Ab initio Gene Finding in Drosophila Genomic DNA (original) (raw)
Abstract
Ab initio gene identification in the genomic sequence of Drosophila melanogaster was obtained using Fgenes (human gene predictor) and Fgenesh programs that have organism-specific parameters for human, Drosophila, plants, yeast, and nematode. We did not use information about cDNA/EST in most predictions to model a real situation for finding new genes because information about complete cDNA is often absent or based on very small partial fragments. We investigated the accuracy of gene prediction on different levels and designed several schemes to predict an unambiguous set of genes (annotation CGG1), a set of reliable exons (annotation CGG2), and the most complete set of exons (annotation CGG3). For 49 genes, protein products of which have clear homologs in protein databases, predictions were recomputed by Fgenesh+ program. The first annotation serves as the optimal computational description of new sequence to be presented in a database. Reliable exons from the second annotation serve as good candidates for selecting the PCR primers for experimental work for gene structure verification. Our results shows that we can identify ∼90% of coding nucleotides with 20% false positives. At the exon level we accurately predicted 65% of exons and 89% including overlapping exons with 49% false positives. Optimizing accuracy of prediction, we designed a gene identification scheme using Fgenesh, which provided sensitivity (Sn) = 98% and specificity (Sp) = 86% at the base level, Sn = 81% (97% including overlapping exons) and Sp = 58% at the exon level and Sn = 72% and Sp = 39% at the gene level (estimating sensitivity on std1 set and specificity on std3 set). In general, these results showed that computational gene prediction can be a reliable tool for annotating new genomic sequences, giving accurate information on 90% of coding sequences with 14% false positives. However, exact gene prediction (especially at the gene level) needs additional improvement using gene prediction algorithms. The Fgenesh program was also tested for predicting genes of human Chromosome 22 (the last variant of Fgenesh can analyze the whole chromosome sequence). This analysis has demonstrated that the 88% of manually annotated exons in Chromosome 22 were among the ab initio predicted exons. The suite of gene identification programs is available through the WWW server of Computational Genomics Group at http://genomic.sanger.ac.uk/gf.html.
Many bacterial, as well as several eukaryotic, complete genomes have been sequenced, and Drosophila, mouse, and human genome sequencing is being pursued aggressively. The first challenge in analyzing sequence data is finding the genes. Knowledge of gene sequences has led to a new way of performing biological studies called functional genomics. The second major challenge is to find out what all of these new genes do, how they interact, and how they are regulated (Wadman 1998). Comparisons among genes of different genomes can provide additional insight into the details of gene structure and function. To meet these challenges we need advanced gene-finding algorithms and computer systems utilizing all available information, such as similarity with known proteins or ESTs to increase the accuracy of genome annotation. We cannot precisely predict all gene components because of limitations in our knowledge of complex biological processes and signals regulating gene expression. In this respect, the analysis of 2.9 Mb of Drosophila sequence by several gene-finding approaches gives us a unique opportunity to define the reliability and limitations of our predictions and provides a strategy for the interpretation of predicted results in the analysis of new genomic sequences. Current gene identification approaches (Burge and Karlin 1998) use dynamic programming and pattern-based or probabilistic scheme for scoring potential gene variants. They employ the best signal and content recognizers and an optimization technique developed previously (Burge and Karlin 1977; Brunak et al. 1991; Fickett and Tung 1992; Guigó et al. 1992; Snyder and Stormo 1993; Krogh et al. 1994; Stormo and Haussler 1994; Solovyev et al. 1994). We tested two gene prediction approaches developed in our group, Fgene (pattern based human gene prediction) and Fgenesh (hidden Markov model(HMM)) based gene prediction with Drosophila gene parameters. The optimal strategy to annotate long genomic sequences and predict new genes was investigated. The best results were produced by organism-specific Fgenesh program that can accurately predict ∼80% of verified exons. The overpredicted exons (∼10%) can be false positives or belong to genes that do not have corresponding ESTs or proteins and have not been predicted by GENSCAN. Some of them represent the retroviruses genes which we included in our annotation.
METHODS
For identification of potential protein-coding regions in the Adh region of Drosophila sequence we have used three gene prediction programs (Fgenes, Fgenesh, and Fgenesh+) developed in our group. Fgenesh is a HMM based algorithm with the parameters trained on the set 1600 of Drosophila genes annotated in GenBank (Benson et al. 1999). Fgenesh+ is a variant of Fgenesh that takes into account some information about similar proteins. Fgenes is the program based on discriminant functions trained to predict human genes. We included the last program because we observed that when predicting human genes, the same exons predicted by the Fgenes and Fgenesh approaches are accurate with a specificity of ∼90%–95% (Solovyev and Salamov,1999). Therefore, using both programs we can find a subset of reliable exons that can be used to start an experimental gene verification study.
Our approach to the annotation was based on applying basic gene prediction tools and using the BLAST program to improve the accuracy of gene prediction when similar proteins are found for ab initio predicted exons. For most genes only small fragments of mRNA sequences are presented in databases and complete cDNAs are known only for a fraction of these genes. Our experience shows that the use of short EST fragments does not improve the accuracy of predictions. Therefore, we decided not to use EST information to make additional improvements testing the system to predict genes in which the information about the transcript sequences is practically absent. The suggested scheme is designed to expedite initial analysis of large-scale genomic sequences and can be the first step in a complex system that might apply additional information to improve the quality of gene annotation.
General Scheme of Analysis
- The large genomic sequence (2.9 Mb) was divided into six contiguous subsequences, ∼0.5Mb each. Fgenesh and Fgenes were run on all regions of the sequence and the points of division were selected within the fragments, which were free of predicted genes. Fgenesh variants were developed to predict genes of a sequence of any practical length and applied to the analysis of human Chromosome 22 (http://genomic.sanger.ac.uk/inf/infodb.shtml).
- Repetitive sequences were masked in the sequence using RepeatMasker (Smit 1999) and using the Repbase data set (Jurka 1998).
- Prediction of genes on masked sequences was done with Fgenes and Fgenesh.
- For each predicted exon similarity searches were run using the BLAST program (Altschul et al. 1997) on protein and EST databases. We used NCBI's nonredundant (nr) protein database and the Berkeley Drosophila Genome Project's EST database.
- For genomic regions containing predicted exons with significant protein similarity, we recomputed gene predictions using a special program Fgenesh+.
- The total pool of predicted genes was based on annotations, with priority given to the genes predicted by Fgenesh+ (i.e., we removed all predictions which overlapped with Fgenesh+ exons).
We have presented three annotations to demonstrate different possibilities to use the predicted genes. The major CGG1 annotation comprised the nonambiguous gene set. The genes were included according to the following criteria (descending in priority): (1) All genes were predicted by Fgenesh+; (2) genes were predicted identically by both Fgenes and Fgenesh programs; and (3) in the regions of overlapped (but not exactly coincide) predictions, only one predicted gene was included with priority given to the genes producing longer proteins. The annotation CGG2 is intended to provide a subset of reliable exons. It comprised the set of all exons predicted by Fgenesh+ augmented by the exons, identically predicted by both programs (Fgenes and Fgenesh). The annotation CGG3 included all exon candidates predicted by Fgenesh+, Fgenes, and Fgenesh genes.
Gene Identification Programs
Fgenes
Fgenes (Find genes) is the multiple gene prediction program based on dynamic programming. It uses discriminant classifiers to generate a set of exon candidates. Similar discriminant functions were developed initially in Fexh (Find exon), Fgeneh (Find gene) programs (h stands for the version that analyzes human genes), and described in detail earlier (Solovyev and Lawrence 1993; Solovyev et al. 1994).
The following major steps describe analysis of genomic sequences by the Fgenes algorithm:
- Create a list of potential exons, selecting all ORF: ATG…GT, AG-GT, AG. . . . .Stop with exon scores higher than the specific thresholds depending on GC content (four groups); 2. Order all exon candidates according to their 3′-end positions; 3. Select for each exon maximal score path (compatible exons combination) ending on the particular exon using dynamic programming approach similar to that of Guigó (1999); 4. Add promoter or poly(A) scores (if predicted) to terminal exons. Run time of the algorithm grows approximately linearly with the sequence length.
Scoring functions of Fgenes is based on usage linear discriminant functions developed for identification splice sites, exons, promoter, and poly(A) sites (Solovyev and Salamov 1997).
Fgenesh
Fgenesh is the HMM-based gene-finding program with the algorithm similar to Genie (Kulp et al. 1996) and GENSCAN (Burge and Karlin 1997). The difference between Fgenesh and analogous programs is that in the model of gene structure a signal term (such as splice site or start site score) has some advantage over a content term (such as coding potentials), reflecting the biological significance of the signals. It means in log-likelihood terms that in the model, splice sites and start sites have an additional score, depending on the environments of the sites, but not on conserved nucleotides. At the same time in computing the coding scores of potential exons, a priori probabilities of exons were taken into account according to Bayes theorem. As a result, the coding scores of potential exons are generally lower than in GENSCAN. Parameters of the program were trained on 1600 D. melanogaster entries from GenBank. Separate coding potentials were calculated for each of two isochores: GC content < 45% and GC content > 45%. The Fgenesh variant for predicting Drosophila genes was prepared 1 year before the GASP experiment; therefore, we did not use a learning set of sequences provided by the organizers. The run time of Fgenesh is approximately linear.
Fgenesh+
Fgenesh+ is a version of Fgenesh, which uses additional information from the available protein homolog. When exons predicted by Fgenesh show high similarity to a protein from the database, it is often advantageous to use this information to improve the prediction accuracy. Fgenesh+ requires an additional file with protein homolog and aligns all predicted potential exons with that protein using the Smith–Waterman algorithm, as implemented in the sim program (Huang and Miller 1991). To decrease the computational time, all overlapping exons in the same reading frame are combined into one sequence and aligned only once against the protein sequence.
The main additions to the algorithm, relative to Fgenesh, include (1) the augmentation of the scores of exons with detected similarity by an additional term proportional to the alignment score, and (2) the additional penalty included for the adjacent exons in dynamic programming (Viterbi algorithm), if their corresponding aligned protein segments are not close in the corresponding (similar) protein.
Fgenesh+ was tested on the selected set of 61 GenBank human sequences, for which Fgenesh predictions were not accurate (correlation coefficient 0.0 ≤ CC < 0.90) and which had protein homologs from another organism. The identity between encoded proteins and homologs varied between 99% and 40%. The prediction accuracy of this set is presented in Table 1. The results show that if the alignment covers the significant parts of both proteins, Fgenesh+ usually increases the accuracy relative to Fgenesh that is not depending significantly on the level of identity (for ID >40%). This property makes knowledge of proteins from even distant organisms useful for improving the accuracy of gene identification.
Table 1.
Comparison of Accuracy of Fgenesh and Fgenesh+ on the Set of Human Genes with Known Protein Homologs from Other Organisms
| CG (%) | Sne (%) | Spe (%) | Snb (%) | Spb (%) | CC (%) | |
|---|---|---|---|---|---|---|
| Fgenesh | 0 | 63 | 68 | 86 | 83 | 0.74 |
| Fgenesh+ | 46 | 82 | 85 | 96 | 98 | 0.95 |
RESULTS AND DISCUSSION
Fgenes predicted 384 genes (202 in reverse chain), with 3.9 being the average number of exons per gene. The average size of the genes was 5.4 kb (from ATG to stop codon, including introns) and the average size of intergenic regions was 7.6 kb. Of these genes, 207 had sequence similarities on both protein and EST levels, 405 exons had similarities with only proteins, and 335 exons had similarities with only ESTs (with _E_-value < 10−5). Fgenesh predicted 530 genes (269 in reverse chain), with 3.2 being the average number of exons per gene. The average size of the genes was 2 kb and the average size of intergenic region was 5.5 kb. Of these exons, 252 had sequence similarities on both protein and EST levels, 601 exons had similarities only with proteins, and 390 exons had similarities with only ESTs (with _E_-value < 10−5).
We used Fgenesh+ to improve the accuracy of prediction for 49 genes. Of these genes, 37 were predicted using D. melanogaster's own proteins already deposited in protein databases. Analysis of these predictions demonstrates that even for such cases, prediction of accurate gene structure may not be trivial, although in most cases Fgenesh+ improved the prediction accuracy relative to ab initio methods. For example, in the region of the Beaten path protein (2505534–2530156 bp) Fgenesh predicts three genes (Fig. 1A). The first and last predicted genes have common exons with the real gene, but the second predicted gene is in reverse strand and located inside of first intron. Such splitting is probably caused by the relatively large size of the first intron (∼20 kb). Prediction becomes completely accurate using a proper protein product of the gene (Fig. 1b). In total, from 49 genes, only 24 coincided completely with the genes annotated in std3 set. Four predicted retrovirus-related genes from transposons were not annotated because annotators excluded transposon sequences. Of the remaining 21 predicted genes, most agreed with annotations, with slight discrepancies in one to two exons. Below are listed some cases where we beleive our predictions are more correct than the annotations in the std3 set.
- Kuzbanian gene (34,358–130,401), a disintegrin-like metalloprotease (Rooke et al. 1996). Annotators probably missed the second exon (52555–52587). Our predicted protein product is closer to the protein shown in GenBank's mRNA entry for this gene (accession no. U60591).
- p38b gene (274,751–275,848), stress-activated MAP kinase (Han et al. 1998). We predicted a single-exon gene where protein product is identical to GenBank's mRNA entry for this gene (accession no. AF035548). In std3 this gene is described as having two coding exons (274,751–275,110 and 275,123–275,848) separated by a small intron of 13 bp.
- Adhr gene (1,111,284–1,112,578) is alcohol dehydrogenase-related gene (Brogna and Ashburner 1997). Our predicted protein is 99.6% accurate (1 mismatch in 256 amino acids) and coincides with the protein in GenBank's mRNA entry for this gene (accession no. X98338).The protein product of the corresponding gene in std3 has an insert of 10 amino acids.
- TfIIS gene (1,549,142–1,550,149), RNA polymerase II elongation factor (Marshall et al. 1990), which is probably an alternative splicing variant. We predicted a gene with two exons, the protein product of which is identical to the TfIIS protein with identifier sp|P20232 in NCBI's nr database (313 amino acids). The corresponding gene in std3 is a single exon and has a protein product identical to the TfIIS protein with identifier pir|S55899 in the nr database (263 amino acids).
Figure 1.


Prediction of beaten gene by Fgenesh (a) and by Fgenesh+(b). Fgenesh+ predictions coincide with the experimentally verified gene structure.
Because the annotators used GENSCAN for unsupported protein and EST gene identification, we can anticipate that the annotation contains some false-positive and false-negative predictions. In fact, 39 genes in std3 were annotated only on the basis of high score predictions in GENSCAN (Ashburner et al. 1999). Table 2 shows the prediction accuracy for our annotation sets (based on combination of Fgenes and Fgenesh), some of the best predictions from another groups (taken from initial analysis of Reeves et al. 2000), along with the results for Fgenesh alone. Our results shows that HMM-based Fgenesh program with Drosophila parameters performed better than pattern-based Fgenes, discriminant functions of which were developed for prediction of human genes. Even though it was technically incorrect to use the human version of Fgenes, we were able to demonstrate that applying two different approaches for prediction can generate a set of exons with expected properties.
- The main annotation CGG1 predicts ∼87% of real coding nucleotides and ∼23% of false positives (there might be some errors in coding due to the absence of experimental data in many regions); 89% of exons are predicted exactly or with overlapping exons. These data show that ab initio predictions can provide information about almost all of the protein coding genes (only 13% of the coding region was not predicted) and can serve as a basis for further experimental analysis.
- The annotation CGG2 contains ∼50% of coding exons but has ∼20% fewer false positive exons. These exons can be used to start experimental gene verification.
- The annotation CGG3 included ∼70% of correct exons and 92% of all coding nucleotides. Such redundant annotation can be useful in identifying some genes with additional selection filters, (i.e., analysis of similarity with some important proteins or some experimental procedures).
Table 2.
Performance of Several Programs on the Adh Region of Drosophila
| CGG1 | CGG2 | CGG3 | Fgenesh | Fgenesh pruned | Genie | Genie EST | MAGPIE | ||
|---|---|---|---|---|---|---|---|---|---|
| Base level | Sn std1 | 89 | 49 | 93 | 98 | 98 | 96 | 97 | 96 |
| Sn std3 | 87 | 46 | 91 | 92 | 88 | 79 | 79 | 94 | |
| Sp std3 | 77 | 86 | 60 | 71 | 86 | 92 | 91 | 63 | |
| Exon level | Pe | 1115 | 598 | 2900 | 1671 | 979 | 786 | 849 | 1835 |
| Ce std1 | 80 | 54 | 92 | 100 | 100 | 86 | 95 | 84 | |
| Sn std1 | 65/89 | 44/55 | 75/94 | 81/97 | 81/97 | 70 | 77 | 68 | |
| Ce std3 | 544 | 405 | 620 | 601 | 565 | 447 | 470 | 705 | |
| Sn std3 | 60/82 | 45/54 | 69/90 | 66/89 | 62/82 | 49 | 52 | 78 | |
| Sp std3 | 49 | 68 | 24 | 36 | 58 | 57 | 52 | 41 | |
| Gene level | Pg | 288 | 201 | 875 | 530 | 262 | 241 | 246 | 549 |
| Cg std1 | 22 | 7 | 26 | 31 | 31 | 24 | 28 | 20 | |
| Sn std1 | 51 | 16 | 60 | 72 | 72 | 56 | 65 | 47 | |
| Cg std3 | 102 | 45 | 113 | 108 | 106 | 86 | 92 | 136 | |
| Sn std3 | 46 | 20 | 51 | 49 | 48 | 39 | 41 | 61 | |
| Sp std3 | 36 | 32 | 14 | 20 | 39 | 37 | 38 | 25 |
It is interesting to note that the use of two programs provided stable prediction accuracy on both (std1 and std3) sets. The Genie program demonstrated a 20% decrease in sensitivity (Table 2). Because we have no version of Fgenes with all parameters computed for Drosophila genes, we tried to find an optimal variant using one program. We discovered that the Fgenesh predictions provided the best accuracy. In this simple variant we took a set of predicted genes and discarded the low-scoring genes (with an average gene score <15). This resulted in 88% accurate coding nucleotide predictions with only 14% false positives on the std3 set (Table 2).
Our results demonstrate that most of the annotated genes in std3 are at least partially covered by predictions. For example, only five genes from std3 do not overlap with Fgenesh predictions (two of them are also included in the std1 set). Of these five genes, four are located inside introns of other genes; four are single-exon genes (three are inside intron genes). Therefore, one of the limitations of current gene-finding programs is that they cannot detect nested genes, that is, genes located inside introns of other genes. This is one in the future directions for improvement in gene-finding software. Although this is probably a rare event for the human genome, for organisms like Drosophila it presents a real problem. For example, annotators identified 17 examples of such cases in the Adh region. (Ashburner et al. 1999). Another drawback of the current gene-finding programs is that predictions of terminal exons are generally much worse than the internal ones. This results in the splitting up of some actual genes and/or joining some other multiple genes into a single gene. Several examples of such situations can be clearly seen in our Genes in Pictures interactive system (Seledtsov and Solovyev 1999) (Fig. 2) developed to present information about gene structures described in GenBank (collecting information about a gene from many entries) or annotated using gene prediction programs. In total, on std3, 63% of internal exons from the CGG1 annotation are predicted exactly, with 54% specificity, whereas the corresponding numbers for initial exons are 58% and 44%, and for terminal exons 53% and 40%, respectively. On std1 Fgenesh predicts all internal exons correctly (100%), whereas only 72% of initial exons and 77% of terminal exons are predicted correctly (Table 3). Thus, methods to better predict terminal exons and the related problem of recognizing the beginnings (transcription start sites) and endings [poly(A) sites] of genes are other possible areas for improvement in the use of gene-finding programs.
Figure 2.
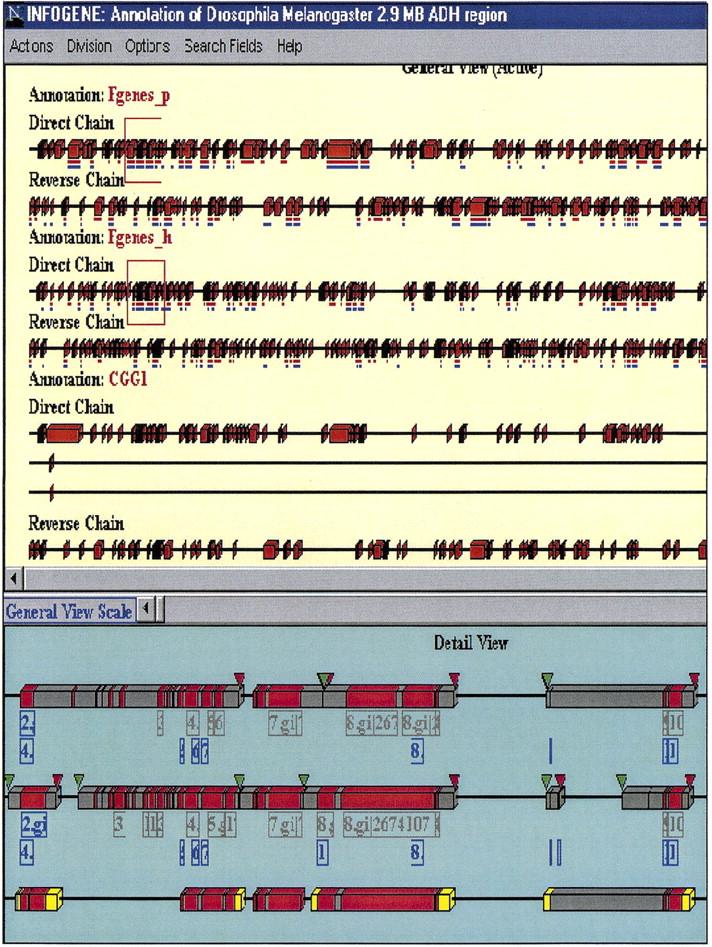
Genes _in_ Pictures (Seledtsov and Solovyev 1999) presentation of Fgenes and Fgenesh predictions in the Adh region. (Bottom) Fragments of annotations that are marked in the top panel. The last level presents std3 manual annotation. Coding exons are marked in red; introns are in gray. Inverted green triangles show the start of transcription; red ones mark the poly(A) signal. Exons having protein or EST similarity are underlined with red and blue lines, respectively.
Table 3.
Prediction Accuracy for Different Types of Exons
| Initial exons | Internal exons | Terminal exons | Single exons | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn std 1 | Sn std 3 | Sp std 3 | Sn std 1 | Sn std 3 | Sp std 3 | Sn std 1 | Sn std 3 | Sp std 3 | Sn std 1 | Sn std 3 | Sp std 3 | |
| CGG1 | 55 | 58 | 44 | 79 | 63 | 54 | 61 | 53 | 40 | 42 | 56 | 45 |
| Fgenesh | 72 | 65 | 26 | 100 | 70 | 51 | 77 | 57 | 23 | 28 | 56 | 26 |
| Fgenesh pruned | 72 | 58 | 51 | 100 | 67 | 66 | 77 | 53 | 46 | 28 | 56 | 41 |
In conclusion, we note that even programs based on similar approaches often produce significantly different results. For example, Fgenesh predicts 5839 exons on human Chromosome 22 (>88% of 3488 manually annotated exons having some EST or protein similarity are among these predictions), whereas GENSCAN predicts 6100 exons. Fgenesh predictions are presented in Infogene database format (Solovyev and Salamov 1999) at http://genomic.sanger.ac.uk. Of these exons, ∼80% are the same or similar when comparing Fgenesh and GENSPAN predictions and further experiments are necessary for verification. A region that has been analyzed experimentally (but with low gene density and unusually difficult for ab initio predictions) provides a good test of the programs accuracy and demonstrates their differences. The results of Fgenesh, Fgenes, and GENSCAN gene predictions on the BRACA2 region are presented in Table 4. We can see that the repeat masked sequence results in fewer false-positive predictions, especially for the GENSCAN program. Exons predicted by different methods might represent alternative splicing variants.
Table 4.
Performance of Gene-finding Programs on the BRCA2 1.4-Mb Region of Human Chromosome 13
| CC | Snb | Spb | Pe | Ce | Sne | Snep | Spe | Pg. | Cg | PCg | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GENSCAN | 0.68 | 90 | 53 | 271 | 109 | 65 | 80 | 40 | 49 | 0 | 19 |
| Fgenesh | 0.80 | 89 | 73 | 188 | 115 | 69 | 80 | 61 | 25 | 0 | 17 |
| Fgenes | 0.69 | 79 | 62 | 298 | 110 | 66 | 86 | 37 | 34 | 0 | 19 |
| GENSCAN masked | 0.76 | 90 | 66 | 217 | 109 | 65 | 80 | 50 | 32 | 0 | 19 |
| Fgenesh masked | 0.84 | 89 | 82 | 172 | 114 | 68 | 79 | 66 | 19 | 0 | 17 |
| Fgenes masked | 0.73 | 80 | 68 | 257 | 107 | 64 | 85 | 42 | 38 | 0 | 19 |
Acknowledgments
We thank Dr. Igor Seledtsov for collaborative work with Infogen visualization. Development of gene prediction approaches was supported by a Wellcome Trust research grant (to V.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL solovyev@sanger.ac.uk; FAX 44-1-2223-494919.
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Milller W, Lipman DJ. Gapped BLAST and PSI_BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Misra S, Roote SE, Lewis SE, Blazej R, Davis T, Doyle C, Galle R, George R, Harris N, et al. An exploration of the sequence of a 2.9Mb region of the genome of Drosophila melanogaster: The Adh region. Genetics. 1999;153:179–219. doi: 10.1093/genetics/153.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF, Rapp RA, Wheeler DL. GenBank. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogna S, Ashburner M. The Adh-related gene of Drosophila melanogaster is expressed as a functional dicistronic messenger RNA: Multigenic transcription in higher organisms. EMBO J. 1997;16:2023–2031. doi: 10.1093/emboj/16.8.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Etzold T, Ulyanov A, Argos P. SRS: Information retrieval system for molecular biology data banks. Methods Enzymol. 1996;266:114–128. doi: 10.1016/s0076-6879(96)66010-8. [DOI] [PubMed] [Google Scholar]
- Fickett JW, Tung CS. Assessment of protein coding measures. Nucleic Acids Res. 1992;20:6441–6450. doi: 10.1093/nar/20.24.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigó R, Knudsen S, Drake N, Smith T. Prediction of gene structure. J Mol Biol. 1992;226:141–157. doi: 10.1016/0022-2836(92)90130-c. [DOI] [PubMed] [Google Scholar]
- Han ZS, Enslen H, Hu X, Meng X, Wu IH, Barrett T, Davis RJ, Ip YT. A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol Cell Biol. 1998;18:3527–3539. doi: 10.1128/mcb.18.6.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Miller W. A time-efficient, linear-space local simularity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
- Jurka J. Repeats in genomic DNA: Mining and meaning. Curr Opin Struct Biol. 1998;8:333–337. doi: 10.1016/s0959-440x(98)80067-5. [DOI] [PubMed] [Google Scholar]
- Krogh A, Mian S, Haussler D. A hidden Markov model that finds genes in E. coli DNA. Nucleic Acids Res. 1994;22:4768–4778. doi: 10.1093/nar/22.22.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. Intell Syst Mol Biol. 1996;4:134–142. [PubMed] [Google Scholar]
- Marshall TK, Guo H, Price DH. Drosophila RNA polymerase II elongation factor DmS-II has homology to mouse S-II and sequence similarity to yeast PPR2. Nucleic Acids Res. 1990;18:6293–6298. doi: 10.1093/nar/18.21.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M.G., N. Harris, G. Hartzell, U. Ohler, and S. Lewis. 2000. Genome annotation assesment in Drosophila melanogaster. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science. 1996;273:1227–1231. doi: 10.1126/science.273.5279.1227. [DOI] [PubMed] [Google Scholar]
- Seledtsov I, Solovyev V. Genes _in_ Pictures: Interactive system of representation and analysis of eukaryotic gene structures. 1999. http://genomic.sanger.ac.uk/infodb.shtml http://genomic.sanger.ac.uk/infodb.shtml. . [Google Scholar]
- Smit, A. 1999. http://ftp/genome.washingtonedu/RM/RM-details.html.
- Snyder EE, Stormo G. Identification of coding regions in genomic DNA sequences: An application of dynamic programming and neural networks. Nucleic Acids Res. 1993;21:607–613. doi: 10.1093/nar/21.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev VV, Lawrence CB. Identification of human gene functional regions based on oligonucleotide composition. Intell Syst Mol Biol. 1993;1:371–379. [PubMed] [Google Scholar]
- Solovyev VV, Salamov AA, Lawrence CB. Predicting internal exons by oligonucleotide composition and discriminant analysis of spliceable open reading frames. Nucleic Acids Res. 1994;22:5156–5163. doi: 10.1093/nar/22.24.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev VV, Salamov AA. The Gene-Finder computer tools for analysis of human and model organisms genome sequences. Intell Syst Mol Biol. 1997;5:294–302. [PubMed] [Google Scholar]
- Solovyev VV, Salamov AA. INFOGENE: A database of known gene structures and predicted genes and proteins in sequences of genome sequencing projects. Nucleic Acids Res. 1999;27:248–250. doi: 10.1093/nar/27.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G, Haussler D. Optimally parsing a sequence into different classes based on multiple types of evidence. Intell Syst Mol Biol. 1994;2:369–375. [PubMed] [Google Scholar]
- Wadman M. “Rough draft” of human genome wins researchers backing. Nature. 1998;393:399–400. doi: 10.1038/30790. [DOI] [PubMed] [Google Scholar]
- Xu Y, Einstein JR, Mural RJ, Shah M, Uberbacher EC. An improved system for exon recognition and gene modeling in human DNA sequences. Proc Intell Syst Mol Biol. 1994;2:376–384. [PubMed] [Google Scholar]