FDA-approved small-molecule kinase inhibitors might similarly block ANDV-induced permeability. The VEGFR2 kinase inhibitor pazopanib as well as SFK inhibitors dasatinib, PP1, bosutinib, and Src inhibitor 1 dramatically inhibited ANDV-induced endothelial cell permeability. Consistent with their kinase-inhibitory concentrations, dasatinib, PP1, and pazopanib inhibited ANDV-induced permeability at 1, 10, and 100 nanomolar 50% inhibitory concentrations (IC50s), respectively. We further demonstrated that dasatinib and pazopanib blocked VE-cadherin dissociation from the AJs of ANDV-infected endothelial cells by >90%. These findings indicate that VEGFR2 and Src kinases are potential targets for therapeutically reducing ANDV-induced endothelial cell permeability and, as a result, capillary permeability during HPS. Since the functions of VEGFR2 and SFK inhibitors are already well defined and FDA approved for clinical use, these findings rationalize their therapeutic evaluation for efficacy in reducing HPS disease. Endothelial cell barrier functions are disrupted by a number of viruses that cause hemorrhagic, edematous, or neurologic disease, and as a result, our findings suggest that VEGFR2 and SFK inhibitors should be considered for regulating endothelial cell barrier functions altered by additional viral pathogens.
Hantaviruses predominantly infect endothelial cells (ECs) and nonlytically cause diseases associated with dramatic increases in vascular permeability (12, 51, 54, 66, 82, 83, 98). Andes virus (ANDV) infection results in acute pulmonary edema and respiratory insufficiency termed hantavirus pulmonary syndrome (HPS) or hantavirus cardiopulmonary syndrome (HCPS) (7, 8, 12, 17, 19, 32, 47, 55, 57, 66, 68, 98). Endothelial cells within vast pulmonary capillary beds provide a primary means for ANDV infection to increase capillary permeability and cause pulmonary edema (7, 8, 32). Interendothelial cell adherens junctions (AJs) form a fluid barrier within capillaries that regulates permeability of the vascular endothelium (11, 53). However, endothelial cell AJs must dissociate in order to permit immune cell extravasation and repair of capillary damage, and thus, opposing signals regulate endothelial cell responses that control AJ disassembly (9, 11, 56). Maintaining vascular integrity is of fundamental importance for preventing edema, and as a result, vascular permeability is tightly regulated by redundant systems that act on a unique set of endothelial cell-specific receptors, AJ proteins, and signaling pathway effectors (11, 13, 20, 24, 90).
Acute pulmonary edema and hypoxia are hallmarks of HPS disease, and hypoxic conditions alone are capable of inducing acute pulmonary edema (5, 8, 12, 18, 32, 42, 47, 64, 66, 89). Hypoxia induces the expression of vascular endothelial growth factor (VEGF) within pulmonary endothelial cells, and VEGF was originally named vascular permeability factor for its ability to induce tissue edema (5, 10, 13, 14, 48, 59, 64, 70, 89). Secreted VEGF acts locally in an autocrine or paracrine manner to activate VEGFR2 receptors on endothelial cells, and VEGFR2 activation induces the internalization of VE-cadherin from AJs and paracellular permeability (11, 13, 15, 22, 23, 53). In fact, even small changes in vascular permeability result in large changes in fluid efflux within pressurized vessels (79). Intracellularly, VEGFR2-induced permeability is directed by Src/Rac/PAK signaling responses (23, 24, 64). Src family kinases (SFKs) are recruited to the cytoplasmic tails of VEGFR2 receptors and link VEGFR2-directed signaling responses to downstream pathway targets that induce changes in VE-cadherin and regulate interendothelial cell adherence. VEGFR2-Src pathway activation directs the disassembly of VE-cadherin from AJs and increases paracellular permeability of the endothelium, which results in edema (23, 34).
Hypoxia causes high-altitude pulmonary edema through the induction of permeabilizing VEGF responses (5, 42). HPS patients are acutely hypoxic, and hyperoxygenation of patients reduces HPS mortality (7, 8, 12, 32, 47, 66, 98). In vitro, endothelial cells are not permeabilized by hantavirus infection alone or by the addition of tumor necrosis factor alpha (TNF-α) to monolayers (27, 31, 44, 87). However, the permeability of endothelial cells infected by Hantaan virus (HTNV), ANDV, and New York 1 virus (NY-1V) is dramatically enhanced in response to VEGF days after infection and this is not observed following infection by nonpathogenic hantaviruses Tula virus (TULV) and Prospect Hill virus (PHV) (26, 27, 31).
VEGFR2-induced endothelial cell permeability is positively or negatively regulated by β3 integrins; Robo1/4, Edg-1, and Tie-2 receptors; and surface proteoglycans, syndecans, and galectin-3, which interact with VEGFR2 or downstream signaling pathways (6, 24, 40, 60, 63, 75-77, 79, 90). Pathogenic hantaviruses uniquely bind inactive αvβ3 integrin conformers, and this suggests a means for cell-associated hantaviruses to alter VEGFR2 signaling responses at late times postinfection (p.i.) (25, 26, 29, 31, 61, 74). VEGFR2 receptors form a complex with αvβ3 integrins that is required for endothelial cell migration (6), and Src is critical for this receptor synergy (75). Pathogenic hantaviruses bind αvβ3 and block αvβ3-directed endothelial cell migration in response to VEGF (27-29, 31, 61, 74). Paradoxically, knocking out β3 or inhibiting αvβ3 also promotes VEGFR2-directed signaling responses and enhances endothelial cell permeability (75). Consistent with this, ANDV-infected endothelial cells are hyperpermeabilized by VEGF (27). This results from hyperphosphorylation of VEGFR2 and the dissociation of VE-cadherin from the AJs of ANDV-infected endothelial cells (26, 31, 58). Thus, ANDV enhances the permeability of infected endothelial cells by augmenting responses of the VEGFR2-Src-VE-cadherin signaling pathway (26, 27, 31).
These findings suggest that ANDV-induced edema may be blocked by inhibiting VEGFR2- and SFK-directed pathway activation and that this edemagenic pathway provides several potential therapeutic targets for regulating ANDV-induced endothelial cell permeability. Although there are no effective therapeutics for treating HPS patients or hantavirus disease (41), VEGFR2 signaling pathways have been prominent targets of anticancer therapies (1-3, 37, 65, 71, 78, 86). These clinically available small-molecule kinase inhibitors may similarly regulate ANDV-induced permeability and have therapeutic utility against HPS.
In this study, we investigate whether FDA-approved small-molecule inhibitors which target VEGFR2 and SFK signaling responses (33, 43, 71, 85) are able to inhibit ANDV-induced endothelial cell permeability. Our findings demonstrate that small interfering RNA (siRNA) knockdown of Src inhibits ANDV-induced endothelial cell permeability, and this finding rationalized testing of small-molecule kinase inhibitors that target the VEGFR2-Src signaling pathway as inhibitors of ANDV-induced vascular permeability. Drugs which inhibit VEGFR2 or SFK responses (1-3, 37, 78, 86) were found to block ANDV-induced endothelial cell permeability. The SFK inhibitor dasatinib (2) blocked ANDV-induced permeability >70% at nanomolar concentrations, and these findings rationalize testing VEGFR2-Src inhibitors as potential therapeutics against hantavirus-induced disease. Since these molecules are already FDA approved for clinical use, they provide the potential for rapid evaluation and implementation in HPS patients.
MATERIALS AND METHODS
Cells and virus.
VeroE6 cells (ATCC CRL-1586) were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS; Sigma), penicillin (100 μg/ml), streptomycin sulfate (100 μg/ml), and amphotericin B (50 μg/ml) (Gibco). Human umbilical vein endothelial cells (HUVECs) were purchased from Cambrex Inc. and grown in endothelial cell basal medium 2 (EBM-2; Clonetics) supplemented with gentamicin (50 μg/ml), amphotericin B (50 μg/ml), and 10% FCS (Sigma). Andes virus (CHI-7913; ANDV) (61) was kindly provided by B. Hjelle (University of New Mexico), cultivated in a biosafety level 3 (BSL-3) facility as previously described (27), and determined to be mycoplasma free (Roche). ANDV titers were determined as previously described (25, 29). HUVEC monolayers were ANDV or mock infected at a multiplicity of infection (MOI) of 0.5, and cells were >90% infected with ANDV as determined by immunoperoxidase staining.
Immunoperoxidase staining of hantavirus-infected cells.
Rabbit polyclonal antinucleocapsid serum directed against the NY-1V nucleocapsid protein was used to detect ANDV-infected cells as previously described (29). Briefly, infected endothelial cell monolayers were fixed with 100% methanol and incubated with antinucleocapsid serum (1:5,000) followed by goat anti-rabbit-horseradish peroxidase (HRP) secondary antibody (1:5,000; Amersham Biosciences). Nucleocapsid protein-expressing cells were identified by staining with 3-amino-9-ethylcarbazole (0.026% in 0.1 M sodium acetate, pH 5.2, and 0.03% H2O2) and quantitated by microscopy (29).
VEGFR2 and Src inhibitors.
The VEGF receptor kinase inhibitor pazopanib {GSK-VEG10003; 5-[[4-[(2,3-dimethyl-2_H_-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzolsulfonamide} (37, 50, 71, 78) and the Src family kinase inhibitors dasatinib {Sprycel; _N_-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide monohydrate} (2, 39, 65), PP1 {pyrazolopyrimidine 1; pyrazolopyrimidine, 4-amino-5-(4-methylphenyl)-7-(_t_-butyl)pyrazolo[3,4-_d_]pyrimidine} (2, 3), Src inhibitor 1 {SKI-1; 4-(4′-phenoxyanilino)-6,7-dimethoxyquinazoline, 6,7-dimethoxy-_N_-(4-phenoxyphenyl)-4-quinazolinamine} (2), and bosutinib {SKI-606; 4-[(2,4-dichloro-5-methoxyphenyl) amino]-6-methoxy-7-[3-(4-methylpiperazin-1-yl) propoxy] quinoline-3-carbonitrile} (2, 93) were purchased from Selleck Chemicals and evaluated for endothelial cell cytotoxicity by trypan blue exclusion. Cytotoxicity for these FDA-approved drugs has been previously reported (33, 43, 71, 85), and inhibitor concentrations used were based on prior studies of drug cytotoxicity and Src inhibition studies. Briefly, serially diluted inhibitors were added to endothelial cells and after 24 h 0.4% trypan blue was added to cells. Cell viability was determined by quantifying the percentage of cells excluding trypan blue, and results were consistent with prior studies of these FDA-approved clinically available drugs (2, 3, 37, 39, 50, 65, 71, 78, 93). Concentrations of inhibitor resulting in <5% change in cell viability were used as maximum inhibitor concentrations in subsequent experiments.
Quantitative RT-PCR (qRT-PCR) and siRNA analysis.
Total cellular RNA was extracted from mock-infected or ANDV-infected HUVECs using the RNeasy minikit (Qiagen) (30, 69). Total RNA (1 μg) was reverse transcribed using the Transcriptor first-strand cDNA synthesis kit (Roche) using oligo(dT)18 primer (25°C for 10 min, 55°C for 30 min, and 85°C for 5 min). TaqMan primers (Applied Biosystems) were used to amplify human Src and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in duplicate using an Applied Biosystems 7300 PCR machine under the following thermocycling parameters: 50°C for 2 min, 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles (69). Src mRNA levels were normalized to GAPDH mRNA levels, and the fold changes in mRNA levels were compared between ANDV- and mock-infected cells (30, 69). The fold change in Src mRNA level was determined using the threshold cycle (2−ΔΔ_CT_) method and plotted (±standard error of the mean [SEM]) using GraphPad Prism 5 software (69).
Src knockdown experiments were performed using SureSilencing small interfering RNAs (siRNAs) against Src or negative-control siRNA (si-NEG2) purchased from SA Biosciences (69). Briefly, endothelial cells were transfected with siRNAs using SureFECT reagent (SA Biosciences). Total RNA was purified as described above and analyzed by qRT-PCR (30).
Endothelial cell permeability assay.
A previously described endothelial cell permeability assay was used to assess the effect of kinase inhibitors on ANDV-induced permeability (26, 27, 31). Briefly, human endothelial cells were plated on Costar Transwell plates (3-μm pores; Corning) and confluent monolayers were infected with pathogenic ANDV at an MOI of 0.5 or mock infected. Prior cytotoxicity of these FDA-approved agents has been previously reported (33, 43, 71, 85), and minimal cytotoxic concentrations of drugs which caused no increase in endothelial cell permeability by themselves were used in assays. Three days postinfection (p.i.), cells were pretreated with kinase inhibitors at noncytotoxic concentrations (33, 43, 71, 85) or mock treated (15 min) prior to the addition of 0.5 mg/ml of fluorescein isothiocyanate (FITC)-dextran and VEGF (100 ng/ml) to the upper chamber. Monolayer permeability was assayed by analyzing FITC-dextran present in the lower chamber (100 μl) at indicated times using a BioTek FLx800 fluorimeter (26, 27, 31). The fold change in FITC-dextran fluorescence intensity over that of mock-treated controls was used as a measure of the paracellular permeability of endothelial cell monolayers. Hantavirus infections were monitored by immunoperoxidase staining as described above (29, 61).
VE-cadherin internalization.
In order to monitor internalization of VE-cadherin, human endothelial cells were grown on gelatin-coated coverslips and were infected with pathogenic ANDV at an MOI of 0.5 or mock infected (31). Three days postinfection, endothelial cells were starved and pretreated for 15 min with the VEGFR2 inhibitor pazopanib (100 nM) or the SFK inhibitor dasatinib (1 nM) or untreated prior to VEGF addition (100 ng/ml, 30 min). Cells were incubated with an antibody to the extracellular domain of VE-cadherin (BV9; Clonetics) at 4°C for 1 h (23, 31). After antibody removal and washing, cells were incubated at 37°C (1 h) to permit intracellular trafficking of antibody-bound VE-cadherin. Cells were subsequently washed with a mild acid solution (2 mM phosphate-buffered saline [PBS]-glycine, pH 2.0; three times for 5 min each) (23, 31) in order to remove VE-cadherin antibody that was not internalized or were left untreated. Cells were paraformaldehyde fixed and Triton X-100 permeabilized prior to incubation with an FITC-tagged anti-mouse secondary antibody and examined on an Olympus IX51 microscope (31).
RESULTS
ANDV infects the endothelial cell lining of capillaries and results in patient hypoxia and acute pulmonary edema leading to respiratory distress (7, 12, 17, 32, 57, 66, 68, 98). Hypoxia transcriptionally induces VEGF, a permeabilizing growth factor that acts on endothelial cells and is 50,000 times more potent than histamine in causing edema (5, 10, 13-15, 18, 48, 89). In vitro, ANDV-infected endothelial cells are hyperresponsive to the permeabilizing effects of VEGF (26, 27, 31). Our recent findings indicate that ANDV infection of endothelial cells results in increased VEGFR2 phosphorylation, enhanced VE-cadherin internalization, and increased paracellular permeability (26, 31, 58). These findings suggest both a mechanism for ANDV-induced edema and a means for therapeutically targeting the VEGFR2-Src-VE-cadherin signaling pathway that directs these responses.
Src knockdown decreases ANDV-induced endothelial cell permeability.
The VEGFR2 cytoplasmic tail recruits Src kinases which activate a specific signaling pathway that results in VE-cadherin internalization and increased paracellular permeability of endothelial cells (23, 52). ANDV-infected endothelial cells are hyperpermeabilized by VEGF (27), suggesting that VEGFR2-Src signaling responses direct ANDV-induced permeability. Here we determine if knocking down Src blocks endothelial cell permeability induced by ANDV infection. We transfected endothelial cells with control or Src-specific siRNAs and assayed ANDV-induced endothelial cell permeability 3 days postinfection. Quantitative RT-PCR analysis indicates that endothelial cells transfected with Src siRNAs specifically reduced Src mRNA levels by ∼70% (Fig. 1 A). Endothelial cells similarly transfected with Src or control siRNAs were subsequently analyzed for their effect on ANDV-induced permeability (27, 69). We found that siRNAs to Src reduced the hyperpermeability of ANDV-infected endothelial cells 55 to 65% compared to control siRNA at all time points after VEGF addition (Fig. 1B). These findings suggest that ANDV-induced hyperpermeability occurs via a VEGFR2-Src pathway (23, 24) and that inhibiting Src is a viable mechanism for reducing ANDV-induced endothelial cell permeability. These findings further suggested that chemical inhibitors of VEGFR2 and Src kinases may similarly inhibit ANDV-induced endothelial cell permeability.
FIG. 1.
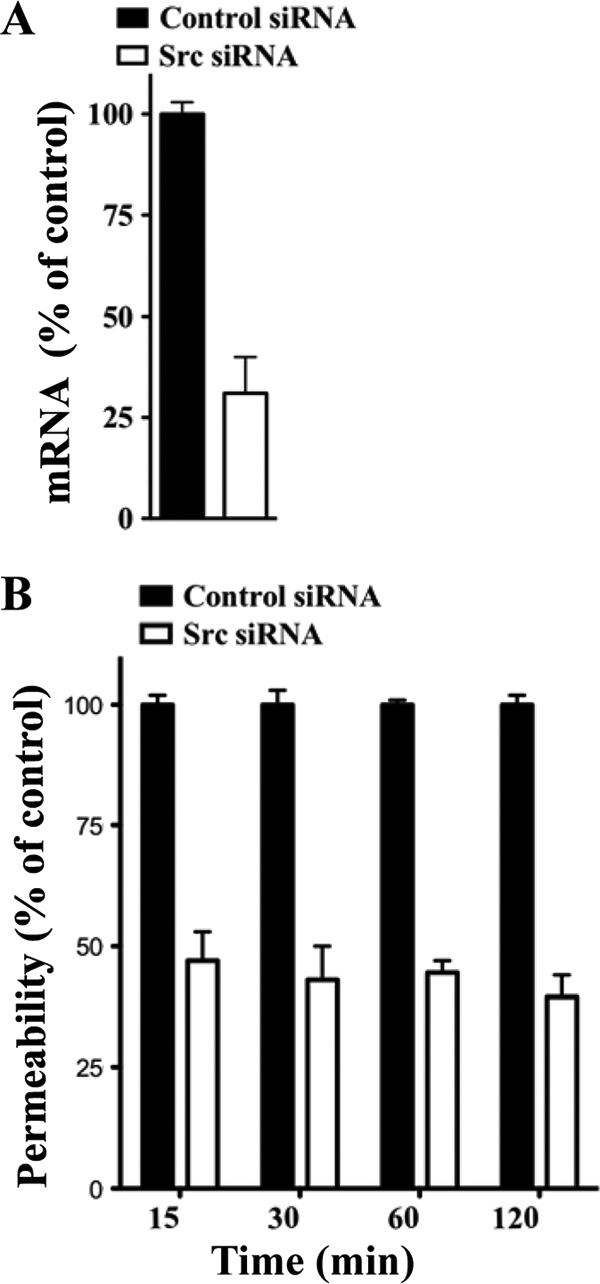
Src knockdown inhibits ANDV-induced EC permeability. (A) Endothelial cells were transfected with Src or scrambled siRNA, and Src mRNAs were analyzed by qRT-PCR (69). mRNA levels were quantitated and standardized to GAPDH mRNA levels and presented as a percentage of controls. (B) Endothelial cells were plated on Transwell inserts and infected at an MOI of 0.5 in triplicate with ANDV. One day postinfection (p.i.), cells were transfected with control or Src-specific siRNAs using SureFECT siRNA transfection reagent. Three days p.i., VEGF (100 ng/ml) and FITC-dextran were added to medium in the upper chamber, and the presence of FITC-dextran in the lower chamber was quantitated at indicated times (26, 27, 31). The percent change in FITC-dextran over controls is presented as a measure of EC monolayer permeability (27).
VEGFR2-Src pathway inhibitors reduce ANDV-induced endothelial cell permeability.
Since VEGF-induced vascularization is required for tumor growth, small molecules which inhibit VEGFR2 signaling responses are already in various stages of human clinical trials as anticancer therapies (2, 37, 65, 71, 78, 86). VEGFR2 signaling responses are inhibited by pazopanib (1, 37, 71, 78, 86), while Src activation is blocked by dasatinib, bosutinib, PP1, and Src inhibitor 1 (2, 65). Interestingly, compounds that target the VEGFR2-Src signaling pathway also have the potential to inhibit ANDV-induced permeability in vitro and reduce edema in HPS patients. Here we address the ability of commercially available drugs which inhibit VEGFR2-Src signaling responses to block ANDV-induced endothelial cell permeability.
Human endothelial cells were grown on Transwell plates and infected with ANDV for 3 days prior to assessment of monolayer permeability in response to VEGF (27). Endothelial cells were treated with increasing concentrations of potentially inhibitory compounds (2, 65, 78, 86), and the permeability of ANDV-infected endothelial cells was determined and compared to that of untreated controls (Fig. 2 A to D). Figure 2 defines concentrations of kinase inhibitors which block ANDV-induced EC permeability approximately 50% (IC50s). Dasatinib and PP1 had the lowest IC50s (1 and 10 nM, respectively), with IC50s of pazopanib and bosutinib being 100 nM and 10 μM, respectively (Fig. 2A to D).
FIG. 2.

IC50s of compounds that block ANDV-induced EC permeability. The concentrations of VEGFR2 and SFK inhibitors required to inhibit ANDV-induced endothelial cell permeability by 50% (IC50s) were determined. Endothelial cells were ANDV infected, and 3 days postinfection the permeability of cells in response to VEGF addition was determined (27) in the presence or absence of increasing amounts of kinase inhibitor. Endothelial cell permeability was determined as described for Fig. 1, and the permeability of ANDV- versus mock-infected controls was determined to be 100%. The effect of inhibitors is presented as the percentage of ANDV-induced permeability of inhibitor-treated monolayers 3 days postinfection and 30 min post-VEGF and FITC-dextran addition (27). Data are derived from at least three different experiments for each condition.
In Fig. 3, we assessed the abilities of VEGFR2 and SFK inhibitors to block ANDV-induced permeability at their IC50s from 15 to 60 min after VEGF addition. The Src family inhibitors dasatinib and PP1, which had the lowest IC50s of the kinase inhibitors tested, inhibited ANDV-induced endothelial cell permeability by 50 to 60% and 40 to 70%, respectively (Fig. 3B and D). The VEGFR2 kinase inhibitor pazopanib also inhibited ANDV-induced permeability by 40 to 60% at nanomolar IC50 levels (Fig. 3A). Src inhibitor 1 and bosutinib inhibited ANDV-induced permeability up to 40 to 50% but only at IC50s of 360 nM and 10 μM, respectively (Fig. 3C and E). Inhibitors blocked ANDV-induced endothelial cell permeability at IC50s consistent with their reported VEGFR2 or SFK inhibitory concentrations (2, 65, 71, 78, 86). These findings demonstrate that commercially available small-molecule VEGFR2 and SFK inhibitors block ANDV-induced permeability at nanomolar concentrations.
FIG. 3.

VEGFR2-Src inhibitors block ANDV-induced permeability. Endothelial cells were plated on vitronectin-coated Transwell inserts and infected at an MOI of 0.5 in triplicate with ANDV. Three days postinfection, the permeability of ANDV- and mock-infected endothelial cell monolayers was determined as described for Fig. 1 (27) at indicated times in the presence or absence of the kinase inhibitors pazopanib (A), dasatinib (B), bosutinib (C), PP1 (D), and Src inhibitor 1 (E). The percent change in FITC-dextran over controls is presented as a measure of EC monolayer permeability (27). Data are derived from two independent experiments performed in triplicate with comparable results.
Dasatinib and pazopanib prevent VE-cadherin internalization in ANDV-infected HUVECs.
The internalization of VE-cadherin from AJs is a downstream effect of VEGFR2-Src activation, and VE-cadherin internalization is increased by ANDV infection of endothelial cells (26, 31). Here we determined whether VEGFR2 and SFK inhibitors are capable of blocking VE-cadherin disassembly following ANDV infection of endothelial cells. We monitored VE-cadherin internalization using a previously described acid wash approach which removes antibody bound to extracellular VE-cadherin and permits quantitation of only intracellularly immunostained VE-cadherin (23, 31). We found that dasatinib and pazopanib inhibited ANDV-induced VE-cadherin internalization 65 to 70% and resulted in VE-cadherin localization nearly identical to that of mock-infected cells (Fig. 4). These data suggest that dasatinib and pazopanib inhibit the permeability of hantavirus-infected endothelial cells by blocking signaling responses that induce VE-cadherin internalization. These results further suggest that VEGFR2-Src signaling responses which direct AJ disassembly may be therapeutically targeted in order to inhibit hantavirus-induced endothelial cell permeability. Our findings rationalize testing of these existing, FDA-approved compounds for therapeutic efficacy in HPS animal models and clinical HPS cases.
FIG. 4.
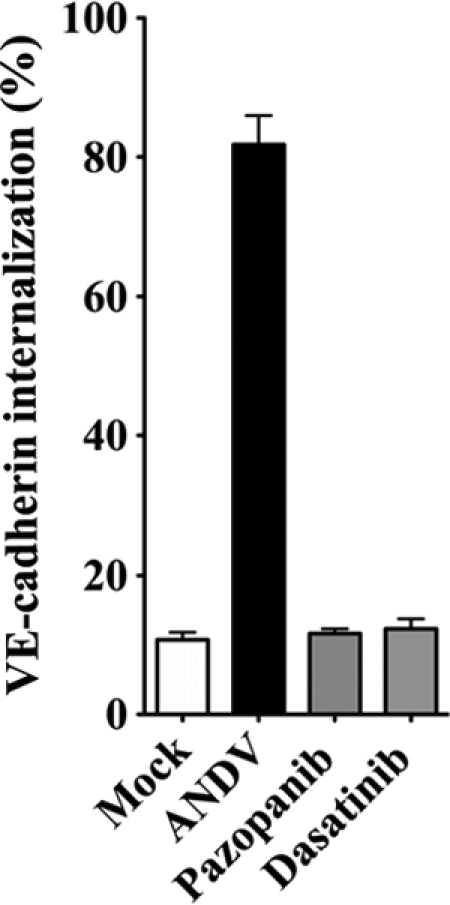
Dasatinib and pazopanib block VE-cadherin internalization in ANDV-infected HUVECs. Human endothelial cells were mock infected or infected with pathogenic ANDV at an MOI of 0.5. Three days postinfection, endothelial cells were pretreated with kinase inhibitors dasatinib (1 nM/liter) and pazopanib (100 nM/liter) or left untreated for 15 min prior to VEGF addition (100 ng/ml, 2 h). Cells were incubated with an antibody to the extracellular domain of VE-cadherin (BV9; Clonetics) at 4°C for 1 h (23, 26, 31). After antibody removal and washing, cells were incubated at 37°C in 5% CO2 (31) (1 h) to permit intracellular trafficking of antibody-bound VE-cadherin (23). Cells were subsequently washed with a mild acid solution (2 mM PBS-glycine, pH 2.0; three times for 5 min each) in order to remove VE-cadherin antibody that was not internalized or were left untreated (23, 26, 31). Cells were paraformaldehyde fixed and Triton X-100 permeabilized prior to incubation with an FITC-tagged anti-mouse secondary antibody and examined on an Olympus IX51 microscope. Data are presented as a percentage of cells with internalized VE-cadherin in ANDV-infected ECs pretreated only with VEGF (control) versus mock-treated infected cells (n = 500; P < 0.001) (31). Data represent the results from two independent experiments.
DISCUSSION
Pulmonary edema and hypoxia are hallmarks of HPS disease and linked to VEGFR2-directed responses of endothelial cells, which are the primary targets of hantavirus infection. Hypoxia induces the secretion of VEGF, a factor which is known for its potent ability to permeabilize capillaries and cause edema. In fact, patient hyperoxygenation has contributed to reducing HPS mortality to ∼40% (7, 8, 12, 32, 47, 66, 98). In vitro, endothelial cells infected by HFRS- and HPS-causing hantaviruses, but not nonpathogenic TULV, result in a dramatic enhancement of permeability in response to VEGF (26, 27, 31). ANDV-enhanced endothelial cell permeability results from the hyperactivation of VEGFR2 signaling pathways, VE-cadherin internalization, and the dissociation of AJs which normally maintain vascular integrity (26, 31, 58).
Here we demonstrate that clinically relevant FDA-approved small-molecule inhibitors of the VEGFR2 signaling pathway are able to block ANDV-induced endothelial cell permeability. Our results indicate that drugs which specifically target the VEGFR2 or Src kinases inhibit ANDV-induced endothelial cell permeability and the internalization of VE-cadherin from AJs. The observed decreases in endothelial cell permeability which we observed are likely to be clinically significant, since even a 2-fold change in permeability results in a dramatic change in fluid flux across pressurized vessels (20, 79, 91). Although there are currently no therapeutics for hantavirus-induced disease (41), these findings directly suggest therapeutic targets and approaches for reducing endothelial cell permeability caused by hantavirus infection.
Pazopanib (GSK-VEG10003) is a VEGF receptor-specific inhibitor which blocks VEGFR2 kinase activity at an IC50 of 30 nM (71, 78). Our findings indicate that pazopanib similarly inhibits ANDV-induced endothelial cell permeability at a nanomolar level and suggest that the VEGFR2 kinase may also be a therapeutic target for regulating hantavirus-induced capillary permeability. However, there are additional approaches for blocking VEGFR2 responses that should similarly be entertained (78). The use of the VEGF antibody bevacizumab (Avastatin), which prevents VEGFR2 activation, has been used clinically to inhibit tumor growth by blocking angiogenesis (78). Suntinib, sorafenib, and asitinib are additional drugs which specifically target the VEGFR2 kinase and have been used clinically to inhibit metastatic renal cell carcinoma (78). Each of these inhibitory drugs is currently used clinically for cancer therapy or is being evaluated in human clinical studies (33, 43, 71, 78, 85). Our findings rationalize testing of these kinase inhibitors for additional indications and clinical application in reducing hantavirus-induced edema.
These studies also demonstrate that FDA-approved drugs that are clinically used as SFK inhibitors are also capable of blocking ANDV-induced permeability. Dasatinib sensitively inhibits all SFK members, including c-Src, Lck, Fyn, and Yes, with an IC50 of 1.1 nM (39). Dasatinib is FDA approved (Sprycel) for clinical treatment of patients with chronic myeloid leukemia to reduce cancer cell growth (33, 43, 65, 72). SFK inhibitors also affect cancer progression by inhibiting angiogenesis and endothelial cell permeability (39, 45). Pharmacologic blockade of SFKs or knocking out Src suppresses tumor cell extravasation and stabilizes endothelial barrier function (39, 45). Our in vitro findings suggest that dasatinib may also have short-term therapeutic utility against hantavirus-induced endothelial cell permeability. Additional SFK inhibitors PP1, Src inhibitor 1, and bosutinib also inhibited ANDV-induced permeability at nanomolar to micromolar concentrations, respectively. As a result, dasatinib and other SFK inhibitors may be rationalized as potential inhibitors of HPS disease and tested in the Syrian hamster HPS disease model (35).
The SFK inhibitor PP1 has been shown to reduce vascular permeability and cerebral edema in response to stroke and to have protective effects on cardiovascular disease (45). PP1 reduced edema and myocardial injury following myocardial infarction via a mechanism that inhibits endothelial cell permeability (45). This is important for hantaviruses since HPS is also referred to as hantavirus cardiopulmonary syndrome (HCPS) for cardiogenic symptoms observed in hantavirus patients (8, 32, 47). The ability of SFK inhibitors to block endothelial cell permeability under ischemic conditions (95, 96) may further suggest the utility of this therapeutic approach in HPS/HCPS patients.
Interestingly, targeting SFKs may also reduce the induction of VEGF in response to hypoxia and thus downregulate secretion of the VEGFR2 permeability activator (5, 18, 48, 64, 70, 89). VEGF expression requires Src activation, and antisense knockdown of Src decreases VEGF expression induced by hypoxia (16, 22, 64). These findings further suggest that drugs which target SFKs may similarly downregulate VEGF induction of hypoxia-inducible factor 1α (HIF1α), which forms an autocrine loop that serves to further induce VEGF under hypoxic conditions (22, 23, 64, 89). One paper suggests that ANDV induces VEGF at early times postinfection, although the low-level early VEGF response suggested was transient and not demonstrated to contribute to permeability or to coincide with VEGF transcription (84). However, VEGF may be secreted from immune, endothelial, or epithelial cells in response to hypoxia and still act on endothelial cells (13, 14, 70, 89). Since HPS patients present with hallmark hypoxia, targeting HIF1α itself may be entertained as a means for reducing capillary permeability in HPS patients alone or in combination with VEGFR2 or SFK inhibitors.
Additional inhibitors of VEGFR2-directed permeability block ancillary signaling pathways that increase AJ stability directly (21, 24, 62, 90). Angiopoietin 1 (Ang-1) and sphingosine-1-phosphate (S1P) inhibit VEGFR2-directed permeability by binding to Tie-2 and Edg-1 receptors, respectively (21). In fact, like VEGF, Ang-1 is an endothelial cell-specific growth factor that enhances vascular stability and dominantly blocks VEGF-directed permeability (24, 27, 31, 38, 56, 90). We have previously shown that both Ang-1 and S1P inhibit ANDV- and HTNV-induced endothelial cell permeability (27, 31), and an S1P analog, FTY720, is already in human clinical studies (81). Collectively, our findings suggest several means for enhancing the capillary integrity of hantavirus-infected endothelial cells that may be considered therapeutically through the use of single or combined therapeutic approaches. Since VEGFR2 and SFK inhibitors are already FDA approved for use in humans, they could be immediately rationalized for use in clinical HPS cases.
Interestingly, the use of SFK inhibitors may be warranted for use against a number of infectious agents that impact vascular permeability, induce edema, or cause cerebral hemorrhage or edematous disease (4, 67, 92, 97). Dengue virus infects human endothelial cells and increases capillary permeability, resulting in hemorrhagic or edematous disease (4, 97). Several additional viruses alter endothelial cell barrier functions or blood-brain barrier (BBB) integrity, an effect that contributes to neurological symptoms at late times postinfection (46, 67, 73, 92, 97). Although with some viruses permeability may result from immune responses that impact the endothelium or BBB, VEGFR2 and SFK inhibitors which enhance endothelial cell integrity may still be of therapeutic utility in stabilizing the vasculature during these and other viral infections.
Studies performed here were not aimed at inhibiting ANDV entry or replication and instead analyzed the effect of inhibitors 3 days post-ANDV infection, when infected endothelial cells are hyperresponsive to VEGF (26, 27, 31). Although antiviral compounds that block virus entry or reduce viral replication may be prophylactic, these approaches may not be efficacious against HPS or hemorrhagic fever with renal syndrome (HFRS) (41, 94). Ribavirin and interferon, which prophylactically inhibit hantavirus replication, have little effect on hantavirus disease once patients are symptomatic (36, 41, 49, 80, 88). This is probably because hantavirus patients already have respiratory (HPS) or hemorrhagic (HFRS) symptoms when they seek medical attention and are at a point when continued hantavirus replication may no longer be required to cause disease (41). As a result, small-molecule inhibitors targeting cellular responses that contribute to disease may have a substantial advantage over antiviral approaches for these and other viruses with long disease onsets or which occur in the midst of high-level neutralizing antibodies. Further, the investigation of potential therapeutics which are already FDA approved for clinical use may provide for the rapid implementation of inhibitors with known functions in regulating specific aspects of viral disease or which may be combined with compounds which reduce disease by restricting inflammatory responses that also impact capillary integrity (20, 79, 91).
Acknowledgments
We thank Valery Matthys and Nadine Dalrymple for helpful discussions and critical review of the manuscript.
This work was supported by National Institutes of Health grants R01AI47873, PO1AI055621, R21AI1080984, and U54AI57158 (Northeast Biodefense Center [director, W. I. Lipkin]).
Footnotes
▿
Published ahead of print on 22 December 2010.
REFERENCES
- 1.Adjei, A. A. 2007. Novel small-molecule inhibitors of the vascular endothelial growth factor receptor. Clin. Lung Cancer 8(Suppl. 2)**:**S74-S78. [DOI] [PubMed] [Google Scholar]
- 2.Aleshin, A., and R. S. Finn. 2010. SRC: a century of science brought to the clinic. Neoplasia 12**:**599-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bain, J., et al. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408**:**297-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu, A., and U. C. Chaturvedi. 2008. Vascular endothelium: the battlefield of dengue viruses. FEMS Immunol. Med. Microbiol. 53**:**287-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger, M. M., et al. 2005. Hypoxia impairs systemic endothelial function in individuals prone to high-altitude pulmonary edema. Am. J. Respir. Crit. Care Med. 172**:**763-767. [DOI] [PubMed] [Google Scholar]
- 6.Borges, E., Y. Jan, and E. Ruoslahti. 2000. Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J. Biol. Chem. 275**:**39867-39873. [DOI] [PubMed] [Google Scholar]
- 7.Bustamante, E. A., H. Levy, and S. Q. Simpson. 1997. Pleural fluid characteristics in hantavirus pulmonary syndrome. Chest 112**:**1133-1136. [DOI] [PubMed] [Google Scholar]
- 8.Chang, B., M. Crowley, M. Campen, and F. Koster. 2007. Hantavirus cardiopulmonary syndrome. Semin. Respir. Crit. Care Med. 28**:**193-200. [DOI] [PubMed] [Google Scholar]
- 9.Corada, M., et al. 1999. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. U. S. A. 96**:**9815-9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dehler, M., E. Zessin, P. Bartsch, and H. Mairbaurl. 2006. Hypoxia causes permeability oedema in the constant-pressure perfused rat lung. Eur. Respir. J. 27**:**600-606. [DOI] [PubMed] [Google Scholar]
- 11.Dejana, E., F. Orsenigo, and M. G. Lampugnani. 2008. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121**:**2115-2122. [DOI] [PubMed] [Google Scholar]
- 12.Duchin, J. S., et al. 1994. Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. The Hantavirus Study Group. N. Engl. J. Med. 330**:**949-955. [DOI] [PubMed] [Google Scholar]
- 13.Dvorak, H. F. 2006. Discovery of vascular permeability factor (VPF). Exp. Cell Res. 312**:**522-526. [DOI] [PubMed] [Google Scholar]
- 14.Dvorak, H. F., L. F. Brown, M. Detmar, and A. M. Dvorak. 1995. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 146**:**1029-1039. [PMC free article] [PubMed] [Google Scholar]
- 15.Dvorak, H. F., et al. 1991. Distribution of vascular permeability factor (vascular endothelial growth factor) in tumors: concentration in tumor blood vessels. J. Exp. Med. 174**:**1275-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eliceiri, B. P., et al. 1999. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4**:**915-924. [DOI] [PubMed] [Google Scholar]
- 17.Enria, D., et al. 1996. Hantavirus pulmonary syndrome in Argentina. Possibility of person to person transmission. Medicina 56**:**709-711. [PubMed] [Google Scholar]
- 18.Forsythe, J. A., et al. 1996. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16**:**4604-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galeno, H., et al. 2002. First human isolate of Hantavirus (Andes virus) in the Americas. Emerg. Infect. Dis. 8**:**657-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamble, J. R., et al. 2000. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ. Res. 87**:**603-607. [DOI] [PubMed] [Google Scholar]
- 21.Garcia, J. G., et al. 2001. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 108**:**689-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavard, J. 2009. Breaking the VE-cadherin bonds. FEBS Lett. 583**:**1-6. [DOI] [PubMed] [Google Scholar]
- 23.Gavard, J., and J. S. Gutkind. 2006. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8**:**1223-1234. [DOI] [PubMed] [Google Scholar]
- 24.Gavard, J., V. Patel, and J. S. Gutkind. 2008. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 14**:**25-36. [DOI] [PubMed] [Google Scholar]
- 25.Gavrilovskaya, I. N., E. J. Brown, M. H. Ginsberg, and E. R. Mackow. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 73**:**3951-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gavrilovskaya, I. N., E. E. Gorbunova, and E. R. Mackow. 2010. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J. Virol. 84**:**4832-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavrilovskaya, I. N., E. E. Gorbunova, N. A. Mackow, and E. R. Mackow. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82**:**5797-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavrilovskaya, I. N., T. Peresleni, E. Geimonen, and E. R. Mackow. 2002. Pathogenic hantaviruses selectively inhibit beta3 integrin directed endothelial cell migration. Arch. Virol. 147**:**1913-1931. [DOI] [PubMed] [Google Scholar]
- 29.Gavrilovskaya, I. N., M. Shepley, R. Shaw, M. H. Ginsberg, and E. R. Mackow. 1998. Beta3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95**:**7074-7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geimonen, E., et al. 2002. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. U. S. A. 99**:**13837-13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbunova, E., I. N. Gavrilovskaya, and E. R. Mackow. 2010. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 84**:**7405-7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hallin, G. W., et al. 1996. Cardiopulmonary manifestations of hantavirus pulmonary syndrome. Crit. Care Med. 24**:**252-258. [DOI] [PubMed] [Google Scholar]
- 33.Hegedus, C., et al. 2009. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 158**:**1153-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes, K., O. L. Roberts, A. M. Thomas, and M. J. Cross. 2007. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19**:**2003-2012. [DOI] [PubMed] [Google Scholar]
- 35.Hooper, J. W., T. Larsen, D. M. Custer, and C. S. Schmaljohn. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289**:**6-14. [DOI] [PubMed] [Google Scholar]
- 36.Huggins, J. W., et al. 1991. Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J. Infect. Dis. 164**:**1119-1127. [DOI] [PubMed] [Google Scholar]
- 37.Iwamoto, F. M., et al. 2010. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 12**:**855-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain, R. K., and L. L. Munn. 2000. Leaky vessels? Call Ang1! Nat. Med. 6**:**131-132. [DOI] [PubMed] [Google Scholar]
- 39.Johnson, F. M., et al. 2010. Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer 116**:**1582-1591. [DOI] [PubMed] [Google Scholar]
- 40.Jones, C. A., et al. 2008. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat. Med. 14**:**448-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonsson, C. B., J. Hooper, and G. Mertz. 2008. Treatment of hantavirus pulmonary syndrome. Antiviral Res. 78**:**162-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaner, R. J., et al. 2000. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am. J. Respir. Cell Mol. Biol. 22**:**657-664. [DOI] [PubMed] [Google Scholar]
- 43.Kantarjian, H., E. Jabbour, J. Grimley, and P. Kirkpatrick. 2006. Dasatinib. Nat. Rev. Drug Discov. 5**:**717-718. [DOI] [PubMed] [Google Scholar]
- 44.Khaiboullina, S. F., D. M. Netski, P. Krumpe, and S. C. St. Jeor. 2000. Effects of tumor necrosis factor alpha on Sin Nombre virus infection in vitro. J. Virol. 74**:**11966-11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim, M. P., S. I. Park, S. Kopetz, and G. E. Gallick. 2009. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 335**:**249-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kortekaas, J., O. Ergonul, and R. J. Moormann. 2010. Interventions against West Nile virus, Rift Valley fever virus, and Crimean-Congo hemorrhagic fever virus: where are we? Vector Borne Zoonotic Dis. 10**:**709-718. [DOI] [PubMed] [Google Scholar]
- 47.Koster, F., et al. 2001. Rapid presumptive diagnosis of hantavirus cardiopulmonary syndrome by peripheral blood smear review. Am. J. Clin. Pathol. 116**:**665-672. [DOI] [PubMed] [Google Scholar]
- 48.Kourembanas, S., et al. 1998. Hypoxic responses of vascular cells. Chest 114**:**25S-28S. [DOI] [PubMed] [Google Scholar]
- 49.Krakauer, T., J. W. Leduc, J. C. Morrill, A. O. Anderson, and H. Krakauer. 1994. Serum levels of alpha and gamma interferons in hemorrhagic fever with renal syndrome. Viral Immunol. 7**:**97-101. [DOI] [PubMed] [Google Scholar]
- 50.Kumar, R., et al. 2007. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 6**:**2012-2021. [DOI] [PubMed] [Google Scholar]
- 51.Lahdevirta, J. 1982. Clinical features of HFRS in Scandinavia as compared with East Asia. Scand. J. Infect. Dis. Suppl. 36**:**93-95. [PubMed] [Google Scholar]
- 52.Lamalice, L., F. Le Boeuf, and J. Huot. 2007. Endothelial cell migration during angiogenesis. Circ. Res. 100**:**782-794. [DOI] [PubMed] [Google Scholar]
- 53.Lampugnani, M. G., and E. Dejana. 2007. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 120(Suppl. 2)**:**S1-S6. [DOI] [PubMed] [Google Scholar]
- 54.Lee, H. W. 1982. Hemorrhagic fever with renal syndrome (HFRS). Scand. J. Infect. Dis. Suppl. 36**:**82-85. [PubMed] [Google Scholar]
- 55.Levis, S., J. Rowe, S. Morzunov, D. Enria, and S. St. Jeor. 1997. New hantaviruses causing hantavirus pulmonary syndrome in central America. Lancet 349**:**998-999. [DOI] [PubMed] [Google Scholar]
- 56.London, N. R., K. J. Whitehead, and D. Y. Li. 2009. Endogenous endothelial cell signaling systems maintain vascular stability. Angiogenesis 12**:**149-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lopez, N., P. Padula, C. Rossi, M. E. Lazaro, and M. T. Franze-Fernandez. 1996. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology 220**:**223-226. [DOI] [PubMed] [Google Scholar]
- 58.Mackow, E. R., and I. N. Gavrilovskaya. 2009. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 102**:**1030-1041. [DOI] [PubMed] [Google Scholar]
- 59.Manalo, D. J., et al. 2005. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105**:**659-669. [DOI] [PubMed] [Google Scholar]
- 60.Markowska, A. I., F. T. Liu, and N. Panjwani. 2010. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 207**:**1981-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matthys, V. S., E. E. Gorbunova, I. N. Gavrilovskaya, and E. R. Mackow. 2010. Andes virus recognition of human and Syrian hamster beta3 integrins is determined by an L33P substitution in the PSI domain. J. Virol. 84**:**352-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McVerry, B. J., and J. G. Garcia. 2004. Endothelial cell barrier regulation by sphingosine 1-phosphate. J. Cell. Biochem. 92**:**1075-1085. [DOI] [PubMed] [Google Scholar]
- 63.Morgan, M. R., M. J. Humphries, and M. D. Bass. 2007. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 8**:**957-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mukhopadhyay, D., et al. 1995. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375**:**577-581. [DOI] [PubMed] [Google Scholar]
- 65.Nam, S., et al. 2005. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 65**:**9185-9189. [DOI] [PubMed] [Google Scholar]
- 66.Nolte, K. B., et al. 1995. Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum. Pathol. 26**:**110-120. [DOI] [PubMed] [Google Scholar]
- 67.Paddock, C. D., et al. 2006. Fatal hemorrhagic fever caused by West Nile virus in the United States. Clin. Infect. Dis. 42**:**1527-1535. [DOI] [PubMed] [Google Scholar]
- 68.Padula, P. J., et al. 1998. Hantavirus pulmonary syndrome outbreak in Argentina: molecular evidence for person-to-person transmission of Andes virus. Virology 241**:**323-330. [DOI] [PubMed] [Google Scholar]
- 69.Pepini, T., E. E. Gorbunova, I. Gavrilovskaya, J. E. Mackow, and E. R. Mackow. 2010. Andes virus regulation of cellular microRNAs contributes to hantavirus-induced endothelial cell permeability. J. Virol. 84**:**11929-11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pham, I., et al. 2002. Hypoxia upregulates VEGF expression in alveolar epithelial cells in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 283**:**L1133-L1142. [DOI] [PubMed] [Google Scholar]
- 71.Podar, K., et al. 2006. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc. Natl. Acad. Sci. U. S. A. 103**:**19478-19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porkka, K., et al. 2010. Dasatinib 100 mg once daily minimizes the occurrence of pleural effusion in patients with chronic myeloid leukemia in chronic phase and efficacy is unaffected in patients who develop pleural effusion. Cancer 116**:**377-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pulzova, L., M. R. Bhide, and K. Andrej. 2009. Pathogen translocation across the blood-brain barrier. FEMS Immunol. Med. Microbiol. 57**:**203-213. [DOI] [PubMed] [Google Scholar]
- 74.Raymond, T., E. Gorbunova, I. N. Gavrilovskaya, and E. R. Mackow. 2005. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc. Natl. Acad. Sci. U. S. A. 102**:**1163-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reynolds, A. R., et al. 2009. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 15**:**392-400. [DOI] [PubMed] [Google Scholar]
- 76.Reynolds, A. R., et al. 2004. Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 64**:**8643-8650. [DOI] [PubMed] [Google Scholar]
- 77.Reynolds, L. E., et al. 2002. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 8**:**27-34. [DOI] [PubMed] [Google Scholar]
- 78.Rini, B. I. 2009. Vascular endothelial growth factor-targeted therapy in metastatic renal cell carcinoma. Cancer 115**:**2306-2312. [DOI] [PubMed] [Google Scholar]
- 79.Robinson, S. D., L. E. Reynolds, L. Wyder, D. J. Hicklin, and K. M. Hodivala-Dilke. 2004. Beta3-integrin regulates vascular endothelial growth factor-A-dependent permeability. Arterioscler. Thromb. Vasc. Biol. 24**:**2108-2114. [DOI] [PubMed] [Google Scholar]
- 80.Rusnak, J. M., et al. 2009. Experience with intravenous ribavirin in the treatment of hemorrhagic fever with renal syndrome in Korea. Antiviral Res. 81**:**68-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanchez, T., et al. 2003. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 278**:**47281-47290. [DOI] [PubMed] [Google Scholar]
- 82.Schmaljohn, C. 2001. Bunyaviridae and their replication, p. 1581-1602. In D. M. Knipe et al. (ed.), Fields virology, 4th ed., vol. 1. Lipppincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 83.Schmaljohn, C., and B. Hjelle. 1997. Hantaviruses: a global disease problem. Emerg. Infect. Dis. 3**:**95-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shrivastava-Ranjan, P., P. E. Rollin, and C. F. Spiropoulou. 2010. Andes virus disrupts the endothelial cell barrier by induction of vascular endothelial growth factor and downregulation of VE-cadherin. J. Virol. 84**:**11227-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sleijfer, S., et al. 2009. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J. Clin. Oncol. 27**:**3126-3132. [DOI] [PubMed] [Google Scholar]
- 86.Sonpavde, G., T. E. Hutson, and C. N. Sternberg. 2008. Pazopanib, a potent orally administered small-molecule multitargeted tyrosine kinase inhibitor for renal cell carcinoma. Expert Opin. Invest. Drugs 17**:**253-261. [DOI] [PubMed] [Google Scholar]
- 87.Sundstrom, J. B., et al. 2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 75**:**6070-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tamura, M., H. Asada, K. Kondo, M. Takahashi, and K. Yamanishi. 1987. Effects of human and murine interferons against hemorrhagic fever with renal syndrome (HFRS) virus (Hantaan virus). Antiviral Res. 8**:**171-178. [DOI] [PubMed] [Google Scholar]
- 89.Tang, N., et al. 2004. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6**:**485-495. [DOI] [PubMed] [Google Scholar]
- 90.Thurston, G., et al. 2000. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 6**:**460-463. [DOI] [PubMed] [Google Scholar]
- 91.Thurston, G., et al. 1999. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 286**:**2511-2514. [DOI] [PubMed] [Google Scholar]
- 92.Verma, S., M. Kumar, U. Gurjav, S. Lum, and V. R. Nerurkar. 2010. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397**:**130-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vultur, A., et al. 2008. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol. Cancer Ther. 7**:**1185-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wahl-Jensen, V., et al. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81**:**7449-7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weis, S., et al. 2004. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest. 113**:**885-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weis, S. M., and D. A. Cheresh. 2005. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 437**:**497-504. [DOI] [PubMed] [Google Scholar]
- 97.Wu-Hsieh, B. A., Y. T. Yen, and H. C. Chen. 2009. Dengue hemorrhage in a mouse model. Ann. N. Y. Acad. Sci. 1171(Suppl. 1)**:**E42-E47. [DOI] [PubMed] [Google Scholar]
- 98.Zaki, S., et al. 1995. Hantavirus pulmonary syndrome: pathogenesis of an emerging infectious disease. Am. J. Pathol. 146**:**552-579. [PMC free article] [PubMed] [Google Scholar]