Nitroarachidonic Acid, a Novel Peroxidase Inhibitor of Prostaglandin Endoperoxide H Synthases 1 and 2 (original) (raw)
Abstract
Prostaglandin endoperoxide H synthase (PGHS) catalyzes the oxidation of arachidonate to prostaglandin H2. We have previously synthesized and chemically characterized nitroarachidonic acid (AANO2), a novel anti-inflammatory signaling mediator. Herein, the interaction of AANO2 with PGHS was analyzed. AANO2 inhibited oxygenase activity of PGHS-1 but not PGHS-2. AANO2 exhibited time- and concentration-dependent inhibition of peroxidase activity in both PGHS-1 and -2. The plot of _k_obs versus AANO2 concentrations showed a hyperbolic function with _k_inact = 0.045 s−1 and K i*app = 0.019 μm for PGHS-1 and _k_inact = 0.057 s−1 and K i*app = 0.020 μm for PGHS-2. Kinetic analysis suggests that inactivation of PGHS by AANO2 involves two sequential steps: an initial reversible binding event (described by Ki) followed by a practically irreversible event (K i*app) leading to an inactivated enzyme. Inactivation was associated with irreversible disruption of heme binding to the protein. The inhibitory effects of AANO2 were selective because other nitro-fatty acids tested, such as nitrooleic acid and nitrolinoleic acid, were unable to inhibit enzyme activity. In activated human platelets, AANO2 significantly decreased PGHS-1-dependent thromboxane B2 formation in parallel with a decrease in platelet aggregation, thus confirming the biological relevance of this novel inhibitory pathway.
Keywords: Arachidonic Acid, Cyclooxygenase (COX) Pathway, Enzyme Inhibitors, Lipid Oxidation, Nitric Oxide, Platelet
Introduction
Prostaglandin endoperoxide H synthase (PGHS)4 is a key enzyme of arachidonic acid (AA) metabolism catalyzing the dioxygenation of AA to prostaglandin G2 (PGG2) and the subsequent reduction of PGG2 to PGH2 (1–3). The final product of PGHS catalysis is metabolized to different products depending on the cell type (2, 4). Two isoforms of PGHS (PGHS-1 and -2) are found in mammalian tissues. PGHS-1 is constitutively expressed, whereas PGHS-2 is an inducible enzyme. Both isoforms are of pharmacological importance because they are targets for nonsteroidal anti-inflammatory drugs (5).
The unique structure of AA, a 20-carbon polyunsaturated fatty acid with four cis double bonds, enables it to be a precursor of potent signaling molecules, i.e. prostaglandins, thromboxanes, and isoprostanes via enzymatic and nonenzymatic oxidative pathways (2, 6–10). Nitrogen dioxide can react with AA in aqueous solution generating a complex mixture of products including cis-trans isomerization derivatives and nitrohydroxyarachidonate (AA(OH)NO2) (6, 11, 12). We have recently reported the synthesis and chemical characterization of nitroarachidonic acid (AANO2) caused by acidic NO2−-dependent nitration (13). Because AA has four _cis_-double bonds, eight positional nitroalkene isomers can be formed from the addition of a NO2 group to AA at the double bonds. Under our experimental conditions, we demonstrated the formation of four mono-nitrated nitroalkenes: 9-nitroeicosa-5,8,11,14-tetraenoic acid (9-AANO2), 12-nitroeicosa-5,8,11,14-tetraenoic acid (12-AANO2), 14-nitroeicosa-5,8,11,14-tetraenoic acid (14-AANO2), and 15-nitroeicosa-5,8,11,14-tetraenoic acid (15-AANO2) (13).
Inflammation contributes to chronic and acute diseases characterized by the production of cytokines, AA-derived eicosanoids and adhesion molecules, as well as reactive oxygen and nitrogen species (14). Inflammatory processes and polyunsaturated fatty acids are related by eicosanoids, which represent mediators and regulators of inflammatory processes formed from 20-carbon-length polyunsaturated fatty acids (14). Thus, nitration of the carbon chain of AA may divert the fatty acid from its normal metabolizing pathways yielding novel compounds with new biological properties different from AA or its oxidized products. We have previously shown peroxynitrite-induced PGHS-1 oxygenase inhibition, in a process stimulated by nitric oxide (•NO) caused by its reaction with AA-derived radicals (15). The aim of this study was to evaluate the capacity of AANO2 to modulate both peroxidase (POX) and oxygenase (COX) activities in PGHS-1 and PGHS-2.
EXPERIMENTAL PROCEDURES
Materials
Deuterated arachidonic acid (AA-_d_8) and 12-hydroxyeicosatetraenoic acid-_d_8 (12-HETE-_d_8) were obtained from Cayman Chemicals (Ann Arbor, MI). Arachidonic acid was purchased from Nu-Check Prep (Elysian, MN). Silica gel HF TLC plates were obtained from Analtech. The solvents used in syntheses were HPLC grade. H2O2, N,N,N′,_N′_-tetramethylphenylenediamine (TMPD), diethylenetriaminepentaacetic acid, TRIZMA base, sodium phosphate, and phenol were from Sigma. Nitrated oleic (OANO2) and linoleic (LNO2) acids were obtained from Bruce Freeman synthesized as described (16, 17). All of the other reagents were obtained at the highest purity available from standard supply sources.
Synthesis of AANO2
Synthesis, purification, and identification of AANO2 was performed as reported previously (13). Quantification was assessed by electrospray ionization MS/MS using AA-_d_8 as an internal standard. A mixture of AANO2 isomers was obtained: 12- and 15-AANO2 (23%), 9-AANO2 (55%), and 14-AANO2 (22%). No differences between batches were observed.
Enzyme Activity
PGHS-1 and PGHS-2 were prepared as described (1, 18, 19), and reconstitution of the apo-PGHS (less than 3% of the holoenzyme present) as well as the generation of the holo-PGHS-indomethacin complex were carried out as described previously (20). Both POX and COX activities were studied. For POX kinetics studies, purified PGHS-1 or -2 in 0.08 m Tris-HCl, 0.1% Tween 20, pH 8.0, were incubated at 4 °C with protoporphyrin IX (PPIX) for 15 min at a 1:1 molar ratio. Peroxidase activity was evaluated spectrophotometrically on a Shimadzu UV-240U by measuring the oxidation of TMPD at 611 nm (ϵ611 = 12,000 _M_−1 cm−1). Experiments were done at 25 °C in 50 mm phosphate buffer, pH 7.4, containing 250 μm TMPD and 300 μm H2O2 in a final volume of 400 μl (21–23). In all cases, nonenzymatic oxidation of TMPD was determined, and the results reported correspond to the difference between enzymatic and nonenzymatic activity. COX activity was evaluated by oxygen consumption at 37 °C in 50 mm phosphate buffer, pH 7.4, containing 500 μm phenol and 15 μm AA, using an OROBOROS O2k instrument (21, 24, 25). Inhibition by indomethacin (IM), a slow, tightly binding, selective inhibitor of COX, was assessed as a positive control for inhibition (15). For both POX and COX, the expressed results correspond to the initial rates of enzyme activity, expressed in mol of oxidized TMPD (POX activity) or mol of consumed O2 (COX activity)/min of reaction/mg of protein.
AANO2-mediated Inhibition of PGHS-1 and -2
The effect of AANO2 on both COX and POX activities was analyzed by using holo-PGHS-1 or -2 in 50 mm phosphate buffer, pH 7.4. Both COX and POX were evaluated in the presence of various amounts of AANO2 (0–30 μm). AANO2 was added immediately before substrate addition, and in all cases, vehicle (methanol) never exceeded 1% of the total reaction mixture. Controls were also performed throughout the experiments to check PGHS self-inactivation in stock solution.
To establish whether AANO2 (up to 110 μm) could act as a competitive inhibitor, COX activity was determined at saturating (110 μm), near Km (15 μm), and intermediate (30 and 70 μm) AA concentrations (4). To elucidate the inhibitory mechanisms in accordance to the classical schemes for time-dependent inhibition (26), PGHS-1 was preincubated with a fixed amount of AANO2 (0.2–10 μm) for various times (0–300 s), and then POX activity was determined. Because PGHS rapidly self-inactivates and to determine _k_obs values, progress curves for TMPD oxidation in the presence of various amounts of inhibitor are not appropriate. Thus, a plot for activity versus preincubation time was obtained for each inhibitor concentration, and data were fitted to a single exponential to obtain the _k_obs values (see supplemental Figs. 5S and 6S for PGHS-1 and -2, respectively).
Reversal of the effect of AANO2 on enzyme activity was also analyzed. Apo-PGHS-1 or -2 (3.5 μm) were incubated for 10 min with AANO2 (1 mm) or IM (1 mm); the samples were subjected to gel filtration using Micro Bio-Spin chromatographic columns and reconstituted with 1 equivalent of Fe3+-PPIX. POX as well as COX activities for holo-PGHS were determined after gel filtration. Alternatively, apo-PGHS was reconstituted with 1 equivalent of Fe3+-PPIX prior to gel filtration, in which case POX activity was determined before and after gel filtration.
Changes in Heme Caused by AANO2
Several approaches were made to determine whether AANO2-mediated inhibition of PGHS-1 involves the heme moiety. The efficiency of heme binding to apo-PGHS-1 and -2 was evaluated spectrophotometrically. The absorbance at 411 nm was determined before and after gel filtration for nontreated enzyme, as well as for enzyme preincubated with AANO2 or IM. Efficiency for heme binding to apo-PGHS-1 under the conditions described above is expressed as the ratio of Abs280/Abs411. Corrections were made for protein dilution after gel filtration. Also, we performed spectrophotometric heme titrations, in 100 mm Tris-HCl, pH 8.0, of apo-PGHS-1 (2 μm) treated for 10 min with AANO2 in a enzyme:AANO2 ratio of 1:5 or 1:18. Fe3+-PPIX was added in consecutive 0.2 μm aliquots up to a final concentration of 2 μm Fe3+-PPIX. Heme content after AANO2 reaction with the enzyme was confirmed by the pyridine hemochrome method (27). Holo-PGHS-1 (5 μm) was incubated with AANO2 (50 μm) for 10 min, followed by gel filtration to remove nonbound heme and inhibitor; then the remaining heme was determined. Briefly, spectrum of pyridine hemochromogen was performed with 70 μl of sample followed by the addition of 2 μl of 5 m KOH, 30 μl of pyridine and finally reduced with dithionite, as reported previously (27). The spectra were recorded after 2 min with the chromophore exhibiting maxima of absorbance at 556 nm (ϵ556 = 34.53 mm−1 cm−1). Heme content was determined using a calibration curve obtained using free protoporphyrin (y = 0.014_x_ + 0.111). Also, potential heme modifications induced by AANO2 were analyzed. To do this, free heme was analyzed with the pyridine hemochrome method before and after its reaction with AANO2 in a 1:5 ratio. Reduction of 15.6 μm Fe3+-PPIX-PGHS to Fe2+-PPIX-PGHS-1 was performed with dithionite as reported (28). An anaerobic condition to keep the enzyme in the reduced form throughout the experiment was obtained by extensive degassing of the buffer with argon. The spectrophotometer cuvette was sealed during the recordings. The absorbance spectra were recorded from 350 to 550 nm for 60 min, in the absence or presence of a 10-fold excess of AANO2. As a control, free protoporphyrin reduction and incubation with the nitro-fatty acid was also performed.
Effect of AANO2 on apo-PGHS-1 Trypsin Proteolysis
The effects of AANO2 on trypsin proteolysis were evaluated as described previously (29). Apo- or holo-PGHS-1 (2 μm) in 100 mm Tris-HCl, pH 8.0, were incubated for 5 min at 25 °C with AANO2 (400 μm) or IM (500 μm). Then trypsin (3.1 μm) was added for 30 s at 25 °C, and trypsinization was quenched by the addition 1.5 mm PMSF for 5 min at 4 °C. PGHS-1 aliquots (2 μg) were heated at 95 °C for 3 min in SDS sample buffer and loaded on a 10% polyacrylamide gel. Protein was stained with Coomassie Brilliant Blue R-250.
Formation of AA-derived Thromboxanes in Platelets
Platelets were isolated from human blood of healthy volunteers in accordance to previous reports (30). Briefly, platelets (2 × 108 cells·ml−1) were activated with thrombin (0.2 unit·ml−1) for 30 min at 37 °C in the presence of 1 mm CaCl2 in Tyrode's buffer (134 mm NaCl, 12 mm NaHCO3, 2.9 mm KCl, 0.34 mm Na2HPO4, 1.0 MgCl2, 10 mm Hepes, 5 mm glucose, pH 7.4). Experiments involving the effect of AANO2 on platelet activation included a 2-min preincubation of the nitro-fatty acid before thrombin addition. Then the lipids were extracted as reported (30) to analyze the formation of thromboxane B2 (TxB2) by LC-MS/MS analysis. Before extraction, 10 ng of 12-HETE-_d_8 was added as an internal standard. In parallel, platelet aggregation was evaluated as described (31).
RESULTS AND DISCUSSION
Inhibition of both COX and POX activities by AANO2 was firstly assessed as a function of nitro-fatty acid concentration (Fig. 1). For kinetics analysis, we used a mixture of AANO2 isomers (9-, 12-, 14-, and 15-AANO2) as reported previously by us (13). Whereas individual isomers exhibited POX inhibitory actions ranging from 30 to 50%, the isomer mixture at 10 μm caused 50% inhibition (supplemental Fig. 1S). AANO2 was able to inhibit POX activity of both PGHS-1 and -2 as well as COX activity in PGHS-1 (Fig. 1, A and B). However, COX activity in PGHS-2 was not affected by the presence of AANO2 (Fig. 1B, inset). Neither PGHS-1 (Fig. 1C and supplemental Fig. 2S) nor PGHS-2 (not shown) were able to use AANO2 as a substrate for COX or POX activity. The observed differences between both isoforms can be ascribed to the different requirements of hydroperoxides to be activated. Because PGHS-1 requires concentrations of hydroperoxides 10-fold higher than those of PGHS-2, PGHS-1 is easier to inhibit by peroxidase inhibitors (2, 32). PGHS-2 might have to get 10-fold higher inhibition of POX to observe an effect on the COX activity. Other nitroalkenes, i.e. LNO2 and OANO2, were unable to affect either PGHS-1 or PGHS-2 POX activities (supplemental Fig. 3S).
FIGURE 1.

Influence of AANO2 in POX and COX activities of PGHS-1 and -2. A and B, to analyze both POX (A) and COX (B) activities, the enzyme was incubated at 25 °C in 50 mm phosphate buffer, pH 7.4, in the presence or absence of AANO2 (0–30 μm). A, POX-dependent oxidation of TMPD (250 μm) with 300 μm H2O2 as substrate and 20 nm PGHS-1 was determined at 611 nm in the presence of increasing AANO2 concentration. B, a representative experiment of the effect of AANO2 (10 μm) on oxygen consumption is shown. The reaction was performed with 25 nm PGHS-1, 15 μm AA, 500 μm phenol, with or without 10 μm AANO2. The results shown are representative of at least four independent experiments. The insets correspond to the effect of AANO2 on PGHS-2. C, PGHS-1 was incubated at 25 °C in 50 mm phosphate buffer, pH 7.4, with AANO2 (50 μm) in the absence (dashed lines) or presence (solid lines) of the corresponding substrate; both COX and POX activities were determined as in A and B. The observed O2 consumption when AANO2 is added in the absence of substrate is due to vehicle addition (data not shown).
It has been shown that a reducing co-substrate (i.e. TMPD) is needed for POX activity (2). We tested whether AANO2 could reduce the enzyme to start a new catalytic cycle competing with TMPD, thus decreasing the observed POX activity. 15-Hydroperoxyeicosatetraenoic acid is a peroxidase substrate, and its reduction to 15-HETE was monitored (supplemental Fig. 4S). In the absence of a reducing co-substrate, no differences were observed in the conversion of 15-hydroperoxyeicosatetraenoic acid to 15-HETE with or without AANO2. However, when phenol was present, an increase in POX activity was observed in the control, whereas a similar extent of inhibition, compared with the TMPD experiments, was observed in the presence of AANO2 (supplemental Fig. 4S).
It is well established in the literature that competitive, rapid, and reversible as well as slow, tightly binding inhibitors of both isoforms of PGHS exist (33). To further characterize the mechanism of AANO2-mediated enzyme inhibition, we preincubated either PGHS-1 or -2 with AANO2 for different amounts of time followed by POX activity determination (supplemental Figs. 5S and 6S). Plots of remaining activity versus preincubation time were obtained for various amounts of AANO2 (0.2–15 μm); TMPD oxidation was fitted to first order decay curves from which we obtained the observed inhibition rate constants (_k_obs). Reversible slow binding inhibitors have a linear relationship between _k_obs and inhibitor concentration; in our case, we observed a hyperbolic dependence between _k_obs and AANO2 concentration for both PGHS-1 and PGHS-2 (Fig. 2). In agreement with previously reported kinetic models of two-step time-dependent inhibition (34), AANO2 reacts with the enzyme and establishes a binding equilibrium (formation of EI) followed by an enzyme modification through a second equilibrium leading to a new enzyme-inhibitor complex, E*I; the forward and reverse rate constant for E*I formation are given by _k_3 and _k_4, respectively (Table 1 and Scheme 1). The plots of _k_obs versus AANO2 concentration were fitted to the equations depicted in Scheme 1 to obtain the values for _k_3, _k_4, and Ki for the first and second steps in the reaction. For both isoforms, _k_4 was different from 0 but smaller compared with _k_3, indicating that AANO2 acts as a poorly reversible inhibitor of the enzyme. This is supported by double-reciprocal plots where 1/_k_obs varies linearly with 1/[AANO2] and intersects the y axis at values greater than zero. Overall, our data indicate that inactivation of PGHS by AANO2 involves two sequential steps: an initial reversible binding event (described by Ki) followed by a practically irreversible event (K i*app) (35). For irreversible or practically irreversible inactivators, _k_3 is termed _k_inact, and the term K i*app is defined as the apparent concentration of inhibitor required to reach half of the maximal rate of inactivation. For both enzyme isoforms, K i*app is smaller than Ki, explaining the slow onset of inhibition observed (Table 1). The overall potency of slow, tightly binding inhibitors is best estimated from the second order rate constant derived from _k_inact/Ki. AANO2 showed slightly greater efficiency for inhibition of PGHS-1 than PGHS-2; however, it displays the same maximal rate of inactivation (as indicated in _k_3) and affinity (as indicated in K i*app) for both isoforms (Table 1). Our kinetic analysis indicates that AANO2 inhibits PGHS-1 and -2 in a kinetically similar way as the reported most potent COX inhibitors (36), through a slow, tightly binding scheme that leads to the formation of a stable binary complex where the dissociation rates of enzyme-inhibitor complexes are so slow that AANO2 appears to be practically irreversible. If AANO2 is formed in vivo (during platelet or macrophage activation) membrane concentrations in the nanomolar range would be enough to potently inhibit PGHS activity.
FIGURE 2.

Time dependent inhibition of PGHS-1 and PGHS-2. PGHS-1 (A) and PGHS-2 (B) were preincubated from 0 to 300 s with different amounts of AANO2 (0.2–15 μm). The remaining POX activity was determined and plotted versus preincubation time where data were fit to a single exponential to obtain _k_obs values. The hyperbolic dependence of _k_obs with [AANO2] follows kinetics for time-dependent enzyme isomerization (34). The data shown correspond to the means ± S.E. with n = 4. Insets, 1/_k_obs versus 1/[AANO2] plot suggesting the occurrence of a two-step mechanism of inhibition. The 1/_k_obs axis intercept higher than 0 also suggests that this is a two-step irreversible inhibition; however, true irreversible inhibition is difficult to differentiate from practically irreversible inhibition when _k_4 is not equal to 0.
TABLE 1.
Kinetic data for the observed AANO2-mediated PGHS inhibition
The observed linear relationship of the 1/_k_obs versus 1/[AANO2] plot observed in Fig. 2 suggests the occurrence of reversible binding of the inhibitor to the enzyme (Ki) followed by practically irreversible binding (_k_3 ≪ _k_4). Because K i*app is ≪ Ki, E*I formation is the limiting step for AANO2-dependent inhibition of PGHS.
| Kinetic parameter | PGHS-1 | PGHS-2 |
|---|---|---|
| _k_3 (_k_inact) | 4.5 × 10−2 s−1 | 5.7 × 10−2 s−1 |
| _k_4 | 8.4 × 10−4 s−1 | 7.2 × 10−3 s−1 |
| Ki | 1.02 μm | 1.76 μm |
| K i*app | 0.019 μm | 0.20 μm |
| Efficiency (_k_inact/Ki) | 4.4 × 104m−1 s−1 | 3.2 × 104m−1s−1 |
SCHEME 1.
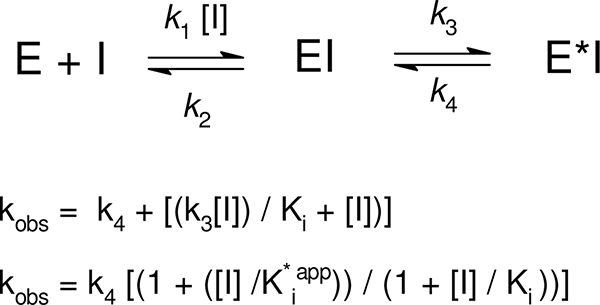
We next compared the inhibitory effect of AANO2 with the slow, tightly binding inhibitor IM. It was previously reported that IM protects apo-PGHS-1 from limited proteolysis induced by trypsin (29); under our experimental conditions, and in contrast to IM, AANO2 did not affect apo-PGHS-1 proteolysis as determined by electrophoresis experiments (Fig. 3A), suggesting that AANO2 interacts differently with PGHS compared with IM. nonsteroidal anti-inflammatory drugs, including IM, bind to the COX active site and inhibit oxygen consumption without affecting the POX activity (36). To determine whether AANO2 binds the COX active site, either PGHS-1 or PGHS-2 was incubated with AANO2 in the presence or absence of a saturating concentration of IM. As expected, IM did not affect POX activity. However, POX activity was inhibited to a similar extent by AANO2 either with or without IM blocking the access to the COX active site (Fig. 3, B and C). Moreover, and in accordance to the results in Fig. 3B, uncompetitive inhibition of PGHS-1 COX activity was observed at concentrations below 110 μm AANO2 (Fig. 3D). Similar results were obtained for PGHS-2 (not shown). Overall, our results indicate that the observed inhibition of both isoforms is not due to a significant reactivity of the nitro-fatty acid at the COX active site. Then AANO2 damage on the apoenzyme was evaluated; apo-PGHS-1 was preincubated with AANO2 or IM, excess inhibitor was removed by gel filtration, and the apoenzyme was reconstituted to holoenzyme with Fe+3-PPIX. Enzyme activity was not affected by incubation with IM, but preincubation of the enzyme with AANO2 led to the inactivation of PGHS-1, as evaluated by POX and COX activities (Table 2). The observed inhibition on PGHS activity was accompanied by a large decrease in the efficiency of the binding of Fe+3-protoporphyrin IX to the apoprotein (Table 2), supporting the possibility that AANO2 is reacting with both the apo- and holoenzyme.
FIGURE 3.

AANO2 binding to PGHS is not at the COX active site. A, apo-PGHS-1 (2 μm) in 100 mm Tris-HCl, pH 8.0, was incubated for 5 min at 25 °C with or without AANO2 (400 μm) or IM (500 μm). Then trypsin (final concentration, 3.1 μm) was added at 25 °C for 30 s. Trypsinization was quenched by the incubation at 4 °C for 5 min with 1.5 mm PMSF. PGHS-1 aliquots (2 μg) were heated at 95 °C for 3 min in SDS sample buffer and loaded on a 10% polyacrylamide gel. The protein was stained with Coomassie Blue. B and C, PGHS-1 (B) and PGHS-2 (C) were incubated with 200 μm IM to block the active site as explained under “Experimental Procedures” (29). Then the effects of 30 μm AANO2 were analyzed and compared with the condition without IM; the nitro-fatty acid was preincubated (prein.) for 2 min with the IM-treated or untreated enzyme or added prior to the initiation of the catalytic reaction. In all cases, POX activity compared with control was determined as previously. The results shown are representative of three independent experiments, each in triplicate. *, p < 0.05 compared with control and enzyme plus IM conditions. D, the inhibitory effect of AANO2 (0–60 μm) was evaluated by oxygen consumption at 25 °C in 50 mm phosphate buffer, pH 7.4. The results correspond to the means, n = 4 representative of at least three independent experiments.
TABLE 2.
AANO2 inhibits enzyme activity concomitant to heme release
Apo-PGHS-1 (3.5 μm) was incubated in 100 mm phosphate buffer, pH 7.4, for 10 min with 1 mm AANO2 or IM. The samples were filtered using Micro Bio-Spin chromatographic columns and reconstituted with 0.5 equivalents of Fe3+-PPIX. POX as well as COX activities for holo-PGHS-1 were determined as described previously. To determine differences in heme content due to AANO2, the absorbance spectra were collected from 250 to 500 nm for each sample before (i) and after (f) gel filtration, and the ratio (Abs411f/Abs411i) was determined. To determine differences in heme content due to AANO2, the absorbance spectra were collected from 250 to 500 nm for each sample before and after gel filtration, and the ratio Abs280/Abs411 was determined. ND, not determined.
| Condition | POX activity | COX activity | Abs280/Abs411 |
|---|---|---|---|
| μ_mol TMPDox/min/mgprot_ | μ_mol O2/min/mgprot_ | ||
| Control | 27.44 ± 1.00 | 71.98 ± 0.086 | 0.97 |
| + AANO2 | 1.08 ± 0.14 | ∼0 | 4.31 |
| + IM | 24.00 ± 0.44 | ND | 1.20 |
Both PGHS-1 and -2 are homodimers of 70 kDa whose dimerization is required for structural integrity and catalytic activity. Each subunit contains a molecule of Fe+3-protoporphyrin IX noncovalently attached to the enzyme; the heme group is essential for both enzyme activities (2). It was recently reported that nitroalkenes are irreversible inhibitors of xanthine oxidase activity because of its reaction with metal centers in the enzyme (37). In view of the results shown in Table 2, we decided to study whether heme is affected by AANO2. As shown in Fig. 4A, after the addition of AANO2 to the holoenzyme, there was a decrease in the absorbance of the heme group at 411 nm. This decay was observed in both isoforms of the enzyme but was faster for PGHS-1 than PGHS-2 (Fig. 4A). The reaction of the AANO2 with the enzyme resulted in the release of the heme moiety from the enzyme in parallel to the AANO2-induced enzyme inhibition. To confirm that AANO2-treated PGHS has a decreased heme content compared with control enzyme, we evaluated total heme content using the pyridine hemochrome method (27). In agreement with Fig. 4A, treatment of PGHS-1 with 10 equivalents AANO2 resulted in a 2-fold decrease in heme content (Fig. 4B). This is not due to a direct reaction of the nitro-fatty acid with heme; reaction of free protoporphyrin with AANO2 did not result in any changes of the spectra of heme by the pyridine hemochrome method (Fig. 4B, inset). Because the nitro-fatty acid reacts with the apo enzyme, heme incorporation was also evaluated (Fig. 4C). Titration of apo-PGHS-1 with heme aliquots leads to an increase in the absorbance at 411 nm. As observed in Fig. 4C, if the apoenzyme was previously incubated with AANO2, heme incorporation was less efficient compared with the control condition. Moreover, this was dependent on the apoenzyme:AANO2 ratio, confirming that AANO2 is modifying the enzyme preventing heme incorporation. HPLC and MS studies in addition to spectrophotometric analysis of either free or PGHS-released heme did not show any covalent modifications induced by AANO2 (not shown). In the case of apoenzyme reacting with AANO2, heme incorporation to PGHS-1 and PGHS-2 was prevented to similar extents (Fig. 4D).
FIGURE 4.

AANO2 displaces the heme from PGHS-1. A, to determine whether there were changes in the heme moiety caused by AANO2, 1 μm holo-PGHS-1 (□) or PGHS-2 (♦) were incubated with 30 μm AANO2, and absorbance at 411 nm was recorded for 4 min. B, determination of heme content of AANO2-treated PGHS-1 by the pyridine hemochrome method. PGHS-1 (5 μm) was preincubated with AANO2 for 10 min and gel-filtrated to remove excess inhibitor and released heme; spectrum of pyridine hemochromogen was performed as explained under “Experimental Procedures.” The chromophore was estimated to be 4.5 and 2.3 μm for the untreated and AANO2-treated enzyme, respectively. The inset shows the spectrum of pyridine hemochromogen of 10 μm free heme (full line) or 10 μm free heme preincubated with 50 μm AANO2 for 5 min (dashed line), showing no differences between both conditions. C, apo-PGHS-1 (2 μm, □) was treated with 5 (■) or 18 (▴) equivalents of AANO2 for 10 min at 25 °C in 100 mm Tris-HCl, pH 8.0. Then consecutive additions of 0.2 μm Fe3+-PPIX were performed, and absorbance at 411 nm was determined. D, apo-PGHS-1 and -2 were incubated with AANO2 for 5 min and then reconstituted with Fe3+-PPIX as described under “Experimental Procedures.” Components under 5,000 Da were separated as previously, and spectra between 200 and 500 nm for the retained and eluted fractions were obtained. The ratio Abs280/Abs411 was determined as an index of heme incorporation. In all cases, the data shown are representative of at least three independent experiments.
We also evaluated the reactivity of AANO2 with the reduced form of the holo-PGHS-1 (Fig. 5). Reduction of hexa-coordinate Fe3+-PPIX-PGHS to the penta-coordinate Fe2+-PPIX-PGHS (28) shifts the Soret band from ∼407 nm to a new maximum at 426 nm (Fig. 5). When incubated with the nitro-fatty acid, there was a time-dependent disappearance of the 426-nm peak concomitant to the formation of a new peak at 412 nm. This was identical to the observed peak for the reduced form of the free protoporphyrin (Fig. 5). Moreover, the spectrum of the reduced form of free protoporphyrin was not affected by the presence of AANO2, suggesting that the nitro-fatty acid is not reacting with the heme (supplemental Fig. 7S). Controls in the absence of AANO2 showed a small decrease in the maximum of absorbance intensity over time for Fe2+-PPIX-PGHS, confirming that AANO2 is reacting with the reduced heme. Overall, these results confirm that AANO2 is releasing the heme moiety from the holoenzyme in its intact form. Heme can be reversibly removed from the enzyme and reattached with little loss in enzyme activity (2). To evaluate whether POX activity in AANO2-treated holo-PGHS can be recovered by the addition of heme, we preincubated the nitro-fatty acid with holo-PGHS-1, added 1 equivalent of Fe+3-PPIX, and incubated for 0.5–4 h; then the remaining POX activity was determined. No recovery of POX activity was observed (Fig. 6). This result, in addition to Fig. 4C, suggests that AANO2 exerts a modification on protein structure/folding, preventing full reconstitution of the active form of the enzyme. Nitroalkenes are potent signaling molecules because of their capacity to covalently modify proteins (38–41). However, LNO2 and OANO2 were unable to inhibit enzyme activity (supplemental Fig. 3S). Thus, ongoing studies are aimed to determine the nature of the modification mediated by AANO2 that could explain the release of heme and prevention of its reattachment to the enzyme.
FIGURE 5.

AANO2 induces heme release from Fe2+-PPIX-PGHS-1. Reduction of resting PGHS-1 (Fe3+-PPIX-PGHS-1) to the pentacoordinate species Fe2+-PPIX-PGHS-1 was obtained by reduction with dithionite (shift of the Soret peak to 426 nm). The reduced form of the enzyme (15.6 μm, solid lines) was incubated at 25 °C in 50 mm phosphate buffer, pH 7.4, in the absence (time, 0 min) or presence of 150 μm AANO2, under anaerobic conditions. The spectrum of the enzyme was continuously recorded from 350 to 550 nm for 60 min. In addition, spectra of the free Fe2+-PPIX (10 μm, dashed line) were determined. Inset, the reduced holoenzyme was not reoxidized during the experiment. The dashed line corresponds to the Fe3+-PPIX-PGHS spectrum.
FIGURE 6.

AANO2 irreversibly inhibits PGHS-1 preventing heme incorporation. Holo-PGHS-1 was incubated as in Fig. 4A, components under 5,000 Da were separated as described, and then 1 equivalent of Fe3+-PPIX was added for different amounts of time. Peroxidase activity was determined for each condition and plotted as a function of incubation time (n = 3). ctrl, control.
To evaluate the potential relevance of enzyme inhibition in vivo, the capacity of AANO2 to modulate PGHS-1 activity in cells was evaluated in platelets. Human washed platelets were isolated and activated with thrombin in the presence or absence of AANO2; lipid extraction was performed, and TxB2 formation was analyzed by LC-MS/MS (Fig. 7A). Thrombin induced an increase of TxB2 that was reduced dose-dependently by AANO2. The nitro-fatty acid itself did not produce any change in TxB2 production by nonactivated platelets (Fig. 7A). In the presence of IM, we observed a similar decrease in TxB2 formation, suggesting that AANO2 is inhibiting PGHS-1 activity in platelets. These results were paralleled by the dose-dependent inhibition (IC50 = 1.3 μm) of platelet aggregation by AANO2 (Fig. 7B and supplemental Fig. 8S). IM, as reported, had no effect on platelet aggregation (42, 43). Overall, our results show that AANO2 critically modulates enzyme activity in human platelets.
FIGURE 7.

AANO2 inhibits PGHS-1 in activated platelets. Washed platelets (2 × 108 cells·ml−1) were preincubated with 1 mm Ca+2 and AANO2 (5 and 7.5 μm) or IM (7.5 μm) for 2 min at 37 °C; then thrombin (0.2 units·ml−1) was added and incubated for 30 min. Controls without thrombin were also performed. Lipid extracts were obtained from organic extraction with 12-HETE-_d_8 (10 ng) as an internal standard. TxB2 formation, as an index of PGHS-1 activity, was analyzed by LC-MS/MS studies following m/z 369/169 transition in the negative ion mode. The results shown are representative of three independent experiments. *, p < 0.05 compared with nonactivated platelets; #, p < 0.05 compared with AANO2 and IM conditions. B, in parallel, platelet aggregation induced by thrombin was also evaluated as described (31).
In summary, we demonstrated that AANO2 inhibited POX activity in both isoforms of the enzyme being a novel poorly reversible POX inhibitor of PGHS-2. Kinetic analysis showed that nitro-fatty acid inhibition was due to a slow, tightly binding mechanism. The most likely reaction mechanism is proposed (Fig. 8) involving the release of heme as a result of AANO2 reaction with the protein, being different from those reported for other well known enzyme inhibitors, e.g. IM (44, 45). The observed effects of AANO2 in platelet function suggest potential physiological and pharmacological relevance under inflammatory conditions.
FIGURE 8.

Proposed mechanism for AANO2-mediated PGHS inhibition. In accordance to kinetic analysis, we propose a reaction of AANO2 (I) with the protein (E) leading to the formation of an enzyme-inhibitor complex (EI). Then in the rate-limiting step of this process, the enzyme-inhibitor complex loses the heme moiety, yielding the functionally inactive form of the enzyme (E*I). The structure of the enzyme was adapted from (5).
Acknowledgments
We thank Dr. Bruce A. Freeman, University of Pittsburgh, for the generous gift of LNO2 and OANO2 and Dr. Carlos Batthyány (Institute Pasteur, Montevideo-Uruguay) and Dr. Gonzalo Peluffo (Universidad de la República-Uruguay) for helpful discussion and technical assistance.
*
This work was supported, in whole or in part, by National Institutes of Health Grant GM15431 (to L. J. M.). This work was also supported by Fondo Clemente Estable-ANII Grant FCE_516 (to A. T.), Programa de Desarrollo Tecnológico-ANII (to H. R.), Wellcome Trust (to H. R. and V. O. D.), and Howard Hughes Medical Institute (to R. R.). L. B. was partially supported by a fellowship from Sistema Nacional de Becas-ANII.
4
The abbreviations used are:
PGHS
prostaglandin H synthase
PPIX
protoporphyrin IX
AA
arachidonic acid
AANO2
nitroarachidonic acid
COX
cyclooxygenase activity
POX
peroxidase activity
TMPD
N,N,N′,_N′_-tetramethylphenylenediamine
TxB2
thromboxane B2
IM
indomethacin
PG
prostaglandin
HETE
hydroxyeicosatetraenoic acid
MS/MS
tandem mass spectrometry.
REFERENCES
- 1.Marnett L. J., Rowlinson S. W., Goodwin D. C., Kalgutkar A. S., Lanzo C. A. (1999) J. Biol. Chem. 274, 22903–22906 [DOI] [PubMed] [Google Scholar]
- 2.Rouzer C. A., Marnett L. J. (2003) Chem. Rev. 103, 2239–2304 [DOI] [PubMed] [Google Scholar]
- 3.van der Donk W. A., Tsai A. L., Kulmacz R. J. (2002) Biochemistry. 41, 15451–15458 [DOI] [PubMed] [Google Scholar]
- 4.Smith W. L., DeWitt D. L., Garavito R. M. (2000) Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 5.Malkowski M. G., Ginell S. L., Smith W. L., Garavito R. M. (2000) Science. 289, 1933–1937 [DOI] [PubMed] [Google Scholar]
- 6.Balazy M., Iesaki T., Park J. L., Jiang H., Kaminski P. M., Wolin M. S. (2001) J Pharmacol. Exp. Ther. 299, 611–619 [PubMed] [Google Scholar]
- 7.Di Rosa M., Ialenti A., Ianaro A., Sautebin L. (1996) Prostaglandins Leukot. Essent Fatty Acids. 54, 229–238 [DOI] [PubMed] [Google Scholar]
- 8.Goodwin D. C., Landino L. M., Marnett L. J. (1999) FASEB J. 13, 1121–1136 [DOI] [PubMed] [Google Scholar]
- 9.Marnett L. J. (2002) Prostaglandins Other Lipid Mediat. 68–69, 153–164 [DOI] [PubMed] [Google Scholar]
- 10.Salvemini D., Currie M. G., Mollace V. (1996) J. Clin. Invest. 97, 2562–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prütz W. A., Mönig H., Butler J., Land E. J. (1985) Arch. Biochem. Biophys. 243, 125–134 [DOI] [PubMed] [Google Scholar]
- 12.Balazy M., Poff C. D. (2004) Curr. Vasc. Pharmacol. 2, 81–93 [DOI] [PubMed] [Google Scholar]
- 13.Trostchansky A., Souza J. M., Ferreira A., Ferrari M., Blanco F., Trujillo M., Castro D., Cerecetto H., Baker P. R., O'Donnell V. B., Rubbo H. (2007) Biochemistry 46, 4645–4653 [DOI] [PubMed] [Google Scholar]
- 14.Calder P. C. (2006) Am. J. Clin. Nutr. 83, 1505S–1519S [DOI] [PubMed] [Google Scholar]
- 15.Trostchansky A., O'Donnell V. B., Goodwin D. C., Landino L. M., Marnett L. J., Radi R., Rubbo H. (2007) Free Radic. Biol. Med. 42, 1029–1038 [DOI] [PubMed] [Google Scholar]
- 16.Baker P. R., Lin Y., Schopfer F. J., Woodcock S. R., Groeger A. L., Batthyany C., Sweeney S., Long M. H., Iles K. E., Baker L. M., Branchaud B. P., Chen Y. E., Freeman B. A. (2005) J. Biol. Chem. 280, 42464–42475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schopfer F. J., Baker P. R., Giles G., Chumley P., Batthyany C., Crawford J., Patel R. P., Hogg N., Branchaud B. P., Lancaster J. R., Jr., Freeman B. A. (2005) J. Biol. Chem. 280, 19289–19297 [DOI] [PubMed] [Google Scholar]
- 18.Marnett L. J., Siedlik P. H., Ochs R. C., Pagels W. R., Das M., Honn K. V., Warnock R. H., Tainer B. E., Eling T. E. (1984) Mol. Pharmacol. 26, 328–335 [PubMed] [Google Scholar]
- 19.Odenwaller R., Chen Y. N., Marnett L. J. (1990) Methods Enzymol. 187, 479–485 [DOI] [PubMed] [Google Scholar]
- 20.Goodwin D. C., Gunther M. R., Hsi L. C., Crews B. C., Eling T. E., Mason R. P., Marnett L. J. (1998) J. Biol. Chem. 273, 8903–8909 [DOI] [PubMed] [Google Scholar]
- 21.Song I., Ball T. M., Smith W. L. (2001) Biochem. Biophys. Res. Commun. 289, 869–875 [DOI] [PubMed] [Google Scholar]
- 22.Kulmacz R. J. (1987) Prostaglandins 34, 225–240 [DOI] [PubMed] [Google Scholar]
- 23.Kulmacz R. J., Pendleton R. B., Lands W. E. (1994) J. Biol. Chem. 269, 5527–5536 [PubMed] [Google Scholar]
- 24.O'Donnell V. B., Coles B., Lewis M. J., Crews B. C., Marnett L. J., Freeman B. A. (2000) J. Biol. Chem. 275, 38239–38244 [DOI] [PubMed] [Google Scholar]
- 25.Landino L. M., Crews B. C., Timmons M. D., Morrow J. D., Marnett L. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 15069–15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Copeland R. A. (2000) Enzymes, 2nd Ed., pp. 305–317, John Wiley & Sons, Inc., New York [Google Scholar]
- 27.Berry E. A., Trumpower B. L. (1987) Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 28.Karthein R., Nastainczyk W., Ruf H. H. (1987) Eur. J. Biochem. 166, 173–180 [DOI] [PubMed] [Google Scholar]
- 29.Kalgutkar A. S., Crews B. C., Marnett L. J. (1996) Biochemistry 35, 9076–9082 [DOI] [PubMed] [Google Scholar]
- 30.Maskrey B. H., Bermúdez-Fajardo A., Morgan A. H., Stewart-Jones E., Dioszeghy V., Taylor G. W., Baker P. R., Coles B., Coffey M. J., Kühn H., O'Donnell V. B. (2007) J. Biol. Chem. 282, 20151–20163 [DOI] [PubMed] [Google Scholar]
- 31.Coles B., Bloodsworth A., Eiserich J. P., Coffey M. J., McLoughlin R. M., Giddings J. C., Lewis M. J., Haslam R. J., Freeman B. A., O'Donnell V. B. (2002) J. Biol. Chem. 277, 5832–5840 [DOI] [PubMed] [Google Scholar]
- 32.Smith W. L., Garavito R. M., DeWitt D. L. (1996) J. Biol. Chem. 271, 33157–33160 [DOI] [PubMed] [Google Scholar]
- 33.Schneider C., Boeglin W. E., Prusakiewicz J. J., Rowlinson S. W., Marnett L. J., Samel N., Brash A. R. (2002) J. Biol. Chem. 277, 478–485 [DOI] [PubMed] [Google Scholar]
- 34.Morrison J. F., Walsh C. T. (1988) Adv Enzymol. Relat. Areas Mol. Biol. 61, 201–301 [DOI] [PubMed] [Google Scholar]
- 35.Kitz R., Wilson I. B. (1962) J. Biol. Chem. 237, 3245–3249 [PubMed] [Google Scholar]
- 36.Blobaum A. L., Marnett L. J. (2007) J. Med. Chem. 50, 1425–1441 [DOI] [PubMed] [Google Scholar]
- 37.Kelley E. E., Batthyany C. I., Hundley N. J., Woodcock S. R., Bonacci G., Del Rio J. M., Schopfer F. J., Lancaster J. R., Jr., Freeman B. A., Tarpey M. M. (2008) J. Biol. Chem. 283, 36176–36184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker L. M., Baker P. R., Golin-Bisello F., Schopfer F. J., Fink M., Woodcock S. R., Branchaud B. P., Radi R., Freeman B. A. (2007) J. Biol. Chem. 282, 31085–31093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batthyany C., Schopfer F. J., Baker P. R., Durán R., Baker L. M., Huang Y., Cerveñansky C., Branchaud B. P., Freeman B. A. (2006) J. Biol. Chem. 281, 20450–20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schopfer F. J., Batthyany C., Baker P. R., Bonacci G., Cole M. P., Rudolph V., Groeger A. L., Rudolph T. K., Nadtochiy S., Brookes P. S., Freeman B. A. (2009) Free Radic. Biol. Med. 46, 1250–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudolph V., Schopfer F. J., Khoo N. K., Rudolph T. K., Cole M. P., Woodcock S. R., Bonacci G., Groeger A. L., Golin-Bisello F., Chen C. S., Baker P. R., Freeman B. A. (2009) J. Biol. Chem. 284, 1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agwu D. E., Holub B. J., Johnstone I. B., Crane S. (1983) Can. J. Comp. Med. 47, 203–206 [PMC free article] [PubMed] [Google Scholar]
- 43.Michibayashi T. (2005) J Atheroscler. Thromb. 12, 154–162 [DOI] [PubMed] [Google Scholar]
- 44.Wu G., Vuletich J. L., Kulmacz R. J., Osawa Y., Tsai A. L. (2001) J. Biol. Chem. 276, 19879–19888 [DOI] [PubMed] [Google Scholar]
- 45.Tsai A., Wei C., Baek H. K., Kulmacz R. J., Van Wart H. E. (1997) J. Biol. Chem. 272, 8885–8894 [DOI] [PubMed] [Google Scholar]