Visualization of Chemokine Receptor Activation in Transgenic Mice Reveals Peripheral Activation of CCR2 Receptors in States of Neuropathic Pain (original) (raw)
Abstract
CCR2 chemokine receptor signaling has been implicated in the generation of diverse types of neuropathology, including neuropathic pain. For example, ccr2 knock-out mice are resistant to the establishment of neuropathic pain, and mice overexpressing its ligand, monocyte chemoattractant protein-1 (MCP1; also known as CCL2), show enhanced pain sensitivity. However, whether CCR2 receptor activation occurs in the central or peripheral nervous system in states of neuropathic pain has not been clear. We developed a novel method for visualizing CCR2 receptor activation in vivo by generating bitransgenic reporter mice in which the chemokine receptor CCR2 and its ligand MCP1 were labeled by the fluorescent proteins enhanced green fluorescent protein and monomeric red fluorescent protein-1, respectively. CCR2 receptor activation under conditions such as acute inflammation and experimental autoimmune encephalomyelitis could be faithfully visualized by using these mice. We examined the status of CCR2 receptor activation in a demyelination injury model of neuropathic pain and found that MCP1-induced CCR2 receptor activation mainly occurred in the peripheral nervous system, including the injured peripheral nerve and dorsal root ganglia. These data explain the rapid antinociceptive effects of peripherally administered CCR2 antagonists under these circumstances, suggesting that CCR2 antagonists may ameliorate pain by inhibiting CCR2 receptor activation in the periphery. The method developed here for visualizing CCR2 receptor activation in vivo may be extended to G-protein-coupled receptors (GPCRs) in general and will be valuable for studying intercellular GPCR-mediated communication in vivo.
Introduction
Neuropathic pain is a widespread health problem involving changes in the properties of primary sensory neurons and their central connections (Ji and Strichartz, 2004). Although the mechanisms that direct these changes are not completely understood, they are believed to involve activation of elements of the innate immune system and the action of inflammatory cytokines (Mogil et al., 2000; Scholz and Woolf, 2007; White et al., 2007; Milligan and Watkins, 2009). For example, it has been demonstrated that in association with chronic pain behaviors, neurons in the dorsal root ganglia (DRG) upregulate the expression of chemokines and their receptors, and that chemokines strongly excite pain-sensing neurons (nociceptors) under these circumstances (Oh et al., 2001; White et al., 2005, 2007; Sun et al., 2006). In particular, expression of the chemokine monocyte chemoattractant protein-1 (MCP1; also known as CCL2) has been shown to be upregulated by DRG neurons in several models of neuropathic pain (Tanaka et al., 2004; Gosselin et al., 2005; White et al., 2005, 2007; Zhang and De Koninck, 2006; Bhangoo et al., 2007; Yang et al., 2007; Jeon et al., 2008; Thacker et al., 2009). Under the same circumstances, it has been reported that the expression of CCR2 receptors, the receptors for MCP1, is upregulated by DRG neurons (White et al., 2005; Sun et al., 2006; Bhangoo et al., 2007) and by microglia in the spinal cord (Abbadie et al., 2003; Zhang and De Koninck, 2006; Zhang et al., 2007; Thacker et al., 2009). It has also been reported that some neurons in the spinal cord that receive GABAergic inputs express CCR2 receptors under some circumstances (Gosselin et al., 2005). Activation of CCR2 receptors is a critical event in the pathogenesis of neuropathic pain, because mice that are deficient in these receptors are resistant to the development of this behavior (Abbadie et al., 2003), and mice that overexpress MCP1 show enhanced pain sensitivity (Menetski et al., 2007). Moreover, drugs that block CCR2 receptors can inhibit established pain hypersensitivity, suggesting that ongoing activation of CCR2 receptors contributes to the maintenance of neuropathic pain (Bhangoo et al., 2007). However, exactly how MCP1–CCR2 signaling is involved in producing pain is not understood, and the locus of action of MCP1 is unclear. We have now used novel transgenic mouse models of MCP1–CCR2 signaling to visualize MCP1-mediated intercellular communication in vivo so as to precisely determine the sites of action of MCP1 in the context of chronic pain behavior. Our results demonstrate that activation of CCR2 expressed in the periphery is sufficient to produce chronic pain behaviors and suggest that peripherally acting CCR2 antagonists may ameliorate pain by inhibiting these mechanisms.
Materials and Methods
Plasmid construction.
To make chemokine fusion protein constructs, MCP1 and stromal cell-derived factor-1α (SDF1α) protein coding sequences were amplified by PCR and cloned into pmRFP1–N1 and pmCherry–N1 (kind gifts from Dr. Roger Tsien, University of California, San Diego, San Diego, CA) using the following primers: for MCP1 upstream, 5′-TTGAATTCATGCAGGTCCCTGTCATGCTT-3′; for MCP1 downstream, 5′-AAGGA-TCCAAGTTCACTGTCACACTGGTCA-3′; for SDF1 upstream, 5′-AAGAATTCATGG-ACGCCAAGGTCGTCG-3′; for SDF1 downstream, 5′-AACCGCGGCTTGTTTAAAGCTTTCTCCAG-3′. The CCR2 constructs were made by cloning the CCR2 protein coding sequence into pIRES2–enhanced green fluorescent protein (EGFP) (Clontech), pEGFP–N1 (Clontech), and pmRFP1–N1 (a kind gift from Dr. Roger Tsien) using the following primers: for upstream, 5′-CACAGATCTAAAGGAAATGGAAGACAA-3′; for downstream, 5′-CTTCTGCAGCAACCCAACCGAGACCTCTT-3′. The CXCR4–enhanced yellow fluorescent protein (EYFP)-expressing vector was a kind gift from Dr. Dongjun Ren (Northwestern University, Chicago, IL). Sequence identity was confirmed by dideoxy-sequencing methods. All chemicals were purchased from Sigma, unless stated otherwise.
Cell culture and transfection.
Human embryonic kidney (HEK293; tSA201 subclone) cells were maintained in DMEM supplemented with 10% fetal bovine serum and 0.5% penicillin–streptomycin at 37°C under 5% CO2. One microgram of plasmid DNA was transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Transfected cells were plated on poly-l-lysine-coated glass coverslips on the following day, and antagonists were added. Materials for cell culture were purchased from Invitrogen, unless stated otherwise.
Recombineering and generation of transgenic mice.
The standard recombineering protocol described by Lee et al. (2001) was used. The EGFP–FRT–kanamycin (KAN)–FRT-targeting cassette was generated by self-ligation of the blunt-ended _Bgl_I/_Sma_I fragment of pIGCN21 (a kind gift from Dr. Neal Copeland, National Cancer Institute–Frederick, Frederick, MD). The monomeric red fluorescent protein-1 (mRFP1)–FRT–KAN–FRT-targeting cassette was generated by replacing the EGFP protein coding sequence with the mRFP1 protein coding sequence. The EGFP- or mRFP1-targeting cassette was amplified by PCR using the chimeric primers, in which the 3′ end was homologous to the targeting cassette, and the 5′ end was homologous to the gene of interest. The amplified cassette was inserted in place of the start or stop codon by λ-Red-mediated recombination to generate the transcriptional reporter (gene∷reporter) and protein level reporter (gene∷gene-reporter), respectively.
The following reporter transgenes were made: (1) CCR2∷CCR2–EGFP (protein level reporter), (2) MCP1∷mRFP1 (transcriptional reporter), and (3) MCP1∷MCP1–mRFP1 (protein level reporter). In case of the MCP1∷mRFP1 transcriptional reporter, the poly-A signal of SV40 (5′-AATAAAGCAATAGCATCACAAAT-TTCACAAATAA-AGCATTTTTTTCACTGC-3′) was added at the end of the mRFP1 protein coding sequence. The CCR2-containing bacterial artificial chromosome (BAC) clone (MSM-529G05) was obtained from RIKEN DNA Bank and the MCP1-containing BAC clone (RP23–328G11) from Invitrogen. The chimeric primers used to generate the targeting cassettes are as follows: for CCR2∷CCR2–EGFP upstream, 5′-TGAGCTCTACATTCA-CTCCTTCCACTGGGGAGCAAGAGGT-CTCGGTTGGGTTGGATGATAATATGG-CCACAACC-3′; for CCR2∷CCR2–EGFP downstream, 5′-CTGTCTTTGAGGCTTGT-TGCTATGTACAAACTGCTCCCTCCTTCCCTGCTCTATTCCAGAAG-TAGTGAGGA-3′; for MCP1∷mRFP1 upstream, 5′-CCAGCACCAG-CCAACTCTCACTGA-AGCCAGCTCTCTCTTCCTCCACCACCC-CGGTCGCCACCATGGCCTCC-3′; for MCP1∷mRFP1 downstream, 5′-CTCCAGCCGGCAACTGTGAACAGCAGGCCCAGAAGCATG-ACAGGGACCTGCTATTCCAGAAGTAGTGAGGA-3′; for MCP1∷MCP1–mRFP1 upstream, 5′-CCACAACCACCTCAAGCACTTCTGTAG-GAGTGACCAGTGTGACAGTGAACCCGGTCGCCACCATGGCCTCC-3′; for MCP1∷MCP1–mRFP1 downstream, 5′-ATAAGTTAAATAAGTTTAATATTAATTAAGGCATCACAGTCCGAGTCACACTATTCCAGAAGTAGT-GAGGA-3′. The CXCR4∷EGFP mouse was a kind gift from the GENSAT project (Rockefeller University, New York, NY).
Lipopolylsaccharide injection.
Mice were injected intraperitoneally with 10 mg/kg lipopolysaccharide (LPS) diluted in saline and were killed after 24 or 72 h. For some experiments, animals were injected intraperitoneally with the CCR2 receptor antagonist (RA) (CCR2–RA; a kind gift from Eli Lilly and Company) diluted in saline. In this case, animals were injected with 50 mg/kg CCR2–RA three times at 4 h intervals during the last 12 h of LPS treatment.
Experimental autoimmune encephalomyelitis.
Detailed methods for the induction of experimental autoimmune encephalomyelitis (EAE) are described previously (Bailey et al., 2007). Briefly, female mice at 12–15 weeks of age were immunized subcutaneously with 100 μl of emulsified incomplete Freund's adjuvant (Difco) supplemented with 200 μg Mycobacterium tuberculosis H37Ra (Difco) and 100 μg MOG (35–55: MEVGWYRSPFSRVVHLYRNGK), and they received intraperitoneal injections of 200 ng pertussis toxin (Sigma) at the time of immunization and 48 h later.
Sciatic nerve demyelination.
Animals were anesthetized with 4% isoflurane and maintained on 2% isoflurane (Halocarbon) in O2. For all demyelination experiments, lysophosphatidylcholine (LPC) (type V, 99% pure; Sigma-Aldrich) was dissolved in buffered sterile saline, pH 7.2, to yield a final concentration of 10 mg/ml. The right sciatic nerve of the mouse was exposed at midthigh level under sterile conditions. A sterile polyvinyl acetal sponge (Ivalon), 2 × 2 mm, was soaked in 7 μl of LPC and then placed adjacent to the sciatic nerve. The dermal incision site was closed with 4.0 suture thread. Sham control animals were prepared as described above, but buffered sterile saline was used in place of LPC. Animals were allowed to survive for 14 d. For some experiments, animals were injected with a CCR2–RA (Eli Lilly and Company). Three injections of CCR2–RA (50 mg/kg) were given intraperitoneally at 4 h intervals during the last 12 h of the experiments. All experiments complied with protocols approved by the Northwestern University and Loyola University Chicago Institutional Animal Care and Use Committees.
Tissue sectioning, immunohistochemistry, and imaging.
To kill, animals were transcardially perfused with 4% paraformaldehyde. Tissues including the brain were postfixed in the same solution overnight before being sectioned. Brains were sectioned at 40 μm using a vibratome (Leica). Free-floating brain sections were immunostained for specific antigens using a standard protocol. DRG, spinal cords, and sciatic nerves were isolated and fixed in 4% paraformaldehyde at 4°C overnight. Tissues were then saturated with 30% sucrose in 1× PBS before sectioning using a cryostat. Tissue samples were sectioned at 20 μm and stored at −70°C until used. The primary antibodies used are as follows: a rabbit polyclonal anti-IBA-1 antibody (WAKO Pure Chemical Industries; 1:400); a rabbit polyclonal anti-GFAP antibody (Sigma; 1:400); and a rat monoclonal anti-PDGFRα (CD140a) antibody (BD Biosciences; 1:400). Corresponding secondary antibodies conjugated with Alexa Fluor 633 (Invitrogen) were used. Fluorescent images were taken by a laser-scanning confocal microscope. The following settings were used to detect each fluorophore: for EGFP, excitation at 488 nm and emission at 500–520 nm; for mRFP1, excitation at 543 nm and emission at 610 nm and above; and for Alexa Fluor 633, excitation at 633 nm and emission at 670 nm and above. When mRFP1 and Alexa Fluor 633 were imaged in the same section, each fluorophore was sequentially excited. A 20× objective and a 60× oil-immersion objective were used, and the thickness of optical sections was 2 μm and 0.4 μm, respectively. To quantify relative intensity of fluorescent signals, the “plot profile” function of ImageJ (http://rsbweb.nih.gov/ij/) was applied to lines in Figure 7. The values were normalized to the maximum value in each profile and expressed as an arbitrary unit.
Figure 7.
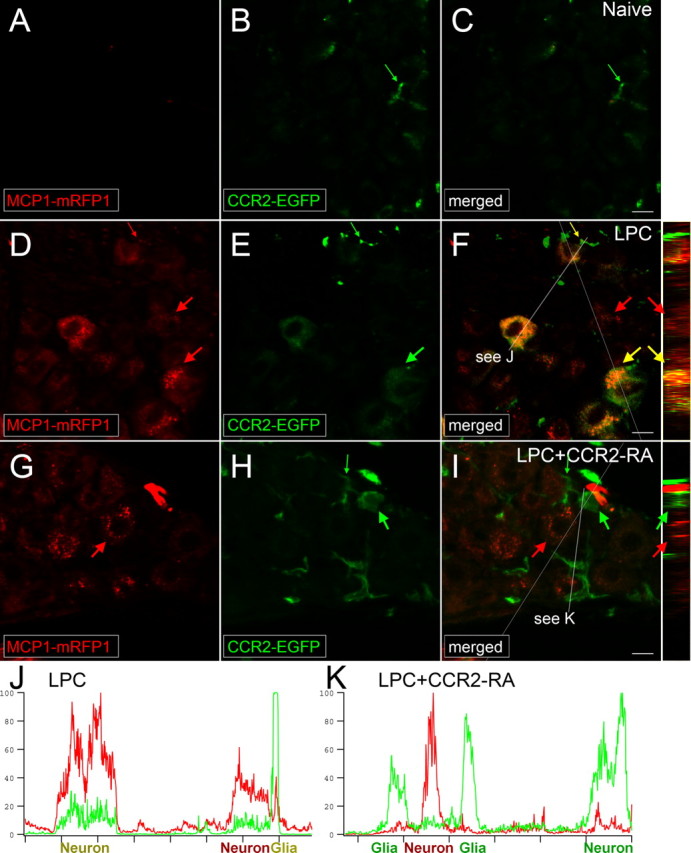
CCR2 signaling is activated in the DRG. MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice were subjected to LPC-induced demyelination of the sciatic nerve. A–F, In the DRG ipsilateral to the injury, expression of both MCP1 and CCR2 increased (D–F), whereas there was little expression of MCP1 or CCR2 under naive conditions (A–C). MCP1–mRFP1 mainly localized to neurons (large red arrow) and, to some extent, to satellite glia (small red arrow; D). CCR2–EGFP localized to neurons (large green arrow) and satellite glia (small green arrow; E). Most CCR2–EGFP-expressing neurons and satellite glia also contained MCP1–mRFP1 (yellow arrow; F). Injection of the CCR2–RA eliminated MCP1–mRFP1 in satellite glia (G–I; small green arrow). Also, after CCR2–RA treatment, MCP1–mRFP1- and CCR2–EGFP-expressing cells existed as separate populations (G–I; large green and red arrows). Cross-sectional images across the white lines are shown right to F and I. J, K, Intensities of mRFP1 and EGFP in shorter white lines in F and I are expressed in arbitrary units to compare relative signal intensities among different cells. J, In the LPC group, most neurons which express CCR2–EGFP also contain MCP1–mRFP1. Also, the CCR2–EGFP signal in neurons is relatively weaker than the signal in satellite glia (J). K, In the LPC plus CCR2–RA group, however, most CCR2–EGFP-expressing cells do not contain significant amount of MCP1–mRFP1 signal. In addition, the CCR2–EGFP signal in neurons is now as strong as the signal in satellite glia (K). Scale bars, 15 μm.
Cell counting and statistics.
For some experiments, CCR2–EGFP-expressing cells which underwent CCR2 receptor activation were counted. Cells in which most cell surface EGFP signal was lost and intracellular vesicular EGFP signal increased were counted as active cells. Cells which had no visible interruption in cell surface EGFP signal were counted as inactive cells. All the tissue sections were obtained from the corresponding anatomical locations, and at least three different sections were counted for each group. The percentages of activated cells out of the total CCR2–EGFP-expressing cells were calculated and compared between two groups using a nonparametric Mann–Whitney U test. To calculate the density of MCP1-expressing cells in the bone marrow, mRFP1-positive cells were counted in 600 × 600 × 10 μm volume from three or more bone marrow sections, and average cell density was calculated. The densities between control and LPS-injected groups were compared by t test. p < 0.05 was considered as significant.
In situ hybridization.
To examine CCR2 expression at the mRNA level in situ, hybridization was performed using a mouse CCR2-specific probe as described previously (Tran et al., 2007). The probe was selected from 3′ untranslated region of mouse CCR2 mRNA to minimize cross-reactivity with closely related genes, including CCR5.
Results
Visualization of MCP1–CCR2 interactions in cell culture
To visualize CCR2 receptor activation, we made use of the widely described property associated with G-protein-coupled receptor (GPCR) signaling that agonist-mediated activation of these receptors results in receptor endocytosis (Hanyaloglu and von Zastrow, 2008). Labeling of GPCR and neuropeptide C termini with fluorescent proteins has been widely used to track subcellular processes such as receptor trafficking and neuropeptide release (Kallal and Benovic, 2000; Levitan, 2004). Indeed, fusion of EGFP or mRFP1 to the C termini of CCR2 receptors did not interfere with either their intracellular processing (i.e., membrane localization) or functions (i.e., ligand-induced endocytosis and coupling to increased intracellular Ca signaling) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Likewise, fusion of EGFP or mRFP1 to the C terminus of the chemokine MCP1 did not interfere with its maturation, intracellular vesicular localization, or release (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results indicate that it may be possible to visualize interactions between MCP1 and CCR2 using the ligand and the receptor labeled with different fluorophores (Fig. 1A).
Figure 1.
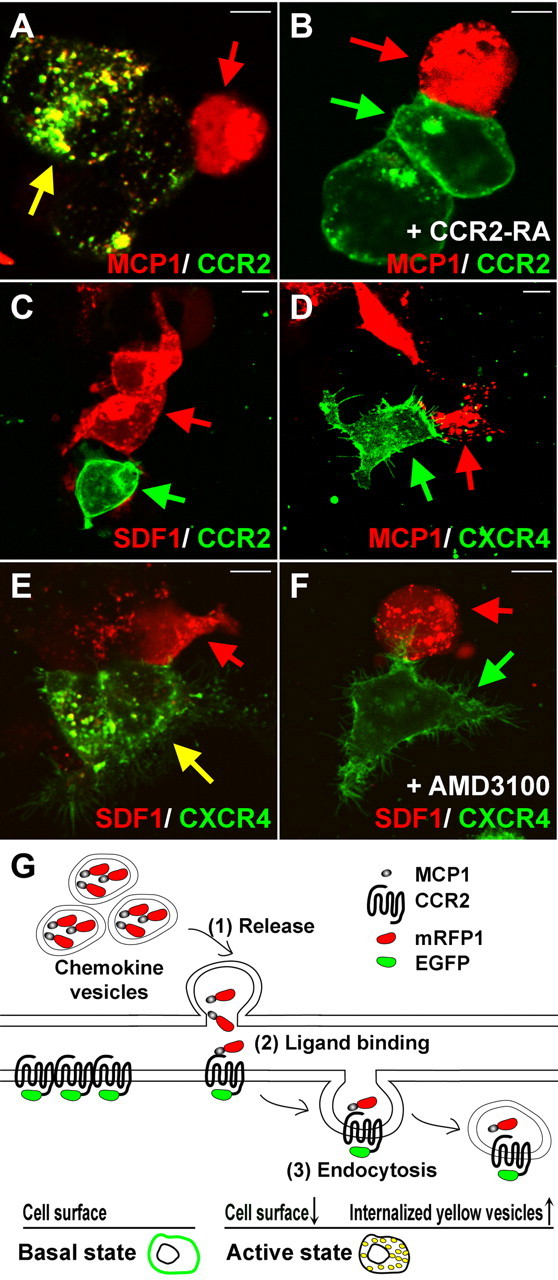
Visualization of MCP1–CCR2 interactions in vitro. A–F, In experiments to demonstrate this phenomenon in cell culture, HEK293 cells were transfected with either a chemokine ligand construct (MCP1–mRFP1 or SDF1–mRFP1) or a chemokine receptor construct (CCR2–EGFP or CXCR4–EYFP). After 24 h, ligand-expressing cells (red arrow) were cocultured with receptor-expressing cells (green arrow). A, Constitutively released MCP1–mRFP1 entered CCR2–EGFP-expressing cells, resulting in the formation of endocytic vesicles containing both MCP1–mRFP1 and CCR2–EGFP. B, The interactions between MCP1–mRFP1- and CCR2–EGFP-expressing cells were completely blocked by a CCR2 receptor antagonist (CCR2–RA; 300 nm). C, D, SDF1–mRFP1 did not interact with CCR2–EGFP (C), and MCP1–mRFP1 did not interact with CXCR4–EYFP (D). E, F, SDF1–mRFP1 did induce the endocytosis of CXCR4–EYFP (E), and this was blocked by a specific CXCR4–RA (AMD3100; 30 nm) (F). Scale bars, 10 μm. G, Proposed model for the assessment of MCP1–CCR2 interactions. MCP1–mRFP1 is localized to secretory vesicles (top space) and CCR2–EGFP to the plasma membrane (bottom space). When signaling occurs, released MCP1–mRFP1 (1) binds to CCR2–EGFP expressed on CCR2-expressing cells (2). Binding induces endocytosis of the MCP1–mRFP1/CCR2–EGFP complexes (3). Indications of CCR2 activation include the loss of membrane-localized EGFP signal and an increase in yellow/orange intracellular vesicles containing both MCP1–mRFP1 and CCR2–EGFP (bottom diagrams).
To investigate this possibility further, cells transfected with either MCP1–mRFP1 or CCR2–EGFP were cocultured. When cultured separately, CCR2–EGFP was primarily localized to the plasma membrane, and MCP1–mRFP1 was localized to intracellular vesicles (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material). In contrast, CCR2–EGFP in cocultures was primarily localized to intracellular vesicles, indicating that the receptors had undergone agonist-induced endocytosis (Fig. 1A). Many of these intracellular vesicles were yellow/orange, demonstrating that they involved the binding of MCP1–mRFP1 (red) to CCR2–EGFP (green) resulting in agonist-induced endocytosis of yellow vesicles (Fig. 1A). In support of this conclusion, the appearance of yellow endocytotic vesicles could be completely blocked by a specific CCR2 RA (Bhangoo et al., 2007) (Fig. 1B). More than 90% of CCR2–EGFP-expressing cells in close proximity to an MCP1–mRFP1-expressing cell exhibited clear yellow vesicles (n = 36). When 300 nm of a CCR2–RA was introduced, any yellow vesicles were observed in <5% of CCR2–EGFP-expressing cells (n = 64). Moreover, the formation of yellow vesicles was found to be ligand-specific. Another chemokine, SDF1–mRFP1, did not induce CCR2–EGFP endocytosis, although it did induce endocytosis of its own cognate receptor, CXCR4–EYFP, a phenomenon which was blocked by the specific CXCR4 receptor antagonist AMD3100 (Fig. 1C,E,F). Moreover MCP1–mRFP1 did not induce endocytosis of CXCR4–EYFP (Fig. 1D). These data indicate that if an endogenous ligand and its cognate receptor are labeled with different fluorescent proteins, it should also be possible to visualize their interactions in vivo (Fig. 1G). In particular, the experiments indicate that MCP1–mRFP1-mediated intercellular communication can be observed from the presence of yellow (or orange or red) (Fig. 1A) vesicles within CCR2–EGFP-expressing cells, whereas in the presence of a receptor antagonist, MCP1- and CCR2-expressing cells should be separated. These predictions were now tested by generating MCP1–mRFP1 and CCR2–EGFP bitransgenic mice.
Generation of transgenic mice using modified BACs
Expression of EGFP-tagged neuropeptides in neurons in vitro and in vivo has been widely used to study dynamics of neuropeptide release (Lou et al., 2005; Shakiryanova et al., 2006). However, most of these experiments used an exogenous promoter to ectopically express the tagged proteins. Recently, it was shown that by using BACs, reliable transcriptional reporter mice can be generated without detailed knowledge of a gene's promoter structure (Lee et al., 2001; Heintz, 2004). It is assumed that a single BAC clone of a sufficiently large size (∼200 kb) contains most regulatory _cis_-elements responsible for expression of a single gene. Using an efficient homologous recombination system in bacteria, a reporter gene (e.g., EGFP) can be inserted into a precise location in a BAC clone, so that endogenous regulatory elements may control the expression of the inserted reporter gene. By combining these two approaches, transgenic reporter mice that express MCP1–mRFP1 and CCR2–EGFP under the control of their respective endogenous regulatory elements were created. The mRFP1 coding sequence was inserted in place of the MCP1 stop codon, and EGFP was inserted in place of the CCR2 stop codon (MCP1∷MCP1–mRFP1 and CCR2∷CCR2–EGFP, respectively) (Fig. 2A). In parallel, a conventional BAC transcriptional reporter mouse for MCP1 (MCP1∷mRFP1) was also generated.
Figure 2.
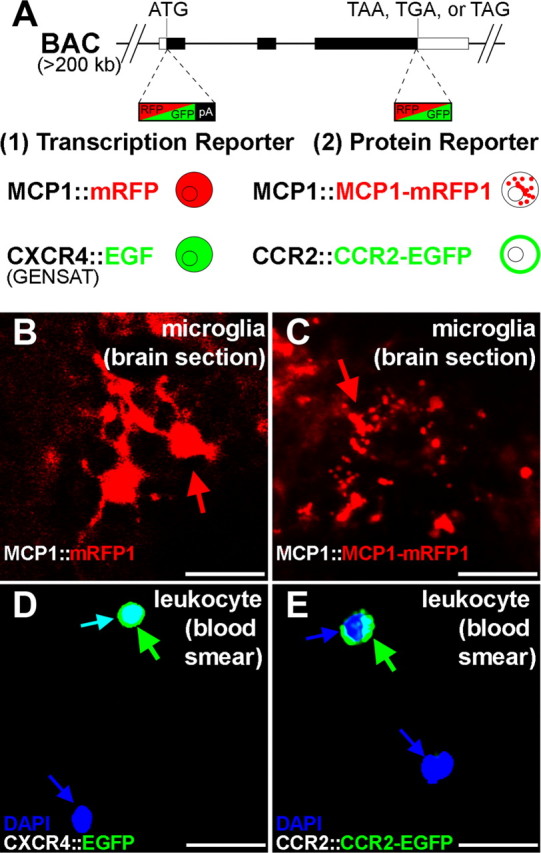
Generation of BAC transgenic reporter mice. A, BAC clones were modified by recombineering. EGFP or mRFP1 was inserted in place of the start codon to generate a transcriptional reporter (1) or the stop codon to generate a protein level reporter (2). CXCR4∷EGFP transcriptional reporter mice were obtained from the GENSAT project. The diagrams indicate expected subcellular localization of reporter proteins. pA, Poly-A signal. B, C, MCP1 reporter genes were expressed by microglia during an inflammatory response elicited by LPS injection (24 h after injection). Brain sections of LPS-injected mice are shown. D, E, The CXCR4 and CCR2 reporter genes were expressed by a subset of leukocytes under normal conditions. Blood smear samples were prepared from naive animals, and nucleated cells were identified by DAPI staining (blue). The reporter gene products (i.e., EGFP or mRFP1) of the transcriptional reporter mice (B, D) diffusely filled entire cells including the nuclei, whereas those of the protein level reporter mice (C, E) were limited to the appropriate subcellular localizations (i.e., MCP1–mRFP1 to vesicles, and CCR2–sEGFP mostly to the plasma membrane). Scale bars, 20 μm.
Faithful expression of reporter genes in BAC transgenic mice
CCR2 receptors are normally expressed by leukocytes (Serbina et al., 2008), although their expression may also be observed in some neurons and microglia under normal and pathological conditions (Banisadr et al., 2002; Abbadie et al., 2003; Gosselin et al., 2005; Jung et al., 2008). In CCR2–EGFP protein reporter mice (CCR2∷CCR2–EGFP), the reporter gene was expressed by cells in the bone marrow and by circulating leukocytes under normal conditions (16.1 and 8.4% of nucleated cells in the bone marrow and in the blood, respectively, as examined by flow cytometry). CCR2–EGFP was also expressed by neurons and glia under pathological conditions (Figs. 2E, 3–7). CCR2–EGFP was mainly localized to the plasma membrane of unstimulated cells (Fig. 2E, note that EGFP signal is absent from the nucleus which is represented as blue). By comparison, a similarly generated transcriptional reporter mouse for the chemokine receptor CXCR4 showed the expected diffuse localization of EGFP throughout the cell (CXCR4∷EGFP) (Fig. 2D, cyan arrow) (EGFP is diffusely localized including in the nucleus in contrast to CCR2∷CCR2–EGFP protein reporter mice in Fig. 2E).
Figure 3.
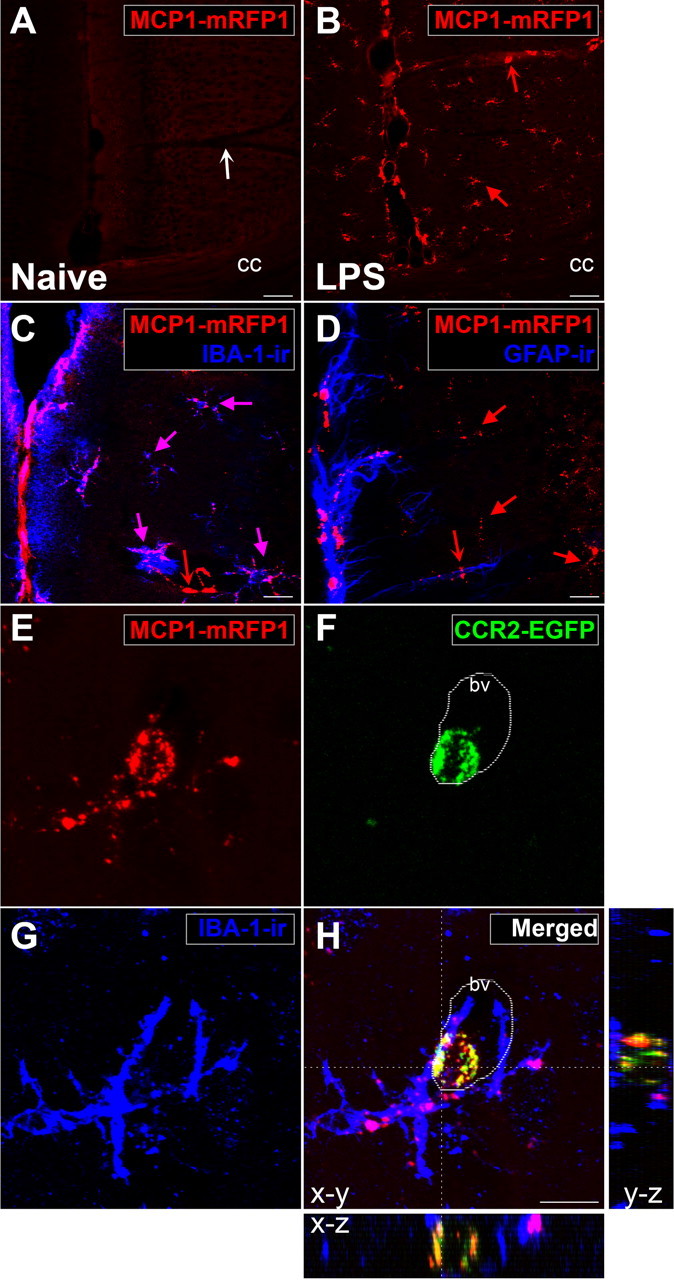
Interactions between MCP1- and CCR2-expressing cells can be visualized using BAC transgenic reporter mice. MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice were injected with LPS. A, B, After the injection (24 h), MCP1 (red) was significantly upregulated across the brains of these animals (B) in contrast to naive animals which showed no MCP1 expression (A). C, MCP1 was upregulated mostly by microglia (purple arrows), which were identified by IBA-1-ir. D, GFAP-expressing astrocytes generally did not upregulate MCP1–mRFP1 (red arrows). A few cells which were closely associated with blood vessels and upregulated MCP1 did not express IBA-1 or GFAP (A, a white concave arrow; B–D, red concave arrows). The pictures were taken from the mediofrontal cortex. cc, Corpus callosum. E, F, After the injection (48 h), interactions between MCP1- and CCR2-expressing cells could be observed in the brain. In this example, two perivascular microglia (G; IBA-1-ir) are interacting with one circulating leukocyte (F; CCR2–EGFP+). Microglia not only upregulated MCP1 but also released MCP1 to activate the CCR2–EGFP-expressing leukocyte (E). H, The internalized vesicles contained both MCP1–mRFP1 and CCR2–EGFP, which indicates that the MCP1-expressing microglia had activated CCR2 receptors expressed by the leukocyte. X-Z and Y-Z cross-sectional images across the white lines are shown for H. bv, Blood vessel. Scale bars: A–D, 20 μm; E, F, 10 μm.
MCP1 is not generally expressed at high levels in the nervous system under normal circumstances. Rather, its expression is induced under various pathological conditions in association with activation of the innate immune response (Mahad and Ransohoff, 2003; Medzhitov, 2008). Cells in the nervous system which upregulate MCP1 expression may include microglia, astrocytes, and neurons (Majumder et al., 1996; Mahad and Ransohoff, 2003; White et al., 2005; Lund et al., 2006). In agreement with known expression patterns, there was little basal expression of mRFP1 in the nervous system of transcriptional and protein level reporter mice (data not shown). However, mRFP1 expression was induced by various manipulations which are known to upregulate MCP1. For example, acute activation of the innate immune response by injection of LPS, a bacterial endotoxin that elicits strong innate immune responses by activating Toll-like receptors, induced upregulation of the reporter gene in microglia across the CNS (Lund et al., 2006) (Fig. 3C) [95% colocalization with IBA-1-immunoreactivity (ir), n = 160]. Several independent lines of mice made using the same construct exhibited similar patterns of expression. Moreover, the cellular localization (i.e., microglia in the case of LPS injection) of the reporter gene products was identical in the transcriptional (MCP1∷mRFP1) and the protein reporter (MCP1∷MCP1–mRFP1) mice, further confirming that expression of the reporter gene products was consistently regulated (Fig. 2B,C). However, as expected, the subcellular localization of the different types of reporter was clearly different. Unlike the transcriptional reporter where mRFP1 filled entire cells in a diffuse manner, MCP1–mRFP1 was localized to vesicular structures in the protein reporter mice as previously shown in cultured cells (Jung et al., 2008) (Figs. 1B,C, 2C).
By crossing these lines of mice, MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP bitransgenic mice were generated so as to report the cellular and subcellular localization of MCP1 and CCR2 in a single animal. For example, we observed the presence of green CCR2–EGFP-expressing cells in the brain, particularly in the region of blood vessels shortly after LPS treatment. Many of these cells were located in close juxtaposition to MCP1-expressing cells and contained yellow/orange/red vesicles (Fig. 3E–H), suggesting active communication between the two cell types as described above in cell culture. These yellow vesicles were not observed in CCR2–EGFP-expressing leukocytes when MCP1 transcriptional reporter mice were used instead (MCP1∷mRFP1; CCR2∷CCR2–EGFP) (data not shown) (see also Figs. 4B and 5B). In addition, using live brain slices of LPS-injected mice, we could monitor the movement of intracellular vesicles containing MCP1–mRFP1 and CCR2–EGFP in CCR2-expressing cells, while these cells continuously extended and retracted cellular processes (supplemental Movie 1, available at www.jneurosci.org as supplemental material). Therefore, these mice may provide a unique opportunity for examining MCP1–CCR2 signaling in real time in live tissue. Such observations support the interpretation that MCP1–mRFP1 released in trans can enter CCR2–EGFP-expressing leukocytes in vivo (Fig. 3H). To further examine whether these observations represent the activation of CCR2 receptors, we used two pathological conditions under which CCR2-mediated signaling is known to be activated: acute inflammation and EAE.
Figure 4.

Activation of CCR2 receptors in bone marrow monocytes during an inflammatory response can be visualized in the BAC transgenic mice. A–D, MCP1∷mRFP1; CCR2∷CCR2–EGFP mice (A, B) and MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP (C, D) mice were injected with LPS (B, D). After 24 h, bone marrow sections were imaged using a confocal microscope. Compared with CCR2 receptor expression under naive conditions (A, C; green arrow), activation of CCR2 receptors was evident after LPS treatment as indicated by internalization and loss of membrane localized CCR2–EGFP (B, D; concave arrow). When MCP1 protein reporter mice were used, MCP1 uptake by CCR2-expressing cells could also be visualized in yellow vesicles (D; yellow concave arrow). E, The activation of CCR2 receptors in the bone marrow was reversed by the CCR2 receptor antagonist (CCR2–RA). MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice were injected with LPS alone (D) or LPS and CCR2–RA (E). The CCR2–RA was injected intraperitoneally (50 mg/kg) three times at 4 h intervals during the last 12 h period of the inflammatory response. The CCR2–RA increased membrane localization and decreased intracellular localization of CCR2–EGFP (green arrow; E). In some cases, MCP1–mRFP1-containing vesicles remained after CCR2–EGFP-containing vesicles had disappeared (white arrow). Scale bars, 10 μm.
Figure 5.

Activation of CCR2 receptors in peripheral leukocytes during the pathogenesis of EAE can be visualized in the BAC transgenic mice. EAE was induced in MCP1:mRFP1; CCR2∷CCR2–EGFP mice (B) and MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice (C). The pictures were taken from the white matter from coronal cerebellar sections. Unlike control conditions (A), there was a marked infiltration of leukocytes (B, C; CCR2–EGFP+) into the parenchyma of the cerebellum (examined on postoperative day 14). Magnified views of the boxed areas are shown below B and C. Although there were close interactions between MCP1 (red arrow, cell body; red arrowhead, cellular process)- and CCR2-expressing cells (green concave arrow) (B, C), only MCP1–mRFP1 (C), but not mRFP1 alone (B), was transferred to CCR2–EGFP-expressing leukocytes as indicated from the appearance of yellow internal vesicles (yellow arrow and arrowhead). Scale bars, 20 μm.
Visualization of MCP1–CCR2 interactions in vivo
Bone marrow: regulated release of MCP1 during inflammation
During an active inflammatory response (e.g., after bacterial infection), CCR2 receptors expressed by bone marrow monocytes are activated, resulting in the egress of these cells into the blood stream (Serbina and Pamer, 2006). We observed that the bone marrow of naive animals contained a number of MCP1-expressing cells surrounded by numerous CCR2-positive cells, which apparently often made physical contact with one another (Fig. 4A,C). However, activation of CCR2 receptors in these cells under normal conditions was minimal, because most of the CCR2–EGFP was localized to the plasma membrane (Fig. 4A,C). Moreover, there were no MCP1–mRFP1-containing vesicles in or around CCR2–EGFP-expressing cells (Fig. 4C). This expression pattern changed dramatically after activation of the inflammatory response. Animals that were injected with LPS showed a significant increase in MCP1 release in the bone marrow (Fig. 4B,D). Two observations supported this conclusion. First, the cell surface expression of CCR2–EGFP was decreased, whereas intracellular CCR2–EGFP was increased (Fig. 4B,D). Second, CCR2-expressing cells now had numerous CCR2–EGFP/MCP1–mRFP1-containing yellow vesicles both inside and around the plasma membrane (Fig. 4D). These data indicate that LPS treatment induced MCP1 release and consequent activation and endocytosis of CCR2 receptors.
The activation of CCR2 receptors during inflammation was likely attributable to rapid regulated release of presynthesized MCP1, because there were already many MCP1–mRFP1-expressing cells in the bone marrow of mice under naive conditions (Fig. 4A,C). When the experiments were performed using MCP1–mRFP1 transcriptional reporter mice in which mRFP1 is not secreted and remains in MCP1-expressing cells, the number of MCP1-expressing cells in both groups were indistinguishable (compare Fig. 4A,B). However, the activation of CCR2 receptors in monocytes during this process was still evident, because the surface expression of CCR2–EGFP was decreased, whereas its expression in endocytotic vesicles increased (Fig. 4B) (percentage activated cells, 7.3% in control vs 72.4% in LPS-injected mice; n = 90 for control and n = 63 for the LPS-injected group; p < 0.0001, Mann–Whitney U test). Appropriately, these endocytotic vesicles were all green rather than yellow. Overall, these data clearly suggest that the activation of CCR2 in monocytes in the bone marrow is attributable to the increased release of presynthesized MCP1 by bone marrow cells.
Finally, the specificity of MCP1–mRFP1 and CCR2–EGFP interactions was confirmed by the systemic injection of a specific CCR2 receptor antagonist (CCR2–RA) (Bhangoo et al., 2007). The CCR2–RA was injected three times at 4 h intervals, 12 h after the injection of LPS. Animals were killed after a total of 24 h. The injection of the CCR2–RA restored the cell surface expression of CCR2–EGFP (Fig. 4E) (percentage cells with strong EGFP signals in intracellular vesicles, 60.4% in LPS-injected vs 15.6% in LPS and CCR2–RA-injected mice; n = 65 for the LPS group and n = 97 for the LPS plus CCR2–RA group; p < 0.0001, Mann–Whitney U test). Some MCP1–mRFP1 containing red vesicles remained inside the CCR2–EGFP-expressing cells (Fig. 4E, white arrow). This may be attributable to different stability or sorting of MCP1–mRFP1 and CCR2–EGFP within the endocytic pathway of target cells. These experiments, using a well established model in which CCR2 signaling plays an important physiological role, confirm that CCR2 receptor activation can be visualized in vivo using bitransgenic mice.
In the brain: interactions between microglia and leukocytes
One well characterized condition under which CCR2 signaling is activated in the brain is experimental autoimmune EAE. In EAE, an animal model of multiple sclerosis, animals develop an autoimmune response characterized by an immune attack against the white matter of the CNS (Karpus and Ransohoff, 1998). The cerebellar white matter is a particularly vulnerable structure. Accordingly, bitransgenic animals that developed EAE exhibited a dramatic infiltration of peripheral leukocytes expressing CCR2–EGFP around blood vessels and in the cerebellar parenchyma (Fig. 5B,C). These cells were closely juxtaposed to MCP1–mRFP1-expressing cells (Fig. 5B,C). Many of the infiltrating cells exhibited yellow subcellular vesicles containing both MCP1–mRFP1 and CCR2–EGFP, confirming that the activation of CCR2-mediated signaling occurs during the pathogenesis of EAE (Mahad et al., 2006) (Fig. 5C, bottom panels) (over 78% CCR2-expressing cells contained yellow vesicles). As in the LPS model, no dual-color vesicles were observed when the transcriptional reporter mice for MCP1 were used (MCP1∷mRFP1; CCR2∷CCR2–EGFP) (Fig. 5B, bottom panels), suggesting that these vesicles represent the MCP1-induced activation of CCR2 receptors.
Overall, therefore, these results suggest that, as in cell culture, MCP1–CCR2 signaling can be monitored in vivo using the fusion protein bitransgenic mice. Using these mice, we now examined the status of CCR2 receptor activation in the context of neuropathic pain.
Visualization of CCR2 receptor activation in states of neuropathic pain
A focal demyelination model of neuropathic pain (Wallace et al., 2003; Bhangoo et al., 2007; Jung et al., 2008) was used in these experiments. In this model, a detergent (LPC)-soaked sponge was implanted near the sciatic nerve at the level of the thigh. The sponge slowly releases LPC which induces focal demyelination of the nerve. As a result, animals develop sustained hyperalgesia in their hindpaws. An injection of the CCR2–RA at the peak of hyperalgesia has been shown to rapidly ameliorate pain hypersensitivity (Bhangoo et al., 2007), suggesting the importance of ongoing activation of CCR2 receptors in this model of neuropathic pain. Using these mice, we examined the peripheral nerves, DRG, and the dorsal horn of the spinal cord for indications of CCR2 activation associated with LPC-induced pain behavior.
In naive mice, we observed that MCP1 was not expressed at appreciable levels in the sciatic nerve, DRG, or spinal cord in keeping with previous findings (Tanaka et al., 2004; White et al., 2005, 2007; Zhang and De Koninck, 2006; Bhangoo et al., 2007; Jeon et al., 2008; Thacker et al., 2009) (Figs. 6A, 7A, 8). CCR2 receptors were also not expressed in the spinal cord or in the sciatic nerve, except for occasional small green cells associated with meninges or perineuria (Figs. 6A, 8). In the DRG, a few small cells surrounding neurons expressed low levels of CCR2 receptors (Fig. 7B). This pattern of expression was greatly altered in mice which had developed sciatic nerve demyelination-induced hindpaw hyperalgesia. First, we noted a large number of CCR2- and MCP1-expressing cells surrounding the point of LPC application and within the nerve (Fig. 6B). Cells expressing MCP1 within the nerve included endoneurial fibroblasts, as has been previously suggested based on the fact that they expressed platelet-derived growth factor (PDGF) receptor-α (Mäurer et al., 2003) and did not express S100 (Carroll and Frohnert, 1998), a marker for Schwann cells (Fig. 6C,D). Interestingly, many CCR2-expressing cells observed in close juxtaposition to MCP1-expressing cells contained yellow/orange vesicles, suggesting active communication between the two cell types (Fig. 6E). We tested this possibility by administering the CCR2–RA to these mice. Under these circumstances, CCR2- and MCP1-expressing cells within the nerve were observed as separate populations, and cells containing yellow vesicles were rarely observed (Fig. 6F) (percentage activated cells, 53.0% in LPC vs 15.3% in LPC and CCR2–RA groups; n = 68 for LPC and n = 44 for LPC–RA group; p < 0.0001, Mann–Whitney U test).
Figure 6.
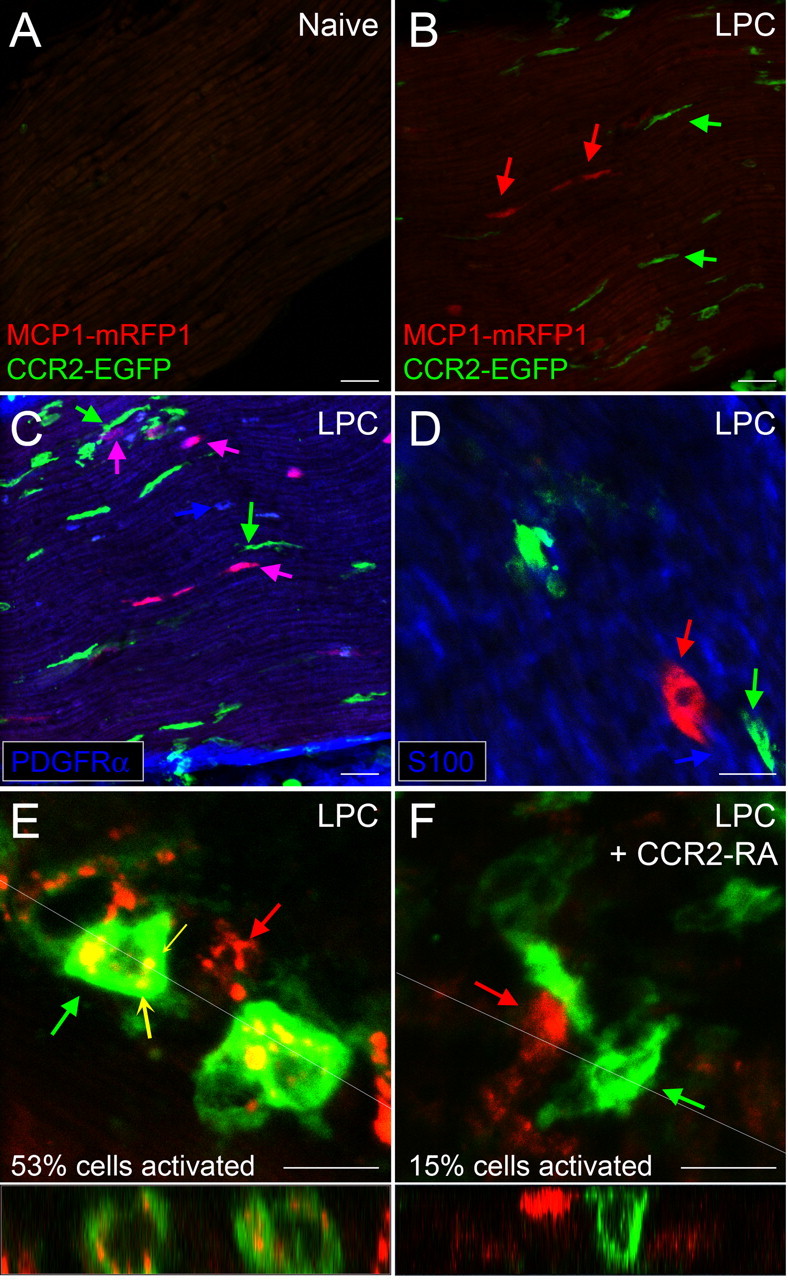
CCR2 signaling is activated in the peripheral nerve after demyelination. MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice were treated with LPC to produce focal demyelination of the sciatic nerve. On POD14 animals were killed, and sciatic nerves were isolated. Longitudinal sections were taken at the level of the midthigh. A, Under control conditions, there were few MCP1- or CCR2-expressing cells in the sciatic nerve. B, At the injury site, MCP1-expressing cells increased significantly in the nerve (red arrow), and there was infiltration of leukocytes both in and around the nerve (green arrow). C, D, Endoneurial fibroblasts upregulated MCP1. Endoneurial fibroblasts were identified as cells not associated with axons, which expressed PDGFR-α (C; blue arrow) and did not express S100 (D), a marker for Schwann cells (blue arrow). Note that some PDGFR-α-expressing cells upregulated MCP1–mRFP1 (C; purple arrow). Expression of PDGFR-α and S100 was examined by immunohistochemistry. The green arrows indicate leukocytes. E, Many of the CCR2-expressing leukocytes were observed undergoing active CCR2 receptor-mediated signaling as evidenced by the internalized yellow vesicles (yellow concave arrow). F, Injection of the CCR2–RA reversed the CCR2 receptor activation. Cross-sectional images across the white lines are shown below E and F. Scale bars: A–D, 30 μm; E, F, 10 μm.
Figure 8.
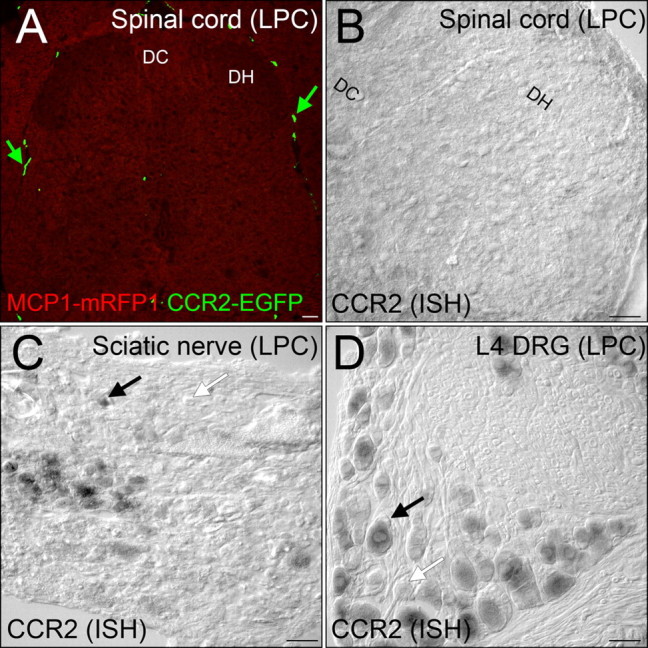
After demyelination, CCR2 signaling was not activated in the spinal cord. A, MCP1∷MCP1–mRFP1; CCR2∷CCR2–EGFP mice were subjected to LPC-induced demyelination of the sciatic nerve. There was no expression of MCP1 or CCR2 at a detectable level. Leukocytes outside the spinal cord were clearly visible (green arrow). B–D, CCR2 expression was also examined at the mRNA level by in situ hybridization. The spinal cord does not contain significant CCR2-expressing cellular components (B), whereas many cells in the sciatic nerve (C) and DRG (D) express CCR2 receptors in the LPC group. DH, Dorsal horn; DC, dorsal column. Scale bars, 60 μm.
As previously observed after LPC treatment and in other chronic pain models, expression of both MCP1 (Tanaka et al., 2004; White et al., 2005; Zhang and De Koninck, 2006; Bhangoo et al., 2007; Jeon et al., 2008; Jung et al., 2008; Thacker et al., 2009) and CCR2 (White et al., 2005; Sun et al., 2006; Bhangoo et al., 2007; Jung et al., 2008) was greatly upregulated in the DRG associated the injured sciatic nerve (i.e., DRG at the levels of L4 and L5). In the L4 DRG ipsilateral to the demyelination injury MCP1 was expressed by many neurons (Fig. 7D) (<1% neurons in the control groups, whereas >30% neurons in the LPC group). CCR2 was also expressed in numerous neurons and also by several small cells (Fig. 7E) (<1% neurons in the control groups, whereas >60% neurons in the LPC group). The apparent levels of CCR2 expression in the neurons appeared generally lower than in the small leukocyte-like cells (Fig. 7E, compare large arrow and small arrow; see also F, J). Interestingly, it was clear that many neurons contained yellow/orange vesicles that were positive for both MCP1 and CCR2, indicating communication between MCP1- and CCR2-expressing elements within the DRG (Fig. 7F,J) (>50% of CCR2-expressing cells). To confirm this conclusion, we examined the DRG from LPC-treated animals after the administration of the CCR2–RA. As in the case of the sciatic nerve and bone marrow discussed above, we again observed that MCP1 and CCR2 were now expressed by separate sets of neurons (Fig. 7G–I; see also see K). We also noted that the levels of CCR2 expressed in neurons were generally higher after the CCR2–RA treatment (Fig. 7H, compare small and large arrows; see also I, K). Our interpretation of this observation is that in the face of ongoing MCP1-induced endocytosis, CCR2 expression is substantially downregulated and that this is relieved after administration of the CCR2–RA. An analogous situation has been observed in the dentate gyrus where the chemokine SDF1 (also known as CXCL12) is tonically released from nerve terminals and downregulates the expression of its receptor CXCR4 on neural progenitor cells (Kolodziej et al., 2008). Administration of the CXCR4 antagonist AMD3100 results in a substantial upregulation of CXCR4 expression in the dentate gyrus under these circumstances.
Although the above observations suggest active communication between MCP1- and CCR2-expressing cellular elements in the sciatic nerve and DRG after demyelination, we found no evidence for this in the spinal cord. As can be observed in Figure 8A, after LPC treatment, MCP1- and CCR2-expressing cells were generally absent from the spinal cord. We did note the presence of a number of rod-shaped cells that expressed CCR2 which were localized around blood vessels and meninges similar to those that have been recently described (Audoy-Rémus et al., 2008), but these were also present in naive animals (Fig. 8A) (data not shown). However, we found no evidence for either MCP1- or CCR2-expressing cells within the parenchyma of the spinal cord. The absence of CCR2-expressing cellular components in the spinal cord parenchyma was further confirmed by in situ hybridization (Fig. 8B). In accordance with the data obtained from the BAC transgenic reporter mice, there was no significant expression of CCR2 receptors, although CCR2 expression in the DRG and sciatic nerve could be detected by the same probe in the same animals (Fig. 8C,D). It is interesting to note that there is evidence that when MCP1 is expressed in DRG neurons, at least some of it is colocalized with calcitonin gene-related peptide (CGRP) (Zhang and De Koninck, 2006; Jung et al., 2008; Knerlich-Lukoschus et al., 2008), a neuromodulator which would be expected to play a role in neurotransmission in the dorsal horn (Zhang and De Koninck, 2006; Jung et al., 2008; Knerlich-Lukoschus et al., 2008; Thacker et al., 2009). Indeed, we observed that numerous nerve terminals in the dorsal horn expressed CGRP (data not shown). However, there was no evidence that these also expressed MCP1, although MCP1 was abundantly expressed in the DRG. Hence, our observations indicate that in association with LPC-induced pain hypersensitivity, ongoing activation of CCR2 receptors is basically localized to the periphery.
Discussion
The data presented in this study allow us to make several conclusions. First, it is clear that by using bitransgenic mice constructed in the manner described here one can monitor intercellular GPCR-mediated communication in vivo, a technique that should prove valuable in many situations. We have used this method to examine the possibility that active MCP1–CCR2 signaling occurs as a mechanism for the maintenance of chronic pain hypersensitivity and observed clear CCR2 activation in the DRG and peripheral nerve under these circumstances.
Indeed, the results we have obtained demonstrate the utility of this method for defining the status of MCP1–CCR2 signaling in numerous tissues. For example, in the bone marrow, MCP1–CCR2 signaling has been implicated in the emigration of monocytes into the circulation (Serbina and Pamer, 2006). In mcp1 and ccr2 knock-out mice, monocyte egress during the inflammatory response is significantly impaired resulting in accumulation of monocytes in the bone marrow and an impaired immune response (Serbina and Pamer, 2006). Although it was known that MCP1–CCR2 signaling plays an important role in regulating monocyte egress, the molecular mechanisms governing this process have not been clear. One important question concerns the source of the MCP1 which activates bone marrow monocytes (e.g., infected tissues or bone marrow). We observed that MCP1 was constitutively expressed and stored in bone marrow cells under normal conditions (Fig. 4). MCP1-expressing cells were often seen making physical contact with CCR2-expressing monocytes, suggesting that they may engage in low levels of basal communication or at least be predisposed for this process to occur. During an inflammatory response, stored MCP1 was rapidly secreted, resulting in activation of CCR2 receptors in closely juxtaposed cells. This type of interaction could encourage egress of CCR2-expressing cells into the circulation as suggested previously (Serbina and Pamer, 2006).
It is also known that MCP1–CCR2 signaling plays a critical role in the pathogenesis of EAE. For example, one study has shown that deletion of the CCR2 gene confers resistance to EAE (Fife et al., 2000). Using the reporter mice developed in this study, it was possible to study the cellular origin of MCP1 upregulation (Fig. 5). It was found that microglia and astrocytes (data not shown) around blood vessels upregulated MCP1 expression in association with the induction of EAE. We observed active communication between infiltrating CCR2-expressing leukocytes and MCP1-expressing cells in the brain so that CCR2-expressing cells contained MCP1/CCR2-positive endocytotic vesicles. This is consistent with recent studies (Mahad et al., 2006) demonstrating that CCR2-expressing cells entering the brain may actively “consume” MCP1 expressed in the brain and consequently reduce its appearance in the CSF. Indeed, this process is also associated with “downregulation” of CCR2 receptors expressed by infiltrating leukocytes for which agonist-induced endocytosis must be a necessary prelude (Tylaska et al., 2002; Mahad et al., 2006).
Having established the veracity of our approach, we investigated the status of MCP1–CCR2 signaling in states of pain hypersensitivity. Numerous studies have indicated that MCP1–CCR2 signaling is important in peripheral injury-induced neuropathic pain behavior (for review, see Abbadie, 2005; White et al., 2007), although exactly how MCP1 produces these effects has not been clear. Sites of MCP1 action in the DRG, peripheral nerve, and spinal cord have all been suggested (White et al., 2007; Knerlich-Lukoschus et al., 2008). Indeed, it is possible that MCP1 may act at multiple sites and that the precise nature of its involvement may differ in different types of pain. Generally speaking, however, it is thought that Schwann cells and/or endoneurial fibroblasts in injured axons upregulate MCP1, which then attracts macrophages into the nerve (Carroll and Frohnert, 1998; Toews et al., 1998; Tofaris et al., 2002; Müller et al., 2008). Infiltrating macrophages may then secrete inflammatory molecules that sensitize the nerve. Second, neurons in the DRG also upregulate both MCP1 (Tanaka et al., 2004; White et al., 2005, 2007; Zhang and De Koninck, 2006; Bhangoo et al., 2007; Yang et al., 2007; Jeon et al., 2008; Jung et al., 2008; Knerlich-Lukoschus et al., 2008; Thacker et al., 2009) and CCR2 (White et al., 2005; Bhangoo et al., 2007; Jung et al., 2008). The activation of CCR2 signaling in DRG neurons is excitatory and therefore pronociceptive (White et al., 2005; Sun et al., 2006; Jung et al., 2008). Third, some reports have suggested that DRG neurons transport MCP1 to central nerve endings in the spinal cord where it is released (Zhang and De Koninck, 2006; Dansereau et al., 2008; Thacker et al., 2009). Once released in the spinal cord, MCP1 may activate microglia, which express CCR2 in the context of neuropathic pain (Abbadie et al., 2003; Zhang et al., 2007). The activation of microglia potentiates synaptic transmission in the dorsal horn of the spinal cord (Milligan and Watkins, 2009). Finally, centrally released MCP1 may act on CCR2-expressing neurons in the spinal cord. Some neurons have been reported to express CCR2, and activation of CCR2 in these neurons inhibits their response to GABAergic input (Gosselin et al., 2005).
The results reported here support the conclusion that, at least in the model used, the major sites of MCP1–CCR2 action are located in the periphery. We observed that in the injured nerve and DRG, CCR2-expressing cells (e.g., DRG neurons, satellite glia, and leukocytes in the nerve) were involved in active ongoing CCR2-mediated signaling (Figs. 6, 7). The activation of CCR2 signaling in these cells was inhibited by the injection of the CCR2–RA, which also blocks chronic pain hypersensitivity in this same model (Bhangoo et al., 2007). These data suggest that CCR2 receptors expressed by leukocytes in the nerve and by neurons and satellite glia in the DRG are the major contributors to pain hypersensitivity in this model (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
There has been no published evidence demonstrating that interneuronal communication between sensory neurons within the DRG takes place in states of pain hypersensitivity, although intersomal communication in the DRG using constitutively expressed neurotransmitters such as substance P has been suggested (Harding et al., 1999). Our findings suggest that nonsynaptic release of MCP1 may modulate DRG neuron excitability after injury by intersomal transmission (Fig. 7). This model suggests that a number of neuronal ion channels expressed by DRG neurons may be the downstream targets of CCR2 activation (Sun et al., 2006). For example, one possible mechanism by which CCR2 signaling in DRG neurons may contribute to pain hypersensitivity is by transactivating TRP channels (Zhang et al., 2005; Jung et al., 2008). Neuronal K and Na channels may also be possible targets (Sun et al., 2006; Wang et al., 2008). Satellite glia in the DRG are a source of many proinflammatory molecules, such as IL-β and NGF (nerve growth factor) (Scholz and Woolf, 2007), and the activation of CCR2 receptors in these cells may induce release of pronociceptive molecules to sensitize DRG neurons. Similar mechanisms may also apply to leukocytes in the nerve (Jin and Gereau, 2006; Scholz and Woolf, 2007).
Thus, the data reported here, together with the effectiveness of the peripherally injected CCR2–RA (Bhangoo et al., 2007), argue that the major sites of drug action, at least in this model, reside in the peripheral nervous system. In keeping with this conclusion, no detectable axonal transport of MCP1 to the dorsal horn was observed, nor were there a significant number of CCR2-expressing cells (e.g., neurons or microglia) observed in the spinal cord (Fig. 8).
It is certainly possible that central MCP1–CCR2 signaling may be important in other types of injury-induced chronic pain behavior. There are several reports which have described chronic pain-related effects of MCP1–CCR2 signaling in the spinal cord. For example, several groups have reported that intrathecal injection of MCP1 induces pain hypersensitivity (Abbadie et al., 2003; Zhang et al., 2007; Thacker et al., 2009). It has been postulated that the activation of CCR2-expressing microglia in the dorsal horn of the spinal cord contributes to central sensitization of pain transmission (Abbadie et al., 2003; Zhang et al., 2007; Thacker et al., 2009). Others have also suggested that MCP1 might contribute to pain hypersensitivity by directly acting on spinal cord neurons to attenuate inhibitory transmission (Gosselin et al., 2005). A couple of possible explanations for this apparent discrepancy exist. One explanation may be that a local increase in the spinal cord recruits circulating monocytes expressing CCR2 receptors, which in turn secrete molecules that activate microglia in the spinal cord or sensitize neuronal transmission. Monocytes may infiltrate into the spinal cord, differentiate into microglia, and then subsequently downregulate CCR2 expression. Indeed, it has been reported that circulating monocytes can enter the spinal cord and differentiate into microglia under some circumstances (Zhang et al., 2007). Use of the bitransgenic mice described here, together with the appropriate models, will be helpful in further examining these possibilities.
In summary, the data reported here support the conclusion that CCR2 activation in the peripheral nervous system can play a critical role in the production of pain hypersensitivity in states of neuropathic pain, supporting the possibility that antagonism of MCP1–CCR2 signaling may be a novel therapeutic intervention for the treatment of chronic pain syndromes.
Footnotes
This work was supported by grants from the National Institutes of Health and the Dana Foundation. We thank Sarah Kay Welch for her help with maintaining mice, Divakar Mithal for his help with flow cytometry experiments, Matthew S. Ripsch for his help with LPC surgery, and Drs. Philip E. Hockberger and Michelle Day for their help with two-photon microscopy.
References
- Abbadie C. Chemokines, chemokine receptors and pain. Trends Immunol. 2005;26:529–534. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoy-Rémus J, Richard JF, Soulet D, Zhou H, Kubes P, Vallières L. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci. 2008;28:10187–10199. doi: 10.1523/JNEUROSCI.3510-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Quéraud-Lesaux F, Boutterin MC, Pélaprat D, Zalc B, Rostène W, Haour F, Parsadaniantz SM. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:38. doi: 10.1186/1744-8069-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SL, Frohnert PW. Expression of JE (monocyte chemoattractant protein-1) is induced by sciatic axotomy in wild type rodents but not in C57BL/Wld(s) mice. J Neuropathol Exp Neurol. 1998;57:915–930. doi: 10.1097/00005072-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Dansereau MA, Gosselin RD, Pohl M, Pommier B, Mechighel P, Mauborgne A, Rostene W, Kitabgi P, Beaudet N, Sarret P, Melik-Parsadaniantz S. Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem. 2008;106:757–769. doi: 10.1111/j.1471-4159.2008.05429.x. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin RD, Varela C, Banisadr G, Mechighel P, Rostene W, Kitabgi P, Melik-Parsadaniantz S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem. 2005;95:1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Harding LM, Beadle DJ, Bermudez I. Voltage-dependent calcium channel subtypes controlling somatic substance P release in the peripheral nervous system. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:1103–1112. doi: 10.1016/s0278-5846(99)00049-4. [DOI] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Park ES, Jeon YH, Cho HJ. Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport. 2008;19:183–186. doi: 10.1097/WNR.0b013e3282f3c781. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Jin X, Gereau RW., 4th Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Miller RJ. Activation of the nuclear factor of activated T-cells (NFAT) mediates upregulation of CCR2 chemokine receptors in dorsal root ganglion (DRG) neurons: a possible mechanism for activity-dependent transcription in DRG neurons in association with neuropathic pain. Mol Cell Neurosci. 2008;37:170–177. doi: 10.1016/j.mcn.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallal L, Benovic JL. Using green fluorescent proteins to study G-protein-coupled receptor localization and trafficking. Trends Pharmacol Sci. 2000;21:175–180. doi: 10.1016/s0165-6147(00)01477-2. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Ransohoff RM. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Knerlich-Lukoschus F, Juraschek M, Blömer U, Lucius R, Mehdorn HM, Held-Feindt J. Force-dependent development of neuropathic central pain and time-related CCL2/CCR2 expression after graded spinal cord contusion injuries of the rat. J Neurotrauma. 2008;25:427–448. doi: 10.1089/neu.2007.0431. [DOI] [PubMed] [Google Scholar]
- Kolodziej A, Schulz S, Guyon A, Wu DF, Pfeiffer M, Odemis V, Höllt V, Stumm R. Tonic activation of CXC chemokine receptor 4 in immature granule cells supports neurogenesis in the adult dentate gyrus. J Neurosci. 2008;28:4488–4500. doi: 10.1523/JNEUROSCI.4721-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- Levitan ES. Using GFP to image peptide hormone and neuropeptide release in vitro and in vivo. Methods. 2004;33:281–286. doi: 10.1016/j.ymeth.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP. Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron. 2005;45:245–255. doi: 10.1016/j.neuron.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Lund S, Christensen KV, Hedtjärn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Pörzgen P, Leist M. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol. 2006;180:71–87. doi: 10.1016/j.jneuroim.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Mahad D, Callahan MK, Williams KA, Ubogu EE, Kivisäkk P, Tucky B, Kidd G, Kingsbury GA, Chang A, Fox RJ, Mack M, Sniderman MB, Ravid R, Staugaitis SM, Stins MF, Ransohoff RM. Modulating CCR2 and CCL2 at the blood-brain barrier: relevance for multiple sclerosis pathogenesis. Brain. 2006;129:212–223. doi: 10.1093/brain/awh655. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Majumder S, Zhou LZ, Ransohoff RM. Transcriptional regulation of chemokine gene expression in astrocytes. J Neurosci Res. 1996;45:758–769. doi: 10.1002/(SICI)1097-4547(19960915)45:6<758::AID-JNR12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Mäurer M, Müller M, Kobsar I, Leonhard C, Martini R, Kiefer R. Origin of pathogenic macrophages and endoneurial fibroblast-like cells in an animal model of inherited neuropathy. Mol Cell Neurosci. 2003;23:351–359. doi: 10.1016/s1044-7431(03)00055-1. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Menetski J, Mistry S, Lu M, Mudgett JS, Ransohoff RM, Demartino JA, Macintyre DE, Abbadie C. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience. 2007;149:706–714. doi: 10.1016/j.neuroscience.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Yu L, Basbaum AI. Pain genes?: natural variation and transgenic mutants. Annu Rev Neurosci. 2000;23:777–811. doi: 10.1146/annurev.neuro.23.1.777. [DOI] [PubMed] [Google Scholar]
- Müller M, Wacker K, Getts D, Ringelstein EB, Kiefer R. Further evidence for a crucial role of resident endoneurial macrophages in peripheral nerve disorders: lessons from acrylamide-induced neuropathy. Glia. 2008;56:1005–1016. doi: 10.1002/glia.20674. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Levitan ES. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006;96:2189–2199. doi: 10.1152/jn.00222.2006. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Toews AD, Barrett C, Morell P. Monocyte chemoattractant protein 1 is responsible for macrophage recruitment following injury to sciatic nerve. J Neurosci Res. 1998;53:260–267. doi: 10.1002/(SICI)1097-4547(19980715)53:2<260::AID-JNR15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Patterson PH, Jessen KR, Mirsky R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci. 2002;22:6696–6703. doi: 10.1523/JNEUROSCI.22-15-06696.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. Chemokine receptor expression by neural progenitor cells in neurogenic regions of mouse brain. J Comp Neurol. 2007;500:1007–1033. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylaska LA, Boring L, Weng W, Aiello R, Charo IF, Rollins BJ, Gladue RP. Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine. 2002;18:184–190. doi: 10.1006/cyto.2002.1031. [DOI] [PubMed] [Google Scholar]
- Wallace VC, Cottrell DF, Brophy PJ, Fleetwood-Walker SM. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci. 2003;23:3221–3233. doi: 10.1523/JNEUROSCI.23-08-03221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Strong JA, Xie W, Yang RH, Coyle DE, Wick DM, Dorsey ED, Zhang JM. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol Pain. 2008;4:38. doi: 10.1186/1744-8069-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Mitchell K, Keller JM, Iadarola MJ. Peripheral inflammation increases Scya2 expression in sensory ganglia and cytokine and endothelial related gene expression in inflamed tissue. J Neurochem. 2007;103:1628–1643. doi: 10.1111/j.1471-4159.2007.04874.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Inan S, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]