Association of the Variants CASP8 D302H and CASP10 V410I with Breast and Ovarian Cancer Risk in BRCA1 and BRCA2 Mutation Carriers (original) (raw)
. Author manuscript; available in PMC: 2011 Nov 1.
Published in final edited form as: Cancer Epidemiol Biomarkers Prev. 2010 Oct 26;19(11):2859–2868. doi: 10.1158/1055-9965.EPI-10-0517
Abstract
Background
The genes caspase-8 (CASP8) and caspase-10 (CASP10) functionally cooperate and play a key role in the initiation of apoptosis. Suppression of apoptosis is one of the major mechanisms underlying the origin and progression of cancer. Previous case-control studies have indicated that the polymorphisms CASP8 D302H and CASP10 V410I are associated with a reduced risk of breast cancer in the general population.
Methods
To evaluate whether the CASP8 D302H (CASP10 V410I) polymorphisms modify breast or ovarian cancer risk in BRCA1 and BRCA2 mutation carriers, we analyzed 7,353 (7,227) subjects of white European origin provided by 19 (18) study groups that participate in the Consortium of Investigators of Modifiers of BRCA1/2 (CIM-BA). A weighted cohort approach was used to estimate hazard ratios (HR) and 95% confidence intervals (95% CI).
Results
The minor allele of CASP8 D302H was significantly associated with a reduced risk of breast cancer (per-allele HR, 0.85; 95% CI, 0.76–0.97; _P_trend = 0.011) and ovarian cancer (per-allele HR, 0.69; 95% CI, 0.53–0.89; _P_trend = 0.004) for BRCA1 but not for BRCA2 mutation carriers. The CASP10 V410I polymorphism was not associated with breast or ovarian cancer risk for BRCA1 or BRCA2 mutation carriers.
Conclusions
CASP8 D302H decreases breast and ovarian cancer risk for BRCA1 mutation carriers but not for BRCA2 mutation carriers.
Impact
The combined application of these and other recently identified genetic risk modifiers could in the future allow better individual risk calculation and could aid in the individualized counseling and decision making with respect to preventive options in BRCA1 mutation carriers.
Introduction
Caspase-8 encoded by the CASP8 gene (OMIM 601763; chromosome 2q33) is an apical caspase involved in receptor-induced apoptosis. Caspase-10, encoded by the CASP10 gene (OMIM 601762; chromosome 2q33) adjacent to CASP8 in the human genome, cooperates with caspase-8 in the transduction of death receptor–mediated apoptotic signals. In addition to deregulated cell proliferation, suppression of apoptosis is one of the major mechanisms underlying the origin and progression of cancer (1–3). Although occurring at low frequencies, inactivating mutations in CASP8 and CASP10 have been identified in different tumor types (4–6). In addition, several coding polymorphisms in selected apoptotic genes, including CASP8 and CASP10, have been investigated with regard to their effect on cancer risk (7–16). Three independent studies found significant associations between the coding variant D302H in CASP8 (rs1045485) and reduced sporadic breast cancer risk (7, 9, 13). In addition, the largest of these studies done on 16,423 cases and 17,109 controls within the Breast Cancer Association Consortium revealed no associations between the variant and age of onset, hormone receptor status, grade, stage, or history of breast cancer in first-degree female relatives (13).
In case-control studies on patients selected for a family history of breast cancer, Frank et al. (9, 10) investigated the effect of the two coding polymorphisms CASP8 D302H (rs1045485) and CASP10 V410I (rs13010627) on breast cancer risk. Whereas there was no significant association between the CASP8 302H allele and breast cancer risk, carriers of the CASP10 410I allele were at a significantly reduced risk of breast cancer [odds ratio (OR), 0.62; 95% confidence interval (CI), 0.43–0.88; _P_trend = 0.0039]. The breast cancer risk was found to be even lower for individuals who also carried the CASP8 H302 allele (OR, 0.35; _P_trend = 0.007).
The associations between these variants and cancer risk for BRCA1 and BRCA2 mutation carriers have not been previously investigated. In the present study, we used data from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) to evaluate the associations of the CASP8 D302H and CASP10 V410I variants and breast and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers.
Materials and Methods
Study population
Female BRCA1 and BRCA2 mutation carriers over the age of 18 years were identified through 19 clinical and population-based research groups mainly from Europe, North America, and Australia. This work was done within the framework of CIMBA. CIMBA is an international collaboration, which was established in 2005 to conduct collaborative analyses of genetic polymorphisms as modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers using sample sizes large enough to provide sufficient power to detect even moderate effects (17). Recruitment of subjects was approved by institutional review boards or ethics committees at all sites. Eligibility was restricted to individuals who were carriers of clearly pathogenic mutations according to defined criteria (http://research.nhgri.nih.gov/projects/bic/; ref. 18). Phenotype data included year of birth, BRCA mutation description, ethnicity, country of residence, age at last follow-up, ages at breast and ovarian cancer diagnosis, age at bilateral prophylactic mastectomy, and age at bilateral prophylactic oophorectomy.
Genotyping
Samples from seven centers were genotyped at the Queensland Institute of Medical Research for the CASP8 and CASP10 single-nucleotide polymorphisms (SNP) using the iPLEX technology. The remaining centers used Taqman allelic discrimination assays (13, 19). The Taqman PCR primers were 5′-ACCACGACCTTTGAA-GAGCTT-3′ (forward) and 5′-GTGGTCCATGAGTTGG-TAGATTTTCA-3′ (reverse) for the CASP8 analysis and 5′-GGCCTGCCAAGGTGAAGAG-3′ (forward) and 5′-GCCTGCTCAGGGTTCAGA-3′ (reverse) for the CASP10 analysis. The probe for the CASP8 C allele was FAM-5′-CCCCACCATGACTG-3′, and for the CASP8 G allele was VIC-5′-CCCCACGATGACTG-3′. For the CASP10 analysis, the probe for the A allele was FAM-5′-CAGCCTTCCATATCC-3′, and for the G allele was VIC-5′-CAGCCTTCCGTATCC-3′. All of the rare CASP8 homozygotes (CC) were confirmed by sequencing using the following primers: 5′-GTGCTCTCCAGCTGTGGTC-3′ (forward) and 5′-CCAGTGAACTGACATGTCAGC-3′ (reverse).
The Deutsches Krebsforschungszentrum (DKFZ) samples were genotyped using PCR-based RFLP analyses. CASP8 D302H was genotyped using newly designed 5′-GCTTTGACCACGACCTTTGAAG-3′ (forward) and 5′-GTTACTGTGGTCCATGAGTTGGTA-GAT-3′ (reverse) primers. The reaction was set up in 10 μL containing 25 ng genomic DNA, 1× PCR buffer (Qiagen), 3.5 mmol/L MgCl2, 0.2 μmol/L of each primer, 200 μmol/L of each deoxynucleotide triphosphate (Promega), and 0.4 unit HotStarTaq DNA polymerase (Qiagen). After an initial denaturation step for 15 minutes at 95°C, 35 cycles of PCRs consisting of 1 minute at 94°C, 1 minute at 62°C, and 1 minute at 72°C were carried out, which were followed by a final extension step for 10 minutes at 72°C. PCR products were digested with 4 units _Hsp_92II (Promega) and separated on a 3.5% agarose gel containing ethidium bromide (Sigma-Aldrich) and scored by UV visualization. Sizes of the labeled fragments were 102 and 12 bp for the G allele and 52, 50, and 12 bp for the C allele. CASP10 V410I was also genotyped with PCR-based RFLP analyses using newly designed 5′-GATCATGTCT-CACTTCACAG-3′ (forward) and 5′-AAGTGGGTGCC-TGCTCAG-3′ (reverse) primers. PCRs and conditions were as for the CASP8 D302H analysis, with the exception of using 2.5 mmol/L MgCl2 and amplifying by 35 cycles of 30 seconds at 94°C, 30 seconds at 56°C, and 30 seconds at 72°C. PCR products were digested with 2.5 units _Bfu_I (Fermentas), separated on a 2.5% agarose gel containing ethidium bromide (Sigma-Aldrich), and scored by UV visualization. Fragment sizes were 112 and 34 bp for the G allele and 146 bp for the A allele.
To ensure consistency in genotyping across studies, all genotyping centers had to adhere to the CIMBA genotyping quality control criteria: for each study site, a call rate of >95% after exclusion of samples that failed amplification of both SNPs was required. At least 2% of the samples had to be included in duplicate and every plate had to be composed of no template controls and random mixture of affected and unaffected carriers. The concordance among duplicates had to be at least 98%. To further validate the accuracy of genotyping across centers, all centers were required to genotype 95 DNA samples from a standard test plate (Coriell Institute, Camden, NJ) for the CASP8 D302H SNP. Details of the CIMBA quality control guidelines are publicly available (http://www.srl.cam.ac.uk/consortia/cimba/eligibility/eligibility.html).
Patient selection
Of 8,890 cases genotyped for CASP8, we excluded 218 cases because they failed genotyping quality control, 35 because they were ascertained by more than one study group, 71 because of insufficient documentation of pathogenic BRCA mutations, 17 because date of birth was not available, 9 because of missing data on age at last observation or age under 18 years, 202 because of non-Caucasian origin, and 985 because of significant deviation from Hardy-Weinberg equilibrium in two study groups. The remaining 7,353 cases were analyzed as described below.
Of 7,842 cases genotyped for CASP10, we excluded 246 cases because they failed genotyping quality control, 35 because they were ascertained by more than one study group, 73 because of insufficient documentation of pathogenic BRCA mutations, 17 because date of birth was not available, 9 because of missing data on age at last observation or because they were under 18 years old, and 235 because of non-Caucasian origin. The remaining 7,227 cases were analyzed as described below.
Statistical analysis
Deviations of observed from expected genotype frequencies under the Hardy-Weinberg equilibrium were assessed among unrelated carriers within each contributing study group using an exact test. Study groups showing a significant deviation of P < 0.005 were excluded from further analysis. Consequently, data from two study groups were excluded from the CASP8 D302H analysis.
The associations between CASP8 D302H or CASP10 V410I genotype and breast or ovarian cancer risk data were analyzed within a Cox proportional hazards framework. To adjust for the nonrandom sampling of mutation carriers with respect to their disease status, we analyzed the data using a weighted cohort approach as described elsewhere (20, 21). This involves assigning differential weights to affected and unaffected mutation carriers such that the breast or ovarian cancer incidence rates observed in the study population are consistent with established BRCA1 and BRCA2 incidences (22). Subjects were followed from birth until the event of interest (first breast or ovarian cancer) or time of censoring. When analyzing breast cancer as the event of interest, subjects without breast cancer were censored at bilateral mastectomy, ovarian cancer, or last observation, whichever occurred first. When ovarian cancer was analyzed, subjects without ovarian cancer were censored at bilateral oophorectomy, first breast cancer, or last observation, whichever occurred first. Follow-up time was censored at age 80 years old. The associations were evaluated by modeling the per-allele hazard ratio (HR; one–degree of freedom model) and by fitting models with separate HR for the heterozygote and homozygote carriers of the minor allele (two–degree of freedom model). Analyses were carried out separately for BRCA1 and BRCA2 mutations carriers (carriers of compound BRCA1 and BRCA2 mutations were included in the BRCA1 subgroup). Analyses were stratified by study group, country of residence (because some study groups contributed samples from more than one country), and year of birth (grouped into <1930, 1930–1939, 1940–1949, 1950–1959, 1960+). To allow for the nonindependence among members of the same family, a robust variance approach was used to estimate the SEs associated with the parameter estimates (23). The heterogeneity of HRs among study groups was tested using Cochran’s Q test based on inverse variance weights. P values of ≤0.05 were considered statistically significant. All analyses were carried out using SPSS 15.0.1.1 (SPSS, Inc.) and R 2.9.1 (The R Foundation for Statistical Computing) using the survival package version 2.35–4.
Results
A total of 7,353 and 7,227 Caucasian subjects could be analyzed for the association of CASP8 D302H and CASP10 V410I with age of onset of breast cancer or ovarian cancer. The cohort comprised 7,782 different individuals, of which 6,798 were genotyped for both SNPs, 555 for only CASP8 D302H, and 429 for only CASP10 V410I.
Tables 1 and 2 give the number of carriers by study, genotype, and disease status. The overall minor allele frequencies (MAF) were 0.12 (range, 0.04–0.16) for CASP8 D302H and 0.07 (range, 0.05–0.09) for CASP10 V410I. The mean age at breast cancer was 42 and 44 years for BRCA1 and BRCA2 mutation carriers, respectively, and the corresponding mean ages of ovarian cancer were 49 and 56 years. Table 3 shows the distribution of genotypes by SNP, mutated BRCA gene, and disease status.
Table 1.
Numbers of carriers included in the analysis (by study group and genotype)
| Study group | CASP8 D302H | CASP10 V410I | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | GG | CG | CC | MAF | Total | GG | GA | AA | MAF | |
| CNIO | 365 | 295 | 66 | 4 | 0.10 | 370 | 332 | 37 | 1 | 0.05 |
| DKFZ | 98 | 76 | 20 | 2 | 0.12 | 98 | 84 | 12 | 2 | 0.08 |
| DNA HEBON | 420 | 335 | 83 | 2 | 0.10 | 481 | 416 | 63 | 2 | 0.07 |
| EMBRACE | 1,081 | 819 | 251 | 11 | 0.13 | 1,086 | 930 | 150 | 6 | 0.07 |
| FCCC | 132 | 108 | 23 | 1 | 0.09 | 137 | 121 | 16 | 0 | 0.06 |
| GEMO | 1,270 | 968 | 266 | 36 | 0.13 | 753 | 657 | 92 | 4 | 0.07 |
| GC-HBOC | 856 | 646 | 196 | 14 | 0.13 | 855 | 737 | 113 | 5 | 0.07 |
| HCSC | 168 | 135 | 29 | 4 | 0.11 | 168 | 151 | 14 | 3 | 0.06 |
| HEBCS | 184 | 141 | 39 | 4 | 0.13 | 184 | 161 | 23 | 0 | 0.06 |
| INHERIT | 154 | 124 | 30 | 0 | 0.10 | 154 | 134 | 19 | 1 | 0.07 |
| kConFab | 767 | 579 | 170 | 18 | 0.13 | 764 | 635 | 125 | 4 | 0.09 |
| MAYO | 160 | 128 | 31 | 1 | 0.10 | 162 | 142 | 19 | 1 | 0.06 |
| NCI | 188 | 148 | 37 | 3 | 0.11 | 195 | 170 | 24 | 1 | 0.07 |
| OCGN | — | — | — | — | — | 306 | 263 | 43 | 0 | 0.07 |
| PBCS | 81 | 63 | 16 | 2 | 0.12 | 84 | 75 | 9 | 0 | 0.05 |
| SMC | 345 | 317 | 28 | 0 | 0.04 | 334 | 304 | 29 | 1 | 0.05 |
| SWE-BRCA | 537 | 411 | 119 | 7 | 0.12 | 551 | 478 | 70 | 3 | 0.07 |
| ModSQuaD | 173 | 122 | 47 | 4 | 0.16 | 174 | 154 | 18 | 2 | 0.06 |
| UPENN | 374 | 304 | 67 | 3 | 0.10 | 371 | 327 | 43 | 1 | 0.06 |
| Total | 7,353 | 5,719 | 1,518 | 116 | 0.12 | 7,227 | 6,271 | 919 | 37 | 0.07 |
Table 2.
Characteristics of study subjects genotyped for the CASP8 D302H and CASP10 V410I polymorphisms
| Characteristic | CASP8 D302H | CASP10 V410I | ||||
|---|---|---|---|---|---|---|
| Total | BRCA1 | BRCA2 | Total | BRCA1 | BRCA2 | |
| Carriers | 7,353 | 4,844 | 2,509 | 7,227 | 4,694 | 2,533 |
| Breast cancer | ||||||
| Not affected | ||||||
| No. | 3,302 | 2,241 | 1,061 | 3,355 | 2,245 | 1,110 |
| Age at censure (mean ± SD) | 43.0 ± 12.6 | 42.7 ± 12.5 | 43.8 ± 12.9 | 43.3 ± 12.6 | 42.8 ± 12.4 | 44.1 ± 13.0 |
| Affected | ||||||
| No. | 4,051 | 2,603 | 1,448 | 3,872 | 2,449 | 1,423 |
| Age at event (mean ± SD) | 42.0 ± 9.7 | 40.8 ± 9.4 | 44.2 ± 9.9 | 42.1 ± 9.7 | 40.8 ± 9.4 | 44.3 ± 9.9 |
| Ovarian cancer | ||||||
| Not affected | ||||||
| No. | 6,628 | 4,268 | 2,360 | 6,503 | 4,122 | 2,381 |
| Age at censure (mean ± SD) | 41.6 ± 10.9 | 40.6 ± 10.7 | 43.3 ± 10.9 | 41.7 ± 11.0 | 40.7 ± 10.8 | 43.5 ± 11.0 |
| Affected | ||||||
| No. | 725 | 576 | 149 | 724 | 572 | 152 |
| Age at event (mean ± SD) | 50.6 ± 9.9 | 49.4 ± 9.5 | 55.6 ± 9.9 | 50.8 ± 10.0 | 49.4 ± 9.5 | 56.0 ± 10.0 |
Table 3.
Genotype distributions for the CASP8 D302H and CASP10 V410I polymorphisms
| Characteristic | CASP8 D302H | CASP10 V410I | ||||||
|---|---|---|---|---|---|---|---|---|
| Total | GG | CG | CC | Total | GG | GA | AA | |
| BRCA1 | 4,844 | 3,784 | 983 | 77 | 4,694 | 4,085 | 586 | 23 |
| Breast cancer | ||||||||
| Not affected | 2,241 | 1,725 | 482 | 34 | 2,245 | 1,946 | 288 | 11 |
| Affected | 2,603 | 2,059 | 501 | 43 | 2,449 | 2,139 | 298 | 12 |
| Ovarian cancer | ||||||||
| Not affected | 4,268 | 3,320 | 876 | 72 | 4,122 | 3,589 | 513 | 20 |
| Affected | 576 | 464 | 107 | 5 | 572 | 496 | 73 | 3 |
| BRCA2 | 2,509 | 1,935 | 535 | 39 | 2,533 | 2,186 | 333 | 14 |
| Breast cancer | ||||||||
| Not affected | 1,061 | 834 | 214 | 13 | 1,110 | 957 | 145 | 8 |
| Affected | 1,448 | 1,101 | 321 | 26 | 1,423 | 1,229 | 188 | 6 |
| Ovarian cancer | ||||||||
| Not affected | 2,360 | 1,816 | 506 | 38 | 2,381 | 2,063 | 308 | 10 |
| Affected | 149 | 119 | 29 | 1 | 152 | 123 | 25 | 4 |
Tables 4 and 5 summarize the results of the association analysis. A significant association was found for BRCA1 mutation carriers (_P_2df = 0.028; _P_trend = 0.011), but not for BRCA2 mutation carriers (_P_2df = 0.794; _P_trend = 0.550). Within the BRCA1 mutation carriers, each copy of the minor allele was estimated to confer a HR (HRper-rare allele) of 0.85 (95% CI, 0.76–0.97). There was also a significant association between CASP8 D302H and ovarian cancer risk for BRCA1 mutation carriers (_P_2df = 0.008; _P_trend = 0.004), but not in the BRCA2 subgroup (_P_2df = 0.718; _P_trend = 0.398). As with breast cancer, the effect of the rare allele was protective for ovarian cancer for BRCA1 mutation carriers (HRper-rare allele 0.69; 95% CI, 0.53–0.89).
Table 4.
Association of CASP8 D302H with breast and ovarian cancer risk
| CASP8 D302H | HR (95% CI) | P* | ||
|---|---|---|---|---|
| Per-rare allele | Heterozygotes† | Rare homozygotes† | ||
| Breast cancer | ||||
| BRCA1 | 0.85 (0.76–0.97) | 0.83 (0.72–0.95) | 0.86 (0.56–1.31) | 0.028 |
| BRCA2 | 1.06 (0.88–1.27) | 1.04 (0.84–1.30) | 1.20 (0.62–2.33) | 0.794 |
| Ovarian cancer | ||||
| BRCA1 | 0.69 (0.53–0.89) | 0.73 (0.55–0.98) | 0.31 (0.10–0.94) | 0.008 |
| BRCA2 | 1.27 (0.73–2.23) | 1.25 (0.67–2.34) | 1.94 (0.25–15.24) | 0.718 |
Table 5.
Association of CASP10 V410I with breast and ovarian cancer risk
| CASP10 V410I | HR (95% CI) | P* | ||
|---|---|---|---|---|
| Per-rare allele | Heterozygotes† | Rare homozygotes† | ||
| Breast cancer | ||||
| BRCA1 | 0.98 (0.84–1.15) | 0.94 (0.80–1.10) | 1.97 (1.04–3.72) | 0.129 |
| BRCA2 | 1.03 (0.80–1.31) | 1.06 (0.82–1.38) | 0.69 (0.21–2.25) | 0.738 |
| Ovarian cancer | ||||
| BRCA1 | 1.12 (0.83–1.52) | 1.08 (0.78–1.52) | 1.86 (0.61–5.72) | 0.601 |
| BRCA2 | 1.78 (1.01–3.12) | 1.60 (0.85–2.98) | 10.23 (3.13–33.39) | 0.097 |
Based on the two–degree of freedom test, there were no significant associations between CASP10 V410I genotype and the risk of breast or ovarian cancer. However, the per-allele effect on ovarian cancer within the BRCA2 carriers was marginally significant (_P_trend = 0.045) with a HRper-rare allele of 1.78 (95% CI, 1.01–3.12).
Figure 1 shows the study-specific estimates of the per-allele HR for the two analyses in which significant associations were detected. There was no significant heterogeneity between the study-specific estimates for the association of CASP8 D302H with breast cancer risk in BRCA1 carriers (_P_heterogeneity = 0.712). However, there was a marginally significant heterogeneity for the association of CASP8 D302H with ovarian cancer risk in BRCA1 carriers (_P_heterogeneity = 0.048). For one study group (MAYO), the estimate of the HR was significantly different and in the opposite direction compared with the overall CIMBA estimate (HR, 6.10; 95% CI, 1.95–19.12), suggesting that the observed heterogeneity is caused by this study group. We therefore repeated the association analysis without the data from the MAYO study group. The overall association remains significant with a similar HR (HRper-rare allele, 0.67; 95% CI, 0.51–0.86; _P_2df = 0.003; _P_trend = 0.002), but heterogeneity among study groups is no longer present (_P_heterogeneity = 0.625).
Figure 1.
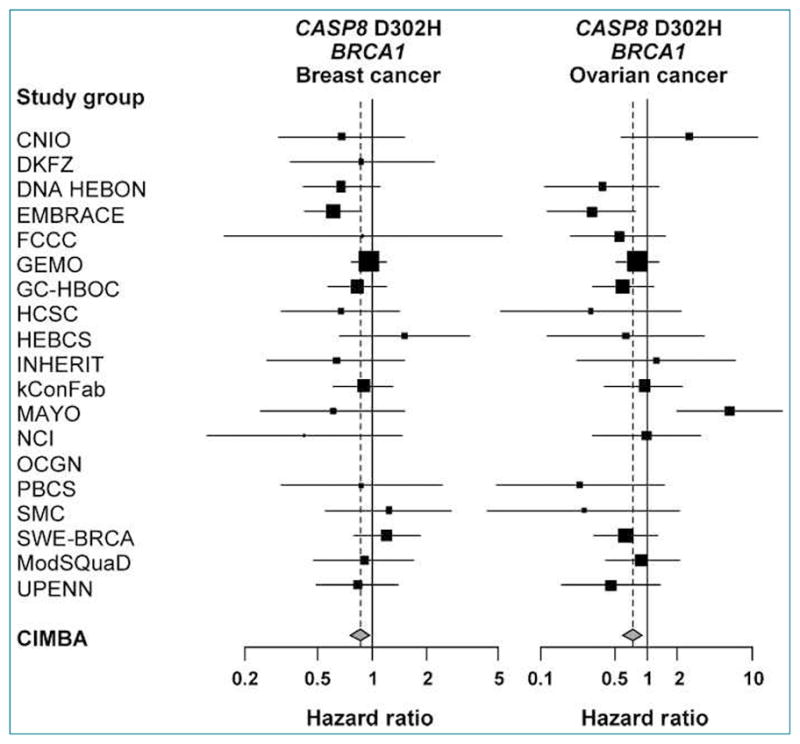
Forest plots of the study-specific estimates of the per-allele HRs for CASP8 D302H in BRCA1 mutation carriers (endpoints breast and ovarian cancer). The area of the squares is proportional to the inverse of the variance of the estimate. Horizontal lines represent the 95% CIs. The vertical dashed line indicates the overall effect estimate.
An analysis of the combined per-allele effects of the two SNPs in a multiplicative (log-additive) model revealed no qualitative changes in the results about the main effects (i.e., the risk-reducing association between CASP8 D302H and breast and ovarian cancer in BRCA1 carriers was still significant). There were no significant interactions between CASP8 and CASP10. However, the marginally significant per-allele effect of CASP10 on ovarian cancer within the BRCA2 carriers was not seen in this combined analysis.
Discussion
In this study, we pooled data from 19 study groups worldwide to investigate the association of CASP8 D302H (CASP10 V410I) genotypes with breast and ovarian cancer risk in >7,000 Caucasian carriers of deleterious mutations in BRCA1 or BRCA2.
We found that the minor allele of CASP8 D302H was associated with a reduced risk of breast cancer (per-allele HR, 0.85; 95% CI, 0.76–0.97) for BRCA1 but not for BRCA2 mutation carriers. Previously published case-control studies in unselected breast cancer cases also described a significantly decreased risk of breast cancer in carriers of the CASP8 302H allele with an OR of 0.83 to 0.89 (7, 13). Thus, the per-allele HR estimate for BRCA1 mutation carriers in our study was consistent with the magnitude of the association in the general population. A recent paper by Palanca Suela et al. (24) reported an OR of 0.40 of CASP8 D302H for breast cancer in a combined analysis of both BRCA1 and BRCA2 mutation carriers. This is partly consistent with our finding for BRCA1 mutation carriers but not for BRCA2, although they had not investigated the associations separately in BRCA1 and BRCA2 mutation carriers, and their study included only 186 BRCA1 and 204 BRCA2 mutation carriers.
The finding that CASP8 D302H is not a risk modifier in both BRCA1 and BRCA2 carriers is consistent with previous findings for other SNPs, which also showed associations for one subgroup only (25). The differential association patterns between BRCA1 and BRCA2 subgroups might be explained by the fact that these groups represent pathologically distinct entities. In our study, we could not analyze whether there are differences in the association by estrogen (ER) or progesterone receptor (PR) status because such data were not available at the time of analysis. Cox et al. did not find significant differences in the CASP8 rs1045485 risk association by ER or PR status of the tumors; however, the statistical power to detect such a difference might have been too low given the weak effect and the relatively low MAF (13). Larger studies will aim to clarify the associations with different disease subtypes in the general population.
Our study was reasonably powered to detect an allele with a MAF of 0.13 and a per-allele relative risk of 0.88 (which are the MAF and association magnitude for CASP8 D302H observed in the general population) at a significance level of 0.05 for BRCA1 mutation carriers (power = 70%), but the power was lower for BRCA2 mutation carriers (power = 37%). Larger studies are needed to elucidate the role of this polymorphism in breast cancer for BRCA2 mutation carriers.
Recent haplotype analysis of the CASP8 locus provided evidence that the D302H variant is not the functionally relevant variant because a haplotype including the D302H variant could be determined that conferred a higher risk than the variant alone. This suggests that the variant cosegregates with one or even several causative variants (26).
This is the first report on a risk-reducing effect of the CASP8 D302H variant for ovarian cancer in BRCA1 mutation carriers (per-allele HR, 0.69; 95% CI, 0.53–0.89). In contrast, there was no association between the variant and ovarian cancer in a large study comprising >4,600 unselected ovarian cancer cases (27), although the effect estimate was in the same direction as in our study. The reason for the lack of association in the general population is not clear. Furthermore, the authors could not find significant associations with different disease subtypes. As indicated above, we could not evaluate in our study whether the association varies with disease subtype. The observed association in our study could potentially reflect a _BRCA1_-specific effect, but future studies with larger numbers of BRCA1 mutation carriers with ovarian cancer should aim to evaluate this further. Palanca Suela et al. (24) also reported a lack of association of ovarian cancer in BRCA mutation carriers with the D302H variant. However, this may be due to the small study sample size, as their cohort comprised only 182 affected mutation carriers without stating the exact number of patients diagnosed with breast or ovarian cancer. Latif et al. (28) found that the D302H variant is associated with a significantly reduced risk of ovarian cancer in a case-control study including 101 women from families with familial ovarian cancer who were BRCA1/2 negative (OR, 0.52; 95% CI, 0.30–0.88). However, they could not detect any association in 52 women from BRCA1/2-positive families, probably because of the small sample size.
The localization of the CASP10 V410I polymorphism five amino acids upstream of the proenzyme cleavage site and seven amino acids downstream of the active-site motif rendered it an interesting SNP as well (29). In a case-control study of 511 familial breast cancer cases who did not carry BRCA1 and BRCA2 mutations and 547 controls, carriers of the CASP10 410I allele had a significantly reduced risk of breast cancer (10). However, a large study in >30,000 breast cancer cases could not confirm the association of V401I with breast cancer risk in the general population (30). This is in line with our results of no association between CASP10 V410I and breast cancer risk in BRCA1 and BRCA2 mutation carriers.
The present study is the largest of its kind in BRCA1/2 mutation carriers and provides significant evidence of association between CASP8 D302H variant and breast and ovarian cancer in BRCA1 but not in BRCA2 mutation carriers. A per-allele HR of 0.85 would result in a risk difference in the absolute risk of breast cancer of ~11% between the common and rare homozygotes by age of 80 years. Based on the present association, this polymorphism is estimated to account for only 0.4% of the genetic variability in breast cancer risk for BRCA1 mutation carriers (31). Thus, the clinical utility of this single variant is limited. However, several other loci, which were originally suggested as genetic risk factors in genome-wide association studies, were recently found by CIMBA to function as risk modifiers in BRCA1 and BRCA2 mutation carriers (31, 32). As further genetic modifiers of risk are identified for BRCA1 mutation carriers, the CASP8 variant in combination with others may be informative for risk assessment purposes. Furthermore, additional consideration of nongenetic risk factors may improve individualized risk prediction.
Grant Support
The CIMBA data management and genotyping is supported by Cancer Research UK.
Epidemiological study of BRCA1 and BRCA2 mutation carriers (EMBRACE)
Douglas F. Easton is the principal investigator of the study. EMBRACE Collaborating Centers are: Coordinating Centre, Cambridge: Susan Peock, Margaret Cook, Clare Oliver, Debra Frost. North of Scotland Regional Genetics Service, Aberdeen: Helen Gregory, Zosia Miedzybrodzka. Northern Ireland Regional Genetics Service, Belfast: Patrick Morrison. West Midlands Regional Clinical Genetics Service, Birmingham: Trevor Cole, Carole McKeown, Laura Boyes. South West Regional Genetics Service, Bristol: Alan Donaldson. East Anglian Regional Genetics Service, Cambridge: Joan Paterson. Medical Genetics Services for Wales, Cardiff: Alexandra Murray, Mark Rogers, Emma McCann. St James’s Hospital, Dublin and National Centre for Medical Genetics, Dublin: John Kennedy, David Barton. South East of Scotland Regional Genetics Service, Edinburgh: Mary Porteous. Peninsula Clinical Genetics Service, Exeter: Carole Brewer, Emma Kivuva, Anne Searle, Selina Goodman. West of Scotland Regional Genetics Service, Glasgow: Rosemarie Davidson, Victoria Murday, Nicola Bradshaw, Lesley Snadden, Mark Longmuir, Catherine Watt. South East Thames Regional Genetics Service, Guys Hospital London: Louise Izatt, Gabriella Pichert, Chris Jacobs, Caroline Langman. North West Thames Regional Genetics Service, Harrow: Huw Dorkins. Leicestershire Clinical Genetics Service, Leicester: Julian Barwell. Yorkshire Regional Genetics Service, Leeds: Carol Chu, Tim Bishop, Julie Miller. Merseyside and Cheshire Clinical Genetics Service, Liverpool: Ian Ellis. Manchester Regional Genetics Service, Manchester: D Gareth Evans, Fiona Lalloo, Felicity Holt. North East Thames Regional Genetics Service, NE Thames: Alison Male, Lucy Side, Anne Robinson. Nottingham Centre for Medical Genetics, Nottingham: Carol Gardiner. Northern Clinical Genetics Service, Newcastle: Fiona Douglas, Oonagh Claber. Oxford Regional Genetics Service, Oxford: Lisa Walker, Diane McLeod. The Institute of Cancer Research and Royal Marsden NHS Foundation Trust: Ros Eeles, Susan Shanley, Nazneen Rahman, Richard Houlston, Elizabeth Bancroft, Lucia D’Mello, Elizabeth Page, Audrey Ardern-Jones, Kelly Kohut, Jennifer Wiggins. Elena Castro, Lisa Robertson. North Trent Clinical Genetics Service, Sheffield: Jackie Cook, Oliver Quarrell, Cathryn Bardsley. South West Thames Regional Genetics Service, London: Shirley Hodgson, Sheila Goff, Glen Brice, Lizzie Winchester. Wessex Clinical Genetics Service, Princess Anne Hospital, Southampton: Diana Eccles, Anneke Lucassen, Gillian Crawford, Emma Tyler, Donna McBride. D.F.E., S.P., M.C., D.F., and C.O. are funded by Cancer Research UK grants C1287/A10118 and C1287/A8874. R.M. is supported by Cancer Research UK grant C8197/A10123. D.G.E. and F.L. are supported by an NIHR grant to the Biomedical Research Centre, Manchester. The Investigators at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust are supported by an NIHR grant to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. R.E., E.B., and L.D’M. are also supported by Cancer Research UK grant C5047/A8385.
Genetic Modifiers of cancer risk in BRCA1/2 mutation carriers study (GEMO)
The GEMO study (Cancer Genetics Network “Groupe Génétique et Cancer,” Fédération Nationale des Centres de Lutte Contre le Cancer, France) is supported by the Ligue National Contre le Cancer, Association for International Cancer Research Grant AICR-07-0454 and the Association “Le cancer du sein, parlons-en!” Award. L.B. is supported by Association for International Cancer Research grant AICR-07-0454. We wish to thank all the GEMO collaborating groups for their contribution to this study.
German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC) GC-HBOC is supported by a grant of the German Cancer Aid (grant 107054) and the Center for Molecular Medicine Cologne (grant TV93) to R.K.S. We thank Juliane Köhler for her excellent technical assistance, and D. Schäfer, Farnoosh Fathali-Zadeh, Claus R. Bartram, and the 12 clinical centers for providing samples and clinical data. GC-HBOC center for data management and biostatistics: Michael Brosig, Ute Enders, Marlies Herold, Markus Loeffler, Jan Schaefer, Marcus Wetzler, Kerstin Wieland, Silke Zachariae.
The Kathleen Cuningham Consortium for Research into Familial Breast Cancer (kConFab)
We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (funded by NHMRC grants 145684, 288704, and 454508) for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by grants from the National Breast Cancer Foundation, the National Health and Medical Research Council (NHMRC), and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania, and South Australia, and the Cancer Foundation of Western Australia.
A.B.S. and G.C.T. are NHMRC Research Fellows.
The Swedish BRCA1 and BRCA2 study (SWE-BRCA)
SWE-BRCA collaborators: Per Karlsson, Margareta Nordling, Annika Bergman, and Zakaria Einbeigi, Gothenburg, Sahlgrenska University Hospital; Marie Stenmark-Askmalm and Sigrun Liedgren, Linkoping University Hospital; Ake Borg, Niklas Loman, Hakan Olsson, Ulf Kristoffersson, Helena Jernstrom, Katja Harbst, and Karin Henriksson, Lund University Hospital; Annika Lindblom, Brita Arver, Anna von Wachenfeldt, Annelie Liljegren, Gisela Barbany-Bustinza, and Johanna Rantala, Stockholm, Karolinska University Hospital; Beatrice Malmer, Henrik Gronberg, Eva-Lena Stattin, and Monica Emanuelsson, Umea University Hospital; Hans Ehrencrona, Richard Rosenquist Brandell, and Niklas Dahl, Uppsala University Hospital. We thank the Swedish Cancer Society.
The Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON)
HEBON Collaborating Centers: Coordinating center: Netherlands Cancer Institute, Amsterdam: Frans Hogervorst, Senno Verhoef, Anouk Pijpe, Laura van ‘t Veer, Flora van Leeuwen, Matti Rookus; Erasmus Medical Center, Rotterdam: Margriet Collée, Ans van den Ouweland, Mieke Kriege, Mieke Schutte, Maartje Hooning, Caroline Seynaeve; Leiden University Medical Center, Leiden: Christi van Asperen, Juul Wijnen, Maaike Vreeswijk, Peter Devilee, Rob Tollenaar; Radboud University Nijmegen Medical Center, Nijmegen: Nicoline Hoogerbrugge, Marjolijn Ligtenberg; University Medical Center Utrecht, Utrecht: Margreet Ausems, Rob B. van der Luijt; Amsterdam Medical Center: Cora Aalfs, Theo van Os; VU University Medical Center, Amsterdam: Hanne Meijers-Heijboer, Hans Gille; University Hospital Maastricht, Maastricht: Encarna Gomez-Garcia, Rien Blok. The HEBON study is supported by the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088, and NKI 2007–3756.
University of Pennsylvania (UPENN) study
K.L.N. is supported by the Breast Cancer Research Foundation (BCRF). S.M.D. is supported by QVC Network, the Fashion Footwear Association of New York, and the Marjorie B. Cohen Foundation.
Spanish National Cancer Centre (CNIO)
Thanks to Rosario Alonso, Alicia Barroso, and Guillermo Pita for their technical support. The samples studied at the CNIO were recruited by the Spanish Consortium for the Study of Genetic Modifiers of BRCA1 and BRCA2 [Spanish National Cancer Centre (Madrid), Sant Pau Hospital (Barcelona), Instituto Catalad Oncologia (Barcelona), Valladolid University (Valladolid), Cancer Research Centre (Salamanca), and Instituto Dexeus (Barcelona)] and the Instituto Demokritos (Greece). The work carried out at the CNIO was partly funded by grants from the Genome Spain Foundation, Mutual Madrilena/06, Asociación Española Contra el Cancer/08, Ministry of Health FIS061090 and RD06/0020/1060, and Ministry of Science and Innovation PI081120.
Sheeba Medical Center Study (SMC)
The SMC study was supported in part by the Israel Cancer Association (ICA)
National Cancer Institute study (NCI)
We acknowledge the contributions of Dr. Jeffery A. Struewing and Marbin A Pineda from the NCI Laboratory of Population Genetics. Drs. Mai and Greene were supported by funding from the Intramural Research Program of the National Cancer Institute, Division of Cancer Epidemiology and Genetics. Their data collection efforts were supported by NIH Support Services Contracts NO2-CP-11019-50 and N02-CP-65504 with Westat, Inc. (Rockville, MD).
Helsinki Breast Cancer Study (HEBCS)
Heli Nevanlinna, Tuomas Heikkinen, Carl Blomqvist, Kirsimari Aaltonen, Kristiina Aittomaki, Helsinki University Central Hospital, Helsinki, Finland.
HEBCS wishes to thank Drs. Kirsimari Aaltonen and Carl Blomqvist and R.N. Hanna Jäntti for their help with the patient data and Tuomas Heikkinen and Kati Kämpjärvi for their help with the genetic analysis. HEBCS gratefully acknowledges the Finnish Cancer Registry for the cancer data. The HEBCS study has been financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (132473), the Finnish Cancer Society, and the Sigrid Juselius Foundation.
MoDSquaD
C.I.S. is supported by the Mayo Rochester Early Career Development Award for Non-Clinician Scientists. We acknowledge the contributions of Petr Pohlreich and Zdenek Kleibl (Department of Biochemistry and Experimental Oncology, First Faculty of Medicine, Charles University, Prague, Czech Republic) and the support of the Grant Agency of the Czech Republic, project no. GP301/08/P103 (to M.Z.). We acknowledge the contribution of Kim De Leeneer and Anne De Paepe. This research was supported by grant 1.5.150.07 from the Fund for Scientific Research Flanders (FWO) to Kathleen Claes and by grant 12051203 from the Ghent University to Anne De Paepe. Bruce Poppe is Senior Clinical Investigator of the Fund for Scientific Research of Flanders (FWO-Vlaanderen). Kim De Leeneer is supported by the Vlaamse Liga tegen Kanker through a grant of the Foundation Emmanuel van der Schueren. L.F., Machackova Eva, and Lukesova Miroslava are supported through the Ministry of Health grant CR-MZ0 MOU 2005.
Hospital Clinico San Carlos (HCSC)
Trinidad Caldes and Miguel de la Hoya were supported by a FMMA/06; FIS 05/0864 and RD06/0020/0021 (RTICC; ISCIII) Spanish Ministry of Science and Innovation.
Mayo Clinic Study (MAYO)
The Mayo Clinic study was supported in part by the Breast Cancer Research Foundation (BCRF), a grant from Susan G. Komen for the Cure, the Mayo Clinic Breast Cancer SPORE (P50-CA116201), and NIH grants CA122340 and CA128978 to F.J.C.
Interdisciplinary Health Research International Team Breast Cancer susceptibility (INHERIT)
Jacques Simard, Francine Durocher, Rachel Laframboise, Marie Plante, Centre Hospitalier Universitaire de Quebec and Laval University, Quebec, Canada; Peter Bridge, Jilian Parboosingh, Molecular Diagnostic Laboratory, Alberta Children’s Hospital, Calgary, Canada; Jocelyne Chiquette, Hôpital du Saint-Sacrement, Quebec, Canada; Bernard Lesperance, Hôpital du Sacré-Cœur de Montréal, Montréal, Canada. J.S. is Chairholder of the Canada Research Chair in Oncogenomics. This work was supported by the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program. This work was supported by the Canadian Breast Cancer Research Alliance grant 019511.
Ontario Cancer Genetics Network Study (OCGN)
We wish to thank Mona Gill, Nayana Weerasooriya, and members of the Ontario Cancer Genetics Network for their contributions to the study. We acknowledge funding from Cancer Care Ontario and the Canadian Institutes of Health Research for the “CIHR Team of Prediction and Communication of Familial Risks of Breast Cancer” program.
Fox Chase Cancer Center (FCCC)
We thank JoEllen Weaver and John Malick for expert technical assistance. A.K.G. was funded by SPORE P50 CA83638, U01 CA69631, and 5U01 CA113916, and the Eileen Stein Jacoby Fund.
Deutsches Krebsforschungszentrum study (DKFZ)
We thank Antje Seidel-Renkert for expert technical assistance. The study was supported by the DKFZ.
Pisa Breast Cancer Study (PBCS)
PBCS acknowledges the Italian Association for Cancer Research.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 2.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–8. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6:308–17. doi: 10.1038/nri1809. [DOI] [PubMed] [Google Scholar]
- 4.Park WS, Lee JH, Shin MS, et al. Inactivating mutations of the caspase-10 gene in gastric cancer. Oncogene. 2002;21:2919–25. doi: 10.1038/sj.onc.1205394. [DOI] [PubMed] [Google Scholar]
- 5.Kim HS, Lee JW, Soung YH, et al. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology. 2003;125:708–15. doi: 10.1016/s0016-5085(03)01059-x. [DOI] [PubMed] [Google Scholar]
- 6.Soung YH, Lee JW, Kim SY, et al. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005;65:815–21. [PubMed] [Google Scholar]
- 7.MacPherson G, Healey CS, Teare MD, et al. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J Natl Cancer Inst. 2004;96:1866–9. doi: 10.1093/jnci/dji001. [DOI] [PubMed] [Google Scholar]
- 8.Imyanitov E, Hanson K, Zhivotovsky B. Polymorphic variations in apoptotic genes and cancer predisposition. Cell Death Differ. 2005;12:1004–7. doi: 10.1038/sj.cdd.4401674. [DOI] [PubMed] [Google Scholar]
- 9.Frank B, Bermejo JL, Hemminki K, et al. Re: Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J Natl Cancer Inst. 2005;97:1012. doi: 10.1093/jnci/dji178. author reply 1012–3. [DOI] [PubMed] [Google Scholar]
- 10.Frank B, Hemminki K, Wappenschmidt B, et al. Association of the CASP10 V410I variant with reduced familial breast cancer risk and interaction with the CASP8 D302H variant. Carcinogenesis. 2006;27:606–9. doi: 10.1093/carcin/bgi248. [DOI] [PubMed] [Google Scholar]
- 11.Son JW, Kang HK, Chae MH, et al. Polymorphisms in the caspase-8 gene and the risk of lung cancer. Cancer Genet Cytogenet. 2006;169:121–7. doi: 10.1016/j.cancergencyto.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Lan Q, Zheng T, Chanock S, et al. Genetic variants in caspase genes and susceptibility to non-Hodgkin lymphoma. Carcinogenesis. 2007;28:823–7. doi: 10.1093/carcin/bgl196. [DOI] [PubMed] [Google Scholar]
- 13.Cox A, Dunning AM, Garcia-Closas M, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39:352–8. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- 14.Sun T, Gao Y, Tan W, et al. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nat Genet. 2007;39:605–13. doi: 10.1038/ng2030. [DOI] [PubMed] [Google Scholar]
- 15.Rajaraman P, Wang SS, Rothman N, et al. Polymorphisms in apoptosis and cell cycle control genes and risk of brain tumors in adults. Cancer Epidemiol Biomarkers Prev. 2007;16:1655–61. doi: 10.1158/1055-9965.EPI-07-0314. [DOI] [PubMed] [Google Scholar]
- 16.Frank B, Rigas SH, Bermejo JL, et al. The CASP8-652 6N del promoter polymorphism and breast cancer risk: a multicenter study. Breast Cancer Res Treat. 2008;111:139–44. doi: 10.1007/s10549-007-9752-z. [DOI] [PubMed] [Google Scholar]
- 17.Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA) Breast Cancer Res. 2007;9:104. doi: 10.1186/bcr1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antoniou AC, Sinilnikova OM, Simard J, et al. RAD51 135G->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet. 2007;81:1186–200. doi: 10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudet M, Fara AG, Beritognolo I, Sabatti M. Allele-specific PCR in SNP genotyping. Methods Mol Biol. 2009;578:415–24. doi: 10.1007/978-1-60327-411-1_26. [DOI] [PubMed] [Google Scholar]
- 20.Antoniou AC, Goldgar DE, Andrieu N, et al. A weighted cohort approach for analysing factors modifying disease risks in carriers of high-risk susceptibility genes. Genet Epidemiol. 2005;29:1–11. doi: 10.1002/gepi.20074. [DOI] [PubMed] [Google Scholar]
- 21.Antoniou AC, Rookus M, Andrieu N, et al. Reproductive and hormonal factors, and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: results from the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol Biomarkers Prev. 2009;18:601–10. doi: 10.1158/1055-9965.EPI-08-0546. [DOI] [PubMed] [Google Scholar]
- 22.Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–30. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin DY, Wej LJ. The robust inference for the Cox proportional hazards model. J Am Stat Assoc. 1989;84:1074–78. [Google Scholar]
- 24.Palanca Suela S, Esteban Cardenosa E, Barragan Gonzalez E, et al. CASP8 D302H polymorphism delays the age of onset of breast cancer in BRCA1 and BRCA2 carriers. Breast Cancer Res Treat. 119:87–93. doi: 10.1007/s10549-009-0316-2. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Pankratz VS, Fredericksen Z, et al. Common variants associated with breast cancer in genome wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2010;19:2886–97. doi: 10.1093/hmg/ddq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shephard ND, Abo R, Rigas SH, et al. A breast cancer risk haplotype in the caspase-8 gene. Cancer Res. 2009;69:2724–8. doi: 10.1158/0008-5472.CAN-08-4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramus SJ, Vierkant RA, Johnatty SE, et al. Consortium analysis of 7 candidate SNPs for ovarian cancer. Int J Cancer. 2008;123:380–8. doi: 10.1002/ijc.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latif A, McBurney HJ, Roberts SA, et al. Breast cancer susceptibility variants alter risk in familial ovarian cancer. Fam Cancer. doi: 10.1007/s10689-010-9349-2. Epub 2010 May 26. [DOI] [PubMed] [Google Scholar]
- 29.Milhas D, Cuvillier O, Therville N, et al. Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem. 2005;280:19836–42. doi: 10.1074/jbc.M414358200. [DOI] [PubMed] [Google Scholar]
- 30.Gaudet MM, Milne RL, Cox A, et al. Five polymorphisms and breast cancer risk: results from the Breast Cancer Association Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18:1610–6. doi: 10.1158/1055-9965.EPI-08-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antoniou AC, Sinilnikova OM, McGuffog L, et al. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2009;18:4442–56. doi: 10.1093/hmg/ddp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antoniou AC, Spurdle AB, Sinilnikova OM, et al. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet. 2008;82:937–48. doi: 10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]