CREB Is a Novel Nuclear Target of PTEN Phosphatase (original) (raw)
. Author manuscript; available in PMC: 2012 Apr 15.
Abstract
PTEN phosphatase is a potent tumor suppressor that regulates multiple cellular functions. In the cytoplasm, PTEN dephosphorylates its primary lipid substrate, phosphatidylinositol 3,4,5-trisphosphate, to antagonize the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway. It has also become increasingly evident that PTEN functions in the nucleus and may play an important part in transcription regulation, but its nuclear targets remain elusive. In this report, we demonstrate the transcription factor cyclic AMP response element-binding protein (CREB) is a protein target of PTEN phosphatase and that PTEN deficiency leads to CREB phosphorylation independent of the PI3K/AKT pathway. Using confocal immunofluorescence and reciprocal immunoprecipitation, we further show that PTEN colocalizes with CREB and physically interacts with CREB. Moreover, we use both in vitro and in vivo experiments to show PTEN can dephosphorylate CREB in a phosphatase-dependent manner, suggesting that CREB is a substrate of PTEN nuclear phosphatase. Loss of Pten results in an elevated RNA level of multiple CREB transcriptional targets and increased cell proliferation, which can be reversed by a nonphosphorylatable CREB mutant or knockdown of CREB. These data reveal a mechanism for PTEN modulation of CREB-mediated gene transcription and cell growth. Our study thus characterizes PTEN as a nuclear phophatase of a transcription factor and identifies CREB as a novel protein target of PTEN phosphatase, which contributes to better understanding of PTEN function in the nucleus.
Introduction
PTEN phosphatase is one of the most frequently lost or mutated genes in human cancers. In addition to inhibiting the phosphatidylinositol 3-kinase (PI3K)/AKT pathway as a lipid phosphatase, PTEN also acts as a protein phosphatase to regulate other cellular functions (1). For example, PTEN interacts with focal adhesion kinase (FAK) and reduces its tyrosine phosphorylation (2). A recent study reported that the regulatory subunit of PI3K, p85, could also serve as a protein substrate of PTEN phosphatase (3), which represents an alternative mechanism for negative regulation of PI3K by PTEN. These findings illustrate the importance of the protein phosphatase function of PTEN for control of multiple cellular processes.
Emerging evidence demonstrates that PTEN has various nuclear functions in addition to its established roles in the cytoplasm (4, 5). In addition to controlling chromosome stability and DNA repair (6, 7), nuclear PTEN is also involved in transcriptional regulation by targeting a variety of transcription factors (8, 9). The phosphorylation-dependent transcription factor, cyclic AMP response element-binding protein (CREB), has been found to mediate PTEN regulation of multiple genes including Bcl-2 (10) and cyclin D2 (11). CREB activation promotes cell survival and phosphorylation at serine 133 is an indicator of CREB activation (12). In this report, we characterize PTEN as a phosphatase acting directly on CREB, and describe a mechanism potentially underlying the involvement of PTEN in transcriptional regulation.
Our study demonstrates that PTEN can dephosphorylate CREB at Ser133 and that PTEN protein phosphatase activity is required for CREB dephosphoryation. Moreover, inhibition of PI3K does not affect the phosphorylation status of CREB in _Pten_-null cells, which excludes the possibility of involvement of the PI3K/AKT pathway in PTEN regulation of CREB. This direct regulatory relationship between PTEN and CREB is further supported by colocalization of these molecules in the nucleus as well as physical association. Our data therefore identify CREB as a protein substrate of PTEN phosphatase and confirm PTEN and CREB function in an enzyme-substrate relationship.
Materials and Methods
Cell culture and plasmids
Primary Pten+/+ and _Pten_−/− mouse embryonic fibroblasts (MEF, <passage 6; ref. 13), Pten+/− (PE2) and _Pten_−/− (PE2C) prostate epithelial cells were a gift from Dr. H. Wu at UCLA and normal human fibroblast NHF3 cells were generated from normal human skin. These cells were cultured in MEM supplemented with 10% FBS. Human cancer cell lines MDA-MB-175, MDA-MB-468, MDA-MB-231, and MDA-MB-415 were obtained and characterized by cytogenetic analysis by American Type Culture Collection (ATCC). All these cells were maintained according to the recommendation of ATCC. The insect cell line SF9 was obtained and characterized by morphological analysis from Invitrogen, and was cultured in Grace’s insect medium (Gibco).
The pSUPER RNAi system (Oligoengine) was used to construct PTEN- or CREB-specific siRNA expression plasmids. A pFastBac1 expression vector (Invitrogen) was used to construct plasmids expressing CREB or different forms of PTEN (wild-type, C124S or G129E) with His-tag.
In vitro binding and dephosphorylation assay
His-CREB, 3 forms of His-tagged PTEN (His-PTEN, His-PTENC124S, and His-PTENG129E), and a control His-tagged protein (His-control) were expressed in SF9 cells and purified using Ni-NTA agarose. For binding assay, purified His-CREB was incubated with His-PTEN or control protein, followed by immunoprecipitation with corresponding antibodies against PTEN or the control protein and immunoblotting detection of CREB. For dephosphorylation assay, purified His-CREB was incubated with either His-PTEN or the control protein, followed by immunoblotting of phospho-CREB (Ser133).
Coimmunoprecipitation, immunofluorescence, and confocal microscopy
Coimmunoprecipitation and immunofluorescence methods were described previously (7). Immunostained cells were analyzed for colocalization of PTEN and CREB using a Leica TCS SP5 confocal microscope with a Plan-Apochromat (60/1.40) oil-immersion objective. Images were captured with a Leica DFC 500 digital camera and processed with the LAS AF software (Leica).
Results
Loss of PTEN leads to upregulation of CREB phosphorylation in a PI3K/AKT-independent manner
In order to determine the effect of PTEN deficiency on the activation status of CREB, we compared the level of CREB phosphorylation at Ser133 in cells with and without Pten. As shown in Figure 1A, Pten deletion results in an elevated level of CREB phosphorylation. To validate this observation, we evaluated CREB phosphorylation in a panel of human breast cancer cell lines with either wild-type PTEN (MB175 and MB231) or mutant PTEN (MB468 and MB415); mouse prostate epithelial cells isolated from Pten conditional knockout mice, PE2 (Pten+/−) and PE2C (_Pten_−/−); and normal human fibroblasts with and without PTEN siRNA. In all these cells, CREB phosphorylation was consistently upregulated in association with PTEN deficiency, as compared with the counterpart control cells (Fig. 1B). These data argue strongly that enhanced phosphorylation of CREB is a feature common to PTEN deficiency.
Figure 1.

Loss of PTEN leads to upregulation of CREB phosphorylation, independent of PI3K/AKT activation. A, Western analysis of CREB phosphorylation in Pten+/+ and _Pten_−/− MEFs. B, phosphorylation status of CREB in different PTEN-deficient cell lines with counterpart controls as indicated. C, inhibition of the PI3K/AKT pathway does not affect CREB phosphorylation in _Pten_-null cells. _Pten_−/− MEFs treated with different doses of LY294002 were evaluated by Western blot for phosphorylation of CREB and AKT.
In order to determine whether activation of the PI3K/AKT pathway mediates CREB phosphorylation in PTEN-deficient cells, we analyzed CREB phosphorylation in _Pten_-null cells following suppression of the PI3K pathway. As expected, LY294002, a PI3K-specific inhibitor, suppresses AKT phosphorylation but fails to reduce CREB phosphorylation (Fig. 1C), showing that CREB phosphorylation is independent of activation of the PI3K/AKT pathway and suggesting it is a direct consequence of PTEN deficiency. These results raise the possibility that there is a direct regulatory relationship between PTEN phosphatase and CREB transcription factor.
PTEN colocalizes with CREB in the nucleus and is associated with CREB
Immunofluorescence and confocal microcopy were used to examine the subcellular distribution and spatial relationship of PTEN and CREB. CREB localizes primarily in the nucleus as punctuate foci as shown in Figure 2A. Although PTEN in Pten+/+ MEFs exhibits a more diffuse distribution pattern in both the cytoplasm and the nucleus, its nuclear staining appears to be aggregated in minute foci and a substantial proportion of these overlap with the CREB staining. These data suggest that PTEN may physically associate with CREB and participate in CREB-related nuclear function.
Figure 2.

PTEN colocalizes and interacts with CREB. A, colocalization of PTEN with CREB in MEFs. Pten+/+ and _Pten_−/− cells were immunostained for CREB (green) and PTEN (red), followed by confocal analysis of their colocalization. The yellow signal in the merged image of Pten+/+ MEFs represents spatial overlap of CREB and PTEN. Scale bars, 7.5 μm. B, reciprocal coimmunoprecipitation of PTEN and CREB in Pten+/+ and _Pten_−/− MEFs and HeLa cells containing PTEN siRNA. C, in vitro binding between PTEN and CREB. A His-tagged irrelevant protein (His-control) was included as a control.
The physical interaction of PTEN and CREB was further examined in MEFs and HeLa cells using reciprocal coimmunoprecipitation. PTEN is detectable in CREB-immunoprecipitates in Pten+/+ but not in _Pten_−/− MEFs (Fig. 2B, top left). In a similar manner anti-PTEN antibody precipitates CREB in cells containing wild-type Pten but not in _Pten_-null cells (Fig. 2B, bottom left). Consistent with findings in the mouse cell system, CREB interacts with PTEN in HeLa cells, and to a significantly reduced extent in HeLa cells containing PTEN siRNA (Fig. 2B, right).
In addition to these in vivo coimmunoprecipitation studies, we also investigated the ability of PTEN to directly interact with CREB in vitro. CREB was detected after incubation with His-PTEN, but not with the control protein (Fig. 2C). These data indicate that PTEN associates directly with CREB, providing a basis for a functional relationship.
PTEN dephosphorylates CREB in a phosphatase-dependent manner
To test our hypothesis that CREB serves as a protein substrate for PTEN phosphatase, we conducted an in vitro dephosphorylation assay using purified His-CREB and His-PTEN protein. As expected, CREB phosphorylation is greatly decreased in the presence of wild-type PTEN, as compared to the control protein (Fig. 3A). To evaluate the importance of PTEN phosphatase activity, we also included 2 naturally occurring mutants with different phosphatase deficiencies. PTENC124S lacks both lipid and protein phosphatase activity (14), whereas PTENG129E is lipid phosphatase deficient but retains protein phosphatase activity (15). It is important to note that the PTENC124S mutant fails to reduce CREB phosphorylation whereas the PTENG129E mutant remains effective in dephosphorylating CREB. These data demonstrate that protein phosphatase activity of PTEN is essential for suppression of CREB phosphorylation.
Figure 3.

PTEN dephosphorylates CREB in vitro and in vivo. A, in vitro phosphatase assay. Purified His-CREB was incubated with 3 forms of His-PTEN proteins or a control His-protein prior to evaluation of CREB phosphorylation. B, Western analysis of CREB phosphorylation in _Pten_−/− MEFs containing ectopic expression of wild-type or phosphatase-deficient PTEN. Phosphorylation of Akt is also shown to indicate PTEN phosphatase activity.
To further confirm the negative effect of PTEN on CREB phosphorylation and the role of PTEN phosphatase activity, we introduced wild-type PTEN as well as 2 phosphatase-deficient PTEN mutants into _Pten_-null cells and analyzed CREB phosphorylation. The elevated level of CREB phosphorylation in _Pten_-null cells is significantly reduced by wild-type PTEN and PTENG129E with deficient lipid phosphatase activity (Fig. 3B). However, CREB phosphorylation is not reduced by PTENC124S which lacks both protein and lipid phosphatase activity. These results argue that dephosphorylation of CREB is dependent on PTEN protein phosphatase activity, and CREB is therefore a protein substrate of PTEN phosphatase.
Pten deletion induces CREB downstream gene expression and cell growth, which can be reversed by CREB knockdown or a CREBS133A mutant
In order to expand upon our observations on PTEN regulation of CREB and assess downstream signaling and phenotype, we examined gene expression of CREB transcriptional targets and evaluated resultant cell growth. We sought to determine whether deletion of Pten leads to upregulation of c-Myc and Bcl2, given these genes are downstream of CREB and involved in cell survival. Quantitative real-time PCR showed an increased level of c-Myc and Bcl-2 in _Pten_−/− MEFs as compared with Pten+/+ cells (Fig. 4A). Interestingly, knockdown of CREB by siRNA or overexpression of a nonphosphorylatable CREB mutant, S133A, significantly reverses the gene upregulation. These results indicate that CREB activation is the reason for the upregulation of c-Myc and Bcl2 in Pten deficient cells.
Figure 4.
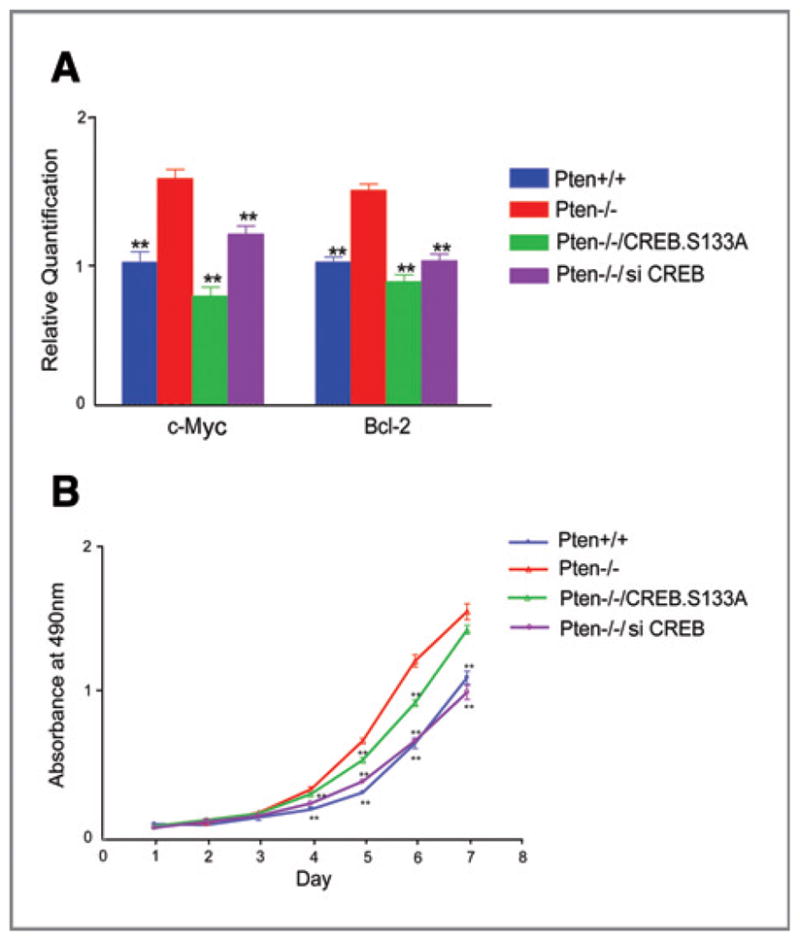
CREB activation is responsible for upregulated CREB transcriptional targets and increased cell growth in Pten null cells. _Pten_−/− MEFs were transfected with CREB siRNA or a nonphosphorylatable CREB mutant, CREBS133A. A, quantitative real-time PCR analysis of Bcl2 and c-Myc expression. The expression levels of c-Myc and Bcl2 were normalized to GAPDH. B, measurement of cell growth by MTT assay. Data represent the mean ± SEM; **, P < 0.01 as compared with _Pten_−/− cells.
Subsequently, we found that _Pten_−/− cells grow much faster than Pten+/+ cells and a significant difference in cell growth was observed in Pten+/+ and _Pten_−/− cells. More interestingly, the higher rate of cell growth in _Pten_-null cells is significantly compromised when CREB is knocked down or when cells overexpress CREBS133A. These results are consistent with the hypothesis that loss of Pten induces expression of CREB downstream targets, and suggest the increased cell growth results from CREB-mediated transcriptional activation and cell proliferation in _Pten_-null cells.
Discussion
In this study, we demonstrate that PTEN uses its protein phosphatase function to regulate the transcription factor CREB. PTEN physically associates with CREB and dephosphorylates CREB in a phosphatase-dependent but PI3K/AKT-independent manner. Our data therefore identify CREB as a protein substrate of PTEN phosphatase in the nucleus, and reveal a novel mechanism wherein PTEN is involved in transcription regulation.
CREB is an important transcription factor as it resides at the center of a complicated regulatory network. CREB can be phosphorylated by various kinases depending on cellular stimuli (16). On the other hand, CREB can also be dephosphorylated by several protein phosphotases, such as PP1 (17) and PP2A (18). PTEN has previously been considered to indirectly regulate CREB through inhibition of AKT (19, 20). However, our study demonstrates a novel mechanism for PTEN regulation of CREB wherein PTEN interacts directly with CREB and dephosphorylates CREB through PTEN protein phosphatase activity. We show that the activation of the PI3K/AKT pathway is not involved in PTEN suppression of CREB phosphorylation. Moreover, our in vivo and in vitro experiments confirm direct interaction between PTEN and CREB. PTEN lipid phosphatase activity is unnecessary for downregulation of CREB phosphorylation, arguing that the PI3K/AKT pathway is not involved in PTEN dephosphorylation of CREB. We therefore conclude that PTEN can act as a nuclear phosphatase that directly targets CREB and thus participate in transcriptional regulation.
In summary, we identify CREB as a novel substrate of PTEN phosphatase in the nucleus and propose a new model in which CREB is dephosphorylated in a manner dependent of PTEN protein phosphatase activity. Our data emphasize the potential importance of the protein phosphatase function of PTEN, which is incompletely understood and clearly warrants further investigation. By demonstration of PTEN regulation of the transcription factor CREB, we highlight a role of PTEN in controlling the transcriptional machinery. Our study may thus serve as a starting point for more extensive exploration of PTEN protein targets in both the cytoplasm and the nucleus, which may ultimately reveal a PTEN driven regulatory signaling network.
Acknowledgments
Grant Support
This study was supported by NIH grant (R01 CA102447), China National Major Scientific Program (973 Project-2010CB912202), National Natural Science Foundation of China (Key Project-30930021), Beijing Natural Science Foundation (Major Project-5100003), and Shu Fan Education and Research Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052–7. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–7. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 3.He J, de la Monte S, Wands JR. The p85beta regulatory subunit of PI3K serves as a substrate for PTEN protein phosphatase activity during insulin mediated signaling. Biochem Biophys Res Commun. 2010;397:513–9. doi: 10.1016/j.bbrc.2010.05.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker SJ. PTEN enters the nuclear age. Cell. 2007;128:25–8. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 5.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 6.Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 8.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 9.Mayo MW, Madrid LV, Westerheide SD, Jones DR, Yuan XJ, Baldwin AS, et al. PTEN blocks tumor necrosis factor-induced NF-kappa B-dependent transcription by inhibiting the transactivation potential of the p65 subunit. J Biol Chem. 2002;277:11116–25. doi: 10.1074/jbc.M108670200. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Cheville JC, Pan Y, Roche PC, Schmidt LJ, Tindall DJ. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J Biol Chem. 2001;276:38830–6. doi: 10.1074/jbc.M103632200. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Chang HY, Fei T, Wu H, Chen YG. GSK3 beta mediates suppression of cyclin D2 expression by tumor suppressor PTEN. Oncogene. 2007;26:2471–82. doi: 10.1038/sj.onc.1210033. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–80. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 13.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–7. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 15.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 16.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 17.Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, et al. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;70:105–13. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- 18.Wadzinski BE, Wheat WH, Jaspers S, Peruski LF, Lickteig RL, Johnson GL, et al. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1993;13:2822–34. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–9. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 20.Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–6. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]