Sequence Mapping by Electronic PCR (original) (raw)
Abstract
The highly specific and sensitive PCR provides the basis for sequence-tagged sites (STSs), unique landmarks that have been used widely in the construction of genetic and physical maps of the human genome. Electronic PCR (e-PCR) refers to the process of recovering these unique sites in DNA sequences by searching for subsequences that closely match the PCR primers and have the correct order, orientation, and spacing that they could plausibly prime the amplification of a PCR product of the correct molecular weight. A software tool was developed to provide an efficient implementation of this search strategy and allow the sort of en masse searching that is required for modern genome analysis. Some sample searches were performed to demonstrate a number of factors that can affect the likelihood of obtaining a match. Analysis of one large sequence database record revealed the presence of several microsatellite and gene-based markers and allowed the exact base-pair distances among them to be calculated. This example provides a demonstration of how e-PCR can be used to integrate the growing body of genomic sequence data with existing maps, reveal relationships among markers that existed previously on different maps, and correlate genetic distances with physical distances.
In recent years mapping strategies have focused on the use of sequence-tagged sites (STSs) as landmarks of the genome (Olson et al. 1989). Operationally, an STS is defined by a pair of oligonucleotide primers that can be used in a PCR assay to detect a site that is unique in the genome. In some cases, the size of the amplified PCR product may be polymorphic, which allows the transmission of allelic variants within families to be studied. This property is essential for STSs used in genetic mapping, whereas any STS can be used for physical mapping. The chief advantage of STSs over other types of markers is that there is no absolute requirement to maintain and distribute any biological materials. Instead, markers can be stored in computer databases and disseminated over electronic networks. This is due to the fact that it is easy and relatively inexpensive to synthesize oligonucleotides, thereby allowing any laboratory around the world to regenerate the necessary reagents to assay for a given marker.
Because STSs are defined by sequence, it is possible to identify these landmarks in DNA sequences by searching for subsequences of a query sequence that match the PCR primers and are in the correct order, orientation, and spacing to be consistent with the PCR product size. We call this procedure electronic PCR (e-PCR). The significance of this technique can be seen by considering that it is possible to determine the map location of a new sequence without performing a single experiment in the laboratory. This report describes a software tool for performing e-PCR in an efficient manner and discusses several potential applications for genomic research.
Sources of STS Data
The STS division of GenBank (dbSTS) is used within the mapping community for bulk submission of STS sequences, PCR reaction conditions, and mapping information (Benson et al. 1996). Figure 1a shows a few database fields from a typical record that are of primary interest for use in e-PCR: various names and identifiers, the sequences of the forward and reverse primers, the size of the PCR product, and the full sequence of the amplified region (the amplicon). Although many of the STSs in the database are of human origin, several other model organisms are represented by smaller numbers of STSs. Additional sources of human STS data include the Genome Data Base (Fasman et al. 1996) and the Radiation Hybrid Database (http://www.ebi.ac.uk/RHdb/).
Figure 1.
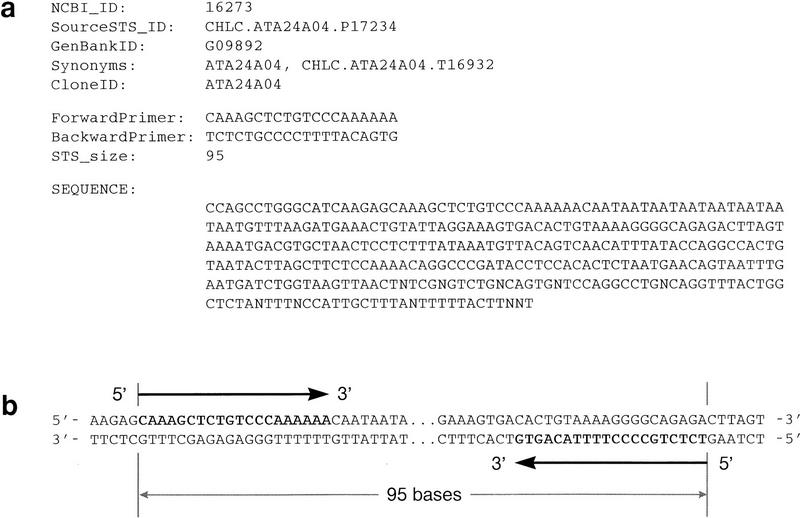
PCR primer sequences from a typical dbSTS record and their relationship to a query sequence that might be searched by e-PCR. (a) A few selected fields are shown from dbSTS record 16273 (GenBank accession no. G09892), including various names and identifiers, the sequences of the forward and reverse primers (both in 5′ → 3′ orientation), the size of the PCR product, and the sequence of the amplicon and flanking regions. (b) For a query sequence that is the same sense as the sequence of the dbSTS record, a successful match will include the forward primer followed by the inverse (i.e., reverse-compliment) of the reverse primer. On the other hand, if the query sequence is of the opposite sense (imagine the lower strand reversed), it will be the reverse primer followed by the inverse of the forward primer.
Within the last 2 years, several “whole-genome” STS-based human mapping projects have been described. One significant milestone is the recent completion of the Généthon genetic map (Dib et al. 1996), which contains 5264 STSs developed from microsatellite sequences. Several physical maps have been published, such as those developed by the Centre d’Etude du Polymorphisme Humain (CEPH), which contain 2601 markers (Chumakov et al. 1995); the Whitehead Institute and Généthon, which contain 15,086 markers (Hudson et al. 1995); Généthon and Cambridge University, which contain 850 markers (Gyapay et al. 1996); and Stanford University, which contains 5994 markers (Stewart et al., this issue). Finally, a transcript map, which contains 20,128 cDNA-based markers representing ∼16,000 distinct genes, has recently been constructed by an international consortium of mapping laboratories (Schuler et al. 1996). Projects such as these, not to mention many chromosome-specific and regional maps, have resulted in a substantial expansion in the number of STSs in the database. Consider, for example, that the GenBank STS division contained 7532 entries when it first appeared in October 1994 while the number has swelled to 44,102 sequences in the December 1996 release—a roughly 6-fold increase in a period of just over 2 years.
Software for e-PCR
The process of PCR is difficult to model in detail because a variety of poorly understood factors affect whether or not a particular pair of primers will lead to a successful outcome (Bangham 1991). Despite this, a computational strategy for identifying the most obvious STSs would be extremely useful. A straightforward attempt would involve searching for subsequences exactly matching the two primers and then checking to see that they are in the correct configuration (i.e., with their 3′ ends pointing toward one another) and that their spacing is consistent with the known size of PCR product. For increased sensitivity, some allowance could be made for mismatches in the primer sites and polymorphism in the amplicon size.
One possible approach, which makes use of widely available software (both public domain and commercial), is to specify each STS as a pattern, or “regular expression,” with the two primer sequences separated by a (possibly variable-length) spacer of arbitrary characters. It should be noted that STS databases store both primers in their 5′ → 3′ orientations, which is perfectly natural for those attempting to synthesize the oligonucleotides but inconvenient for the purposes of sequence analysis because the reverse primer must be inverted before attempting to match it against the sequence (see Fig. 1b). Moreover, it is usually necessary to construct two expressions per STS, one for each of the DNA strands, because most regular expression search programs consider only one strand at a time. Using “standard” regular expression syntax, it is not possible to specify the exact length of the spacer between the primers, although some programs may use an extended syntax to allow this. Search speed may also be an issue in situations where it is desirable to search large collections of sequences against the STS database. Regular expression programs are designed for flexibility and the ability to handle sophisticated expressions and may not be maximally efficient when searching for the relatively simple patterns required by e-PCR.
For both convenience and performance considerations, it was useful to develop a special-purpose program for performing e-PCR. This program (simply called e-PCR) requires one file containing the PCR primers and amplicon sizes for the STSs of interest and one file containing arbitrarily large numbers of query sequences to be searched en masse. To be reported, matches to both primers must be found and the order and orientation of the primer sequences must be consistent with their role in priming the PCR reaction. That is, either the forward primer must be followed by the inverse of the reverse primer (for a plus strand hit) or the reverse primer must be followed by the inverse of the forward primer (for a minus strand hit; see also Fig. 1b). The portion of the amplicon falling between the primers is not considered when evaluating a match.
A word-based strategy is used to significantly speed up the search for the primers within the query sequence. This is done by extracting a string of W consecutive letters (a “word”) from the 3′ end of each primer and converting it to a unique integer (a “hash value”). Using the hash value for indexed access to a table of STS information allows potential matches to be evaluated efficiently as the query sequence is scanned. Candidate STSs are recognized as occurrences of the two words taken from the forward and reverse primers that are spaced such that the predicted amplicon size is within a “margin” of M bases on either side of the expected size. When these criteria are satisfied, a secondary comparison is triggered in which the complete primers are compared against the query sequence, allowing up to N mismatching bases for each primer. In the default mode of operation, no mismatches are allowed (N = 0), which allows the program to report only sites that are certain to be authentic STSs. This is useful for automated analysis of large volumes of data because the results need not be inspected manually. However, it should be recognized that some STSs could be missed either because the primers do not match exactly or because the query sequence may contain errors. Increasing the value of N will allow more potential STSs to be found, but doing so may also result in some false positives being reported. It should be noted that when mismatches are allowed, they may not be within the W bases used to compute the hash value. This apparent limitation is justified by the fact that words are extracted from the 3′ ends of the primers where mismatches cannot be tolerated easily by PCR (Sommer and Tautz 1989). The value of W can be reduced from its default value of 7 to reduce the chance of missing a true STS, but speed is sacrificed in the process. The default value of M is 50, which should accommodate length variations for most polymorphic STSs. However, a larger setting might be desired for certain applications, for instance, to handle the case where primers are designed from cDNA sequences but turn out to be in different exons in genomic DNA.
The e-PCR program is sufficiently rapid that on one common computer architecture it was possible to test all human sequences in GenBank (excluding the STS division; 609,257 sequences) against all of the human STSs (36,973 primer pairs) in <1 hr. The running time scales linearly with the aggregate query sequence length. The memory requirement is modest and increases linearly with the number of STSs searched. The source code for the e-PCR program is freely available (ftp://ncbi.nlm.nih.gov/pub/schuler/e-PCR/).
Why Not Just Use BLAST?
One might wonder why a special search tool need be created at all, considering the widespread availability of general-purpose database search tools such as BLAST (Altschul et al. 1990). dbSTS is one of the standard databases available for searching when using the BLAST network service. However, attempting to use BLAST to identify STSs can lead to many false positives in some cases. With gene-based STSs for instance, there will be sufficient sequence similarity among related gene family members and pseudogenes to result in confounding matches being reported. But perhaps the worst case is encountered when the query sequence contains simple sequence repeats, such as those that are the basis for most polymorphic markers.
To demonstrate the problem posed by repetitive sequences, the mRNA sequence of Br-cadherin (GenBank accession no. L33477) (Selig et al. 1995) was used as the query in a BLAST search against the dbSTS database. This sequence happens to contain a (CA)n microsatellite sequence in its 3′-untranslated region (3′ UTR) that corresponds to the Généthon marker D5S411. The output from this search was quite voluminous, but a portion of it has been reproduced in Figure 2. The best match was to the sequence corresponding to D5S411 (GenBank accession no. Z16831), but thousands of additional hits were also observed, including a few to sequences from organisms other than human (by default BLAST shows only the first 500 hits, but relaxing this limit resulted in a list of >8000). A list of the best 20 matches is shown in Figure 2a. All but the first one are false positives, showing sequence similarity only to the (CA)n repeats and not to any flanking unique sequence (see Fig. 2b for one example). The problem of simple sequence repeats (also known as “low-complexity regions”) causing false positives in database searches has been noted previously and is dealt with typically by “masking” such regions (by converting them to Ns) prior to performing the search (Altschul et al. 1994; Wootton and Federhen 1996). Although this does reduce the problem, it does not eliminate it completely so some manual inspection of the results must still be performed.
Figure 2.
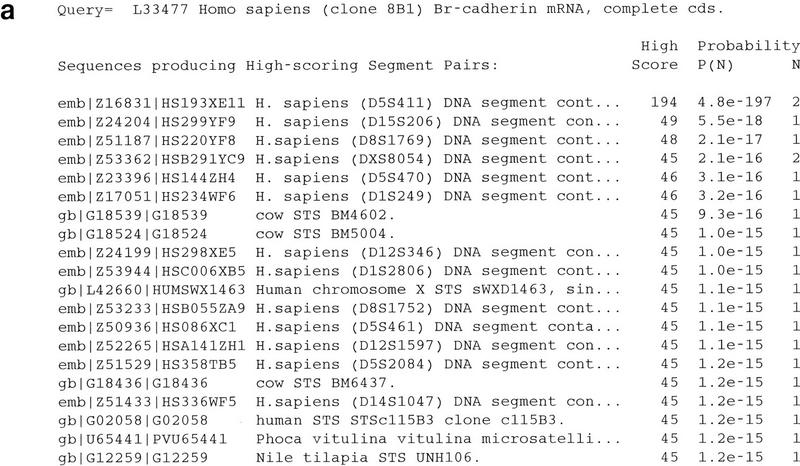

BLAST search with a microsatellite-containing query sequence. The BLASTN program was used to search the dbSTS database with the sequence corresponding to GenBank entry L33477 (Br-cadherin) as the query sequence using a match score (M parameter) of 1 and a mismatch score (N parameter) of −2. The query sequence was not filtered for low-complexity sequences. (a) The first 20 sequences listed on the resulting “hit list” are shown, sorted by statistical significance. Altogether, >8000 sequence matches were found (by default, only the first 500 are shown, but the complete list can be obtained by setting the V parameter to a very large number). The best match was observed against the sequence corresponding to GenBank entry Z16831, which contains the Généthon marker D5S411. When low-complexity filtering is used, the problem is reduced dramatically, but 10 false positives remain so manual inspection of the results is still required. (b) The sequence alignment generated by BLAST for the sequence corresponding to GenBank entry Z24204, which was the second-best hit reported, contains the Généthon marker D15S206. The alignment includes only the (CA)n microsatellite repeats.
It should be noted that BLAST was designed to solve the somewhat different problem of finding sequences related to the query, allowing for some level of mismatching, but extending over a long enough region that there is sufficient information to distinguish an observed similarity from a chance occurrence. BLAST is more analogous to “electronic hybridization,” with the scoring parameters taking the place of the hybridization temperature in determining stringency. Extending the sequence comparison to the longer sequence of the amplicon, instead of focusing on the PCR primers alone, results in a loss of specificity—just as hybridization is less specific than PCR in the laboratory.
How Many Hits Can Be Expected?
An important question for users of the e-PCR tool is how many hits can be anticipated for a “typical” search. This is a difficult question to answer because the results depend on many factors such as the length of the query sequence, the size of the STS database, and other properties of both the query sequences and the STSs that might predispose them to matching.
When using STSs that are randomly distributed throughout the genome, it is easy to see that longer query sequences would have an increased likelihood of containing a matching STS than would shorter sequences. To demonstrate this effect, human genomic sequences of various size classes were tested for the presence of microsatellite-based STSs from the Généthon genetic map (Dib et al. 1996). As expected, larger sequences were found to have proportionately greater frequencies of containing sites (see Table 1). For a sequence in the 30- to 40-kb size range (about the size of a typical cosmid insert), 5% of the sequences were found to contain a Généthon marker. This increases steadily with sequence length to a level of 34% for a sequence >100 kb (a typical size range for bacterial artificial chromosome inserts). This is roughly consistent with expectations: A random arrangement of 5264 markers (Dib et al. 1996) over a genome of 3200 Mb (Morton 1991) should result in ∼1.65 sites per Mb; on average, 1 STS every 608 kb. This suggests about a 1:6 chance of a 100-kb sequence containing a site or about 1:3 chance for a 200-kb sequence. In one instance, a 223-kb sequence (GenBank accession no. U47924) was found to contain two Généthon markers. The apparent number of sites per megabase was lower with shorter sequences, but this is most likely attributable to a bias in this fraction toward single gene-oriented entries in this fraction, as opposed to random large-insert clones that predominate the larger size categories; microsatellites may be less likely to occur within genes than in random DNA.
Table 1.
Numbers of Généthon Microsatellite Markers Found in Human GenBank Sequences of Different Lengths
| Sequence length (kb) | Sequences searched | Sequences with sites (%) | Sites per Mb |
|---|---|---|---|
| ⩾100 | 41 | 14 (34) | 2.30 |
| 50–100 | 46 | 5 (11) | 1.47 |
| 40–50 | 87 | 5 (6) | 1.34 |
| 30–40 | 165 | 8 (5) | 1.35 |
| 20–30 | 87 | 1 (1) | 0.46 |
Dependencies on sequence length and database size may be obvious, but factors such as source of the material can be of even greater consequence. For example, large numbers of STSs have been developed from transcribed sequences (Schuler et al. 1996). Thus, data sources that are enriched for gene sequences, cDNAs for instance, would be expected to have an increased likelihood of bearing a match. To illustrate this effect, several categories of human sequences were compared against databases consisting of STSs derived from random DNA fragments, microsatellite repeats, and transcribed sequences (see Table 2). The genomic sequences used in this test are the same as those described above (Table 1) and, when normalized for the differing sizes of the STS collections, show nearly identical hit rates for all three types of STSs. As expected, however, sources of sequence data derived from mRNAs, including single-pass expressed sequence tag (EST) sequences, show substantially higher numbers of matches with transcript-derived STSs compared to those derived from random DNA fragments and microsatellite repeats. The most pronounced effect was seen when searching with EST sequences from 3′ reads of oligo(dT)-primed cDNA clones, in which the normalized hit rate was about three orders of magnitude greater than was observed with random STSs. Furthermore, the numbers of transcript sites for 3′ ESTs were much greater than for 5′ ESTs, whereas no significant difference between these two sequence categories was observed for random and microsatellite markers. All of these observations can be explained by the fact that 3′ ESTs have been the primary source of material used in development of the transcript-based STSs.
Table 2.
Numbers of Matches Found with Various Types of Sequences and STSs
| Sequence type/ STS type | Sequences searched | Sequences with matching sites (%) | Sequences with sites/1000 STSs |
|---|---|---|---|
| Genomic | 426 | ||
| random | 24 (6) | 6.8 | |
| microsatellite | 31 (7) | 5.9 | |
| transcript | 133 (31) | 8.3 | |
| mRNA | 16,691 | ||
| random | 3 (<0.01) | 0.85 | |
| microsatellite | 7 (<0.01) | 1.3 | |
| transcript | 3,553 (21) | 221 | |
| ESTs, 5′ reads | 248,589 | ||
| random | 18 (<0.01) | 5.1 | |
| microsatellite | 4 (<0.01) | 0.76 | |
| transcript | 8,591 (3) | 534 | |
| ESTs, 3′ reads | 211,942 | ||
| random | 16 (<0.01) | 4.5 | |
| microsatellite | 3 (<0.01) | 0.57 | |
| transcript | 49,617 (23) | 3,082 |
An Example Application
GenBank entry U47924 was chosen to illustrate some practical applications of e-PCR. As noted above, this 223-kb sequence contains two Généthon markers. It originates from a gene-rich region at 12p13 and contains 17 complete protein-coding genes (plus one partial gene, one pseudogene, and one snRNA gene) (Ansari-Lari et al. 1996). Several of them are novel, but four of them correspond to previously known chromosome 12 genes: cell-surface antigen CD4, the B3 subunit of G proteins (GNB3), triose phosphate isomerase (TPI), and ubiquitin isopeptidase T (ISOT). The results of analyzing this sequence by e-PCR using a database consisting of all STSs from the human transcript map (Schuler et al. 1996) and the Généthon genetic map (Dib et al. 1996) are shown in Table 3. Hits to Généthon markers D12S1623 and D12S1625 reveal two polymorphic sites and firmly place the sequence on chromosome 12 at a genetic position of 17.1–17.9 cM (see Fig. 3). Furthermore, assuming the marker order of the Généthon map to be correct, the orientation of the U47924 sequence with respect to the centromere and the 12p telomere can be established. Among the nine gene-based markers detected, all but one are consistent with the chromosome 12 assignment, and they additionally indicate the positions of expressed genes. In Figure 3a, the locations of the gene coding sequences and the direction of transcription are indicated. It can be seen that the sites reported for the transcript-based STSs very often correspond to the 3′ ends of genes, which is consistent with the strategy that was used in the development of these markers (Schuler et al. 1996). In the case of the CD4 gene, two markers (SHGC-12737 and A007D38) were found, spaced ∼1 kb apart but still within the 3′ UTR. Overall, 8 of the 17 genes in this region contained a match to a marker from the human transcript map. The results of this analysis suggest that e-PCR could be used for automated sequence annotation. It should be noted that the annotation of GenBank entry U47924 does include the locations of Généthon markers D12S1263 and D12S1265 (using alternate identifiers) but understandably lacks the transcript-based markers that were not published until after the sequence was submitted.
Table 3.
Analysis of The Sequence Corresponding to GenBank Entry U47924 by e-PCR
| Base range | Marker | Map position |
|---|---|---|
| 24658–24878 | D12S1623 | Chr. 12, 17.1 cM, Généthon AFMa240zf5 |
| 31351–31627 | SHGC-12737 | Chr. 12, between D12S328 and D12S1695 |
| 32580–32799 | A007D38 | Chr. 12, between D12S328 and D12S89 |
| 78482–78629 | SGC32489 | Chr. 12, between D12S93 and D12S77 |
| 82714–82824 | WI-9250 | Chr. 1, between D1S216 and D1S500 |
| 135029–135182 | SHGC-10753 | Chr. 12, between D12S328 and D12S1695 |
| 154204–154327 | stSG2394 | Chr. 12, linked to D12S328 at LOD 4.5 |
| 157865–158008 | SHGC-31976 | Chr. 12, between D12S328 and D12S1695 |
| 177396–177550 | Cda14d08 | Chr. 12, between D12S328 and D12S89 |
| 191757–191847 | Cda19d08 | Chr. 12, between D12D328 and D12S89 |
| 208292–208565 | D12S1625 | Chr. 12, 17.9 cM, Généthon AFMa247yc9 |
Figure 3.
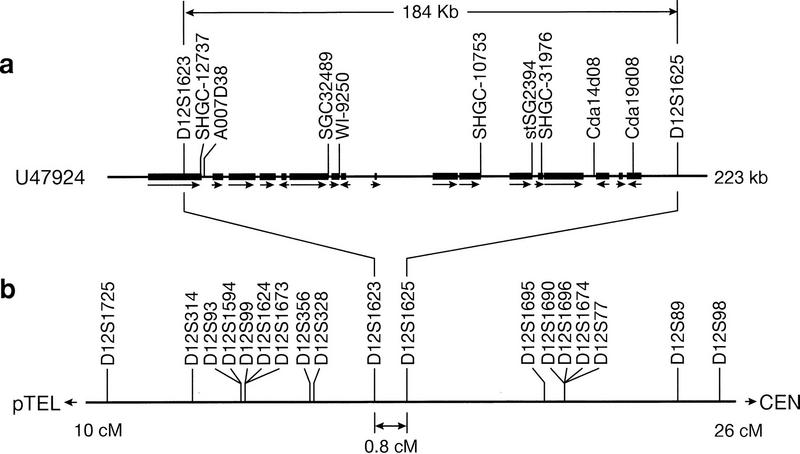
Alignment of the sequence corresponding to GenBank entry U47924 to the Généthon map. (a) A schematic representation of the 223-kb sequence is shown, with solid boxes showing the extent of the coding sequences for each gene (for clarity, the exon/intron structure is not indicated) and arrows showing the direction of transcription. One partial gene with its 3′ UTR, but no coding sequence, spanning the right boundary of the sequence, was not shown. In addition, a pseudogene and an snRNA gene documented in the region are not shown. STSs identified by e-PCR analysis with microsatellite and gene-based markers are indicated. Eight of the nine gene-based markers were found within the 3′ UTR, which is consistent with the strategy that was used in their development. (b) A portion of the Généthon genetic map of chromosome 12 (from 10 to 26 cM) is reproduced, with lines drawn to show how the sequence (GenBank entry U47924) can be aligned to it based on the presence of markers D12S1623 (at 17.1 cM) and D12S1625 (at 17.9 cM).
Looking at the problem from the other point of view, analysis of the sequence provides a useful way to validate the map, at least in a localized region. In the construction of the human transcript map, participating laboratories assigned gene-based STSs to various intervals defined by Généthon microsatellite markers to allow the results to be integrated with each other and with the genetic map (Schuler et al. 1996). Based on the e-PCR analysis of accession no. U47924, the true interval for all of the transcript markers shown in Table 3 is between D12S1623 and D12S1625. It was therefore of interest to see whether the transcript map positions reported for these markers were consistent with this observation. One error is clearly apparent involving the interval for marker WI-9250, which is listed as being on chromosome 1, whereas the remaining body of evidence points to a chromosome 12 assignment. It is perhaps noteworthy that this marker is the only one in the region to have been placed by yeast artificial chromosome (YAC) contig mapping; all of the others in this region were mapped using radiation hybrid panels. Consequently, this inconsistency may be of use in diagnosing mapping artifacts, for instance, those caused by doubly chimeric YACs. However, apart from this one error, the intervals reported for the remaining cDNA markers are all correct, albeit at lower resolution than can be deduced by sequence analysis. This may be seen by comparing the intervals in Table 3 with the portion of the Généthon map of chromosome 12 shown in Figure 3b.
DISCUSSION
This report describes the concept of e-PCR and a software tool that provides an efficient implementation of the basic search strategy. The number of expected STS hits depends on a variety of factors, but those that are reported (using the default parameters) are unequivocal. This differs from the experience using BLAST, in which many false positives were reported. The use of this program in the analysis of one large sequence record demonstrated some practical applications of the program, but several others may be envisioned.
One straightforward application of e-PCR is the large-scale assignment of sequence database records to map positions. This is especially useful for functionally cloned genes and ESTs, for which mapping information may not be initially available. In the case of large-scale genomic sequencing, it is common to use a “sequence-ready map” to select large-insert clones for sequencing so that to some extent the map position will be known in advance. Nonetheless, it is always encouraging to verify that STSs that should be present can be detected in the final sequence.
To simplify the process and make it more widely available, an e-PCR search facility recently has been added to the National Center for Biotechnology Information (NCBI) site on the World Wide Web (http://www.ncbi.nlm.nih.gov/cgi-bin/STS/nph-sts). It allows the user to insert one or more DNA sequences, which are then compared against all PCR primer pairs in dbSTS. In the output of such a search, nucleotide positions of the STSs within the query sequence are given, together with expected and observed amplicon sizes, marker names, and chromosome numbers. Hypertext links to GenBank and dbSTS records are provided for more detailed mapping information and PCR reaction conditions.
When developing new markers for mapping studies, e-PCR can be used to test potential primers in various ways before actually incurring the expense of oligonucleotide synthesis. For instance, one could determine whether a new STS is essentially a duplicate of one already in hand by searching the sequence database for entries that contain them both in close proximity. In addition, potential cross-reactivity with other members of a gene family (which would violate uniqueness of the site in human) or in their rodent homologs (which would be a concern for mapping techniques involving somatic hybrids with rodent cells) could be tested, provided that the relevant sequences are available. One common source of mapping failures is unwittingly selecting PCR primers in repetitive DNA. Although it would be trivial to screen candidate primers against a database of known repeats, these collections are likely to be incomplete considering that new classes of repeats are continually being discovered. An alternative test would be to match the proposed primers against all human genomic sequences in the database to determine whether the hits are greater in number than expected on statistical grounds or involve sequences from several different chromosomes.
With the accelerating pace of large-scale genomic sequencing, there is significant interest in methods for annotating sequences that can be fully automated. Most of the attention has, appropriately, been focused on predicting genes. However, annotating the locations of STSs would also be a valuable activity. In addition to the fact that they are established landmarks of the genome, some STSs provide additional information because they have been developed from specific sequence sources, such as microsatellites (which indicate polymorphic sites), CpG islands (which are often associated with the 5′ ends of genes), and 3′ UTRs (which mark the 3′ gene boundaries). Moreover, the use of e-PCR for STS annotation would be quite easy to automate because the unequivocal nature of the results obviates the need for human intervention.
In this study, localized map validation was demonstrated using e-PCR analysis of a single GenBank entry, but this will become increasingly feasible to do in a more widespread fashion as it becomes more common to have large sequence contigs spanning perhaps 1 Mb or more. This is somewhat analogous to common validation practices involving comparisons of restriction maps determined experimentally with those generated by computer analysis of the sequence. Furthermore, it will be possible, at least in localized regions, to integrate different STS-based maps with each other. Traditionally, map integration has been made difficult because of an insufficient number of markers that are shared across all maps. But once the sequence becomes known, this ceases to be a problem because all STSs will be detectable in the sequence regardless of the source.
One might argue that the completion of the human genomic sequence will make today’s physical maps obsolete. But this is not true of genetic maps, which are the starting point for localizing disease susceptibility and other phenotypes to specific chromosomal regions. Thus, integrating genetic maps and sequence data will be of considerable interest for years to come. Moreover, identifying the positions of genetic markers in sequences will reveal the precise physical distances corresponding to genetic intervals, allowing “hot” and “cold” spots of meiotic recombination to be discerned.
Even before the complete sequence of the human genome is known, we can begin to assemble a composite sequence map consisting of islands of sequence tethered to physical and genetic maps. This is precisely the strategy used in the construction of the human sequence map presented in the Entrez Genomes division (http://www3.ncbi.nlm.nih.gov/Entrez/). By making use of positional data from two physical maps (from the Whitehead Institute and Stanford University) and two genetic maps (from Généthon and the Cooperative Human Linkage Consortium), GenBank sequences have been anchored using STSs identified by e-PCR. It can be anticipated that this technique will continue to play a role in assembly and validation of the human genomic sequence as the Human Genome Project approaches completion.
Acknowledgments
Special thanks go to Eric Green and Gerry Bouffard for helpful discussions and to Mark Boguski for critical review of the manuscript. Many suggestions for improvement of the software have been provided by Jinghui Zhang, Sergei Shavirin, and Gerry Bouffard. The NCBI’s World Wide Web resource for performing e-PCR against dbSTS was developed by Sergei Shavirin. Maintenance of the dbSTS database is performed by Jane Weissman and Carolyn Tolstoschev. The integrated genetic, physical, and sequence maps in the Entrez Genomes division are maintained by Jinghui Zhang.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Boguski MS, Gish W, Wootton JC. Issues in searching molecular sequence databases. Nature Genet. 1994;6:119–129. doi: 10.1038/ng0294-119. [DOI] [PubMed] [Google Scholar]
- Ansari-Lari MA, Muzny DM, Lu J, Lu F, Lilley CE, Spanos S, Malley T, Gibbs RA. A gene-rich cluster between the CD4 and triosephosphate isomerase genes at human chromosome 12p13. Genome Res. 1996;6:314–326. doi: 10.1101/gr.6.4.314. [DOI] [PubMed] [Google Scholar]
- Bangham CRM. The polymerase chain reaction: Getting started. In: Mathew CG, editor. Protocols in human molecular genetics. Clifton, NJ: Humana Press; 1991. pp. 1–8. [Google Scholar]
- Benson DA, Boguski M, Lipman DJ, Ostell J. GenBank. Nucleic Acids Res. 1996;24:1–5. doi: 10.1093/nar/24.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumakov IM, Rigault P, Le Gall I, Bellanne-Chantelot C, Billault A, Guillou S, Soularue P, Guasconi G, Poullier E, Gros I, Belova M, Sambucy J-L, Susini L, Gervy P, Glibert F, Beaufils S, Bui H, Massart C, De Tand M-F, Dukasz F, Lecoulant S, Ougen P, Perrot V, Saumier M, Soravito C, Bahouayila R, Cohen-Akenine A, Barillot A, Bertrand S, Codani J-J, Caterina D, Gorges I, Lacroix B, Lucotte G, Sahbatou M, Schmit C, Sangouard M, Tubacher E, Dib C, Faure S, Fizames C, Gyapay G, Millasseau P, NGuyen S, Muselet D, Vignal A, Morrissette J, Menninger J, Lieman J, Desai T, Banks A, Bray-Ward P, Ward D, Hudson T, Gerety S, Foote S, Stein L, Page DC, Lander ES, Weissenbach J, Le Paslier D, Cohen D. A YAC contig map of the human genome. Nature. 1995;377:175–297. doi: 10.1038/377175a0. [DOI] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Fasman KH, Letovsky SI, Cottingham RW, Kingsbury DT. Improvements to the GDB Human Genome Data Base. Nucleic Acids Res. 1996;24:57–63. doi: 10.1093/nar/24.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyapay G, Schmitt K, Fizames C, Jomes H, Vega-Czarny N, Spillett D, Muselet D, Prud’homme J-F, Dib C, Auffray C, Morissette J, Weissenbach J, Goodfellow PN. A radiation hybrid map of the human genome. Hum Mol Genet. 1996;5:339–346. doi: 10.1093/hmg/5.3.339. [DOI] [PubMed] [Google Scholar]
- Hudson TJ, Stein LD, Gerety S, Ma J, Castle AB, Silva J, Slonim DK, Baptista R, Kruglyak L, Xu S-H, Xintong H, Colbert AME, Roserberg C, Reve-Daly MP, Rozen S, Hui L, Ganiatsas S, Evans, D.M.M. CA, Ingalls KA, Nahf RW, Horton LT, Anderson MO, Collymore AJ, Ye W, Kouyoumjian V, Zemsteva IS, Tam J, Devine R, Courtney DF, Renaud MT, Nguyen H, O’Connor TJ, Fizames C, Faure S, Gyapay G, Dib C, Morissette J, Orlin JB, Birren BW, Goodman N, Weissenbach J, Hawkins TL, Foote S, Page DC, Lander ES. An STS-based map of the human genome. Science. 1995;270:1945–1954. doi: 10.1126/science.270.5244.1945. [DOI] [PubMed] [Google Scholar]
- Morton NE. Parameters of the human genome. Proc Natl Acad Sci. 1991;88:7474–7476. doi: 10.1073/pnas.88.17.7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M, Hood L, Cantor C, Botstein D. A common language for physical mapping of the human genome. Science. 1989;245:1434–1435. doi: 10.1126/science.2781285. [DOI] [PubMed] [Google Scholar]
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, White RE, Rodriguez-Tomé P, Aggarwal A, Bajorek E, Bentolila S, Birren BB, Butler A, Castle AB, Chiannilkulchai N, Chu A, Clee C, Cowles S, Day PJR, Dibling T, Drouot N, Dunham I, Duprat S, East C, Edwards C, Fan J-B, Fang N, Fizames C, Garrett C, Green L, Hadley D, Harris M, Harrison P, Brady S, Hicks A, Holloway E, Hui L, Hussain S, Louis-Dit-Sully C, Ma J, MacGilvery A, Mader C, Maratukulam A, Matise TC, McKusick KB, Morissette J, Mungall A, Muselet D, Nusbaum HC, Page DC, Peck A, Perkins S, Piercy M, Qin F, Quackenbush J, Ranby S, Reif T, Rozen S, Sanders C, She X, Silva J, Slonim DK, Soderlund C, Sun W-L, Tabar P, Thangarajah T, Vega-Czarny N, Vollrath D, Voyticky S, Wilmer T, Wu X, Adams MD, Auffray C, Walter NAR, Brandon R, Dehejia A, Goodfellow PN, Houlgatte R, Hudson JRJ, Ide SE, Iorio KR, Lee WY, Seki N, Nagase T, Ishikawa K, Nomura N, Phillips C, Polymeropoulos MH, Sandusky M, Schmitt K, Berry R, Swanson K, Torres R, Venter JC, Sikela JM, Beckmann JS, Weissenbach J, Myers RM, Cox DR, James MR, Bentley D, Deloukas P, Lander ES, Hudson TJ. A gene map of the human genome. Science. 1996;274:540–546. [PubMed] [Google Scholar]
- Selig S, Bruno S, Scharf JM, Wang CH, Vitale E, Gilliam TC, Kunkel LM. Expressed cadherin pseudogenes are localized to the critical region of the spinal muscular atrophy gene. Proc Natl Acad Sci. 1995;92:3702–3706. doi: 10.1073/pnas.92.9.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer R, Tautz D. Minimal homology requirements for PCR primers. Nucleic Acids Res. 1989;17:6749. doi: 10.1093/nar/17.16.6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, E.A., K.B.M Kusick, A. Aggarwal, E. Bajorek, S. Brady, A. Chu, N. Fang, D. Hadley, M. Harris, S. Hussain et al. 1997. An STS-based radiation hybrid map of the human genome. Genome Res. 7: (this issue). [DOI] [PubMed]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]