Genetic and Pharmacological Inhibition of the Ca2+ Influx Channel TRPC3 Protects Secretory Epithelia from Ca2+-Dependent Toxicity (original) (raw)
. Author manuscript; available in PMC: 2012 Jun 1.
Published in final edited form as: Gastroenterology. 2011 Feb 24;140(7):2107–2115.e4. doi: 10.1053/j.gastro.2011.02.052
Abstract
Background & Aims
Excessive Ca2+ influx mediates many cytotoxic processes, including those associated with autoimmune inflammatory diseases such as acute pancreatitis and Sjögren's syndrome. TRPC3 is a major Ca2+ influx channel in pancreatic and salivary gland cells. We investigated whether genetic or pharmacological inhibition of TRPC3 protects pancreas and salivary glands from Ca2+-dependent damage.
Methods
We developed a Ca2+-dependent model of cell damage for salivary gland acini. Acute pancreatitis was induced by injection of cerulein into wild-type and Trpc3−/− mice. Mice were also given the Trpc3-selective inhibitor pyrazole 3 (Pyr3).
Results
Salivary glands and pancreas of Trpc3−/− mice were protected from Ca2+-mediated cell toxicity. Analysis of Ca2+ signaling in wild-type and Trpc3−/− acini showed that Pyr3 is highly specific inhibitor of Tprc3; it protected salivary glands and pancreas cells from Ca2+-mediated toxicity by inhibiting the Trpc3-mediated component of Ca2+ influx.
Conclusions
TRPC3-mediated Ca2+ influx mediates damage to pancreas and salivary glands. Pharmacological inhibition of TRPC3 with the highly selective TRPC3 inhibitor Pyr3 might be developed for treatment of patients with acute pancreatitis and Sjögren's syndrome.
Keywords: Ca2+ influx, inflammation, cell death, therapeutics
Background and Aims
Ca2+ signaling regulates virtually all cell functions and thus aberrant Ca2+ signaling is associated with many diseases. Particularly, impaired Ca2+ signaling is the underlying cause in diseases that involve cell stress, endoplasmic reticulum (ER) stress, oxidative stress, and inflammation that lead to autophagy and cell death1–4. Prominent gastrointestinal diseases associated with Ca2+ stress are acute pancreatitis5, 6 and Sjögren's syndrome7. These are multifactorial diseases that are caused by generation of toxins within the pancreas and salivary glands in response to inflammatory insults. In the pancreas, this results in mistargeting of digestive enzymes to lysosomes that damage the pancreatic parenchyma8. In salivary glands, the inflammatory mediators, which include cytokines9 and nitric oxides10, induce apoptotic and necrotic cell death11, 12 that are associated with activation of Ca2+ signaling. In addition, patient with Sjögren's syndrome express anti M3 muscarinic receptors that have profound effect on heseacute and chronic damage in salivary gland cells. Because so many key functions are regulated by Ca2+, impaired Ca2+ signaling is intimately associated with acute pancreatitis 13 and likely salivary glands dysfunction.
The receptor-evoked Ca2+ signal involves Ca2+ release from the ER, which leads to activation of the store-operated Ca2+ influx channels (SOCs) at the plasma membrane 14. ER Ca2+ is rapidly exhausted if it is not replenished by the SOCs, which also sustain the physiological Ca2+ oscillations to determine their amplitude and frequency14, 15. In fact, Ca2+ influx provides most of the Ca2+ that regulates exocytosis, fluid secretion and gene regulation14, 16–18. This scenario changes under cell stress, at which the cells are continuously over-stimulated, resulting in a pathological Ca2+ signal. The persistent strong stimulus depletes the ER Ca2+ store, resulting in an uncontrolled activation of the SOCs and prolonged increase in [Ca2+]i6, 13, 14, 19 that is responsible for the pathological effects of Ca2+.
The SOC in pancreatic and salivary gland cells is mediated by TRPC1, TRPC3 and TRPC62, 20 and the Orai channels21–23. The role of the native Orais in these cells has not been examined yet. The TRPC channels (TRPCs) mediate part of the receptor stimulated Ca2+ influx in many cells24–26. Indeed, deletion of Trpc127 and Trpc315 in mice strongly reduces receptor- and store-mediated Ca2+ influx in salivary glands and pancreatic acini.
In a recent work, we demonstrated that part of the pathological effect of excessive Ca2+ influx is mediated by the Trpc315, with deletion of Trpc3 in mice reduced the severity of acute pancreatitis. These findings raised several important questions: Does Trpc3 have a role in the function of other secretory glands, like the salivary glands? Does excessive Ca2+ influx by Trpc3 contribute to Ca2+ toxicity in other secretory glands? Can pharmacological inhibition of Trpc3 reduce the severity of Ca2+-dependent cell stress and damage toward developing treatment for acute pancreatitis and Sjögren's syndrome? To address these questions, we developed a cell model of Ca2+ toxicity in salivary glands based on intense and persistent stimulation of receptor-mediated Ca2+ signaling. We then used the salivary gland Ca2+ toxicity and acute pancreatitis models to show that Trpc3 has prominent role in salivary gland function and dysfunction. Most notably, deletion of Trpc3 in mice and pharmacological inhibition of Trpc3 by the selective Trpc3 inhibitor Pyrazole3 (Pyr3) similarly protected salivary glands and the pancreas from Ca2+-mediated cell toxicity. These findings establish Ca2+ influx by Trpc3 in the function and pathogenesis of Ca2+ in secretory cells. Since Pyr3 given acutely and chronically is well tolerated by mice with no known toxicity28, our findings describe a promising drug for the treatment of Ca2+ toxicity as observed in acute pancreatitis and salivary gland damage.
Materials and methods
Detailed methods are given in the supplementary information. Here only key methods are described in details.
Induction of acute pancreatitis in mice
Mice fasted overnight were injected hourly in the abdominal cavity over 4 hours with cerulein (40 ng/g body weight) and with or without pyrazole3 (Pyr3) (0.1 μg/g body weight), as indicated15. Collection and processing of blood, measurement of serum amylase, hematoxylin and eosin (H&E) staining and evaluation of edema were by standard methods and are detailed in supplement.
Determination of intracellular Trypsin activation and [Ca2+]i
Intracellular trypsin activity was measured using the cell permeate synthetic substrate, rhodamine 110-(CBZ-Ile-Pro-Arg)28, and [Ca2+]i was determined with the Ca2+-sensitive dye Fura2, as described before15 and in supplementary methods.
Western Blot
Protein levels were analyzed by standard western blot analysis, as detailed in supplementary methods. Proteins were probed with a 1:500 dilution of phospho-PERK and 1:1000 dilutions of β-actin.
Immunohistocytochemistry
Tissue sections and acini were stained by incubation with anti-LC3 (1:100), α-amylase (1:100), LAMP2 (1:100) and ceramide (1:50) antibodies as detailed in Supplementary methods.
Measurement of whole saliva secretion
Prior to the experiment, the body weight of each mouse was measured. To collect the whole saliva, mice were anesthetized with the mixture of 75 mg/kg body weight ketamine and 1 mg/kg body weight dexmedetomidine. The mice were endotracheal intubated to maintain open airway path. Salivation was stimulated by injection of pilocarpine (10 mg/kg, body weight) intraperitoneally and saliva was collected through the tube placed close to the duct exist into the oral cavity that was under minimal vacuum to ensure collection of all the saliva. Saliva was collected every five minutes over 35 min and salivary secretion was determined by its weight. Salivation was normalized and presented as salivary weight/body weight. To determine the effect of Pyr3 on salivation, Pyr3 (0.1 μg/g body weight) was injected into the abdominal cavity for 1 hr prior to anesthesia and salivary secretion measurement. After anesthetizing the mice, they were injected with a mixture of pilocarpine and Pyr3 to induce salivation.
Determination of Cytotoxicity
Freshly prepared acini from the pancreas and salivary glands were stimulated with the indicated agonist for 20 min at 37°C in a shaking water bath. The acini were spun down for 20 sec at 3000xg and the supernatants were collected. Cell damage was assayed with the VybrantTM Cytotoxicity assay kit (Molecular Probe) following the manufacturer instructions. Briefly, glucose 6-phosphate dehydrogenase (G6PD) released by damaged cells was determined in each sample. Samples were incubated with lyophilized mixture of diaphorase, glucose 6-phosphate, NADP+ and resazurin in 0.5M Tris buffer (pH7.5) for 15 min at 37°C. Resazurin fluorescence was measured at excitation wavelength of 545 nm and emission wavelength of 590 nm. Fluorescence intensity in each sample was calculated as percentage of total fluorescence released by lysing the cell.
Cell damage was also assayed by determining accumulation of ceramide. Pancreatic and salivary gland acini stimulated with supermaximal agonist concentration were fixed by incubation with cold MeOH and processed for staining with anti-ceramide antibodies as described above under immunohistochemistry.
Statistics
Results are expressed as mean±s.e.m of the indicated number of observations obtained from the indicated number of independent experiments and mice. Statistical significance was determined by analysis of variance.
Results and Discussion
Pyr3 is a selective inhibitor of Trpc3
Previous work reported that Pyr3 is a selective TRPC3 inhibitor since it did not or poorly inhibited other TRPC and TRP channels28. However, the specificity of Pyr3 for the native TRPC3 was not determined. We use pancreatic and parotid acini from Trpc3−/− mice to test the selectivity of Pyr3. Preliminary experiments showed that maximal inhibition of receptor-stimulated Ca2+ signaling is observed with 3 μM Pyr3, with 10–50 μM Pyr3 inhibiting Ca2+ signaling to the same extent as 3 μM. Therefore, in most experiments below we used 3 μM Pyr3.
Acini were stimulated with 10 pM CCK8 to evoke Ca2+ oscillations. Figs. 1A, E show that 3 μM Pyr3 reduced the frequency of oscillations by about 50%. Notably, deletion of Trpc3 reduced the frequency of the Ca2+ oscillations to the same extent as Pyr3 and Pyr3 had no further effect on these oscillations (Figs. 1B, E). Moreover, deletion of Trpc3 and Pyr3 had the same effect on the sustained Ca2+ signal induced by supra-maximal stimulation with 10 nM CCK8 (Fig. 1C). As reported before15, deletion of Trpc3 selectively reduced Ca2+ influx and the plateau phase (Fig. 1D). Pyr3 reduced Ca2+ influx in wild-type acini to the same extent as deletion of Trpc3 and had no effect on the Ca2+ signal in Trpc3−/− acini (Fig. 1C, D, F). Finally, Pyr3 partially inhibited the CRAC-like current in divalent-free media activated by passive store depletion (Fig. 1G), as was found before for deletion of Trpc315.
Fig. 1. Deletion of Trpc3 and Pyr3 similarly inhibit receptor-evoked Ca2+ oscillations and Ca2+ influx.

Fura2-loaded pancreatic acini from wild-type (A, C) or _Trpc3_−/− mice (B, D) were used to measure [Ca2+]i in response to stimulation with 10 pM CCK8 to induce Ca2+ oscillations (A, B) or 10 nM CCK8 to evoke a sustained response. The upper traces in (A, B) are the control and the lower traces are from acini that were treated with 3 μM Pyr3. In (C, D), the dark traces are the controls and the gray traces are acini treated with Pyr3. Panels (E) and (F) show the mean±s.e.m of the frequency and plateau, respectively, of the Ca2+ signals. Panel (G) shows example traces of the time course and I/V of the CRAC-like current in the absence and presence of Pyr3 and the mean±s.e.m of 5 experiments. * denotes P<0.05 or better with n=6–8 experiments with acini from 3 mice of each phenotype.
Ca2+ influx can be activated also by inhibition of the SERCA pumps to passively deplete ER Ca2+ and activate the SOC. Figs. 2A, B confirm the previous findings that deletion of Trpc3 reduces the SOC activity in pancreatic acini15, which are extended in Fig. 2C, D to submandibular gland acini (SMG). Significantly, Pyr3 reduced the SOC activity in wild-type acini to the same extent as deletion of Trpc3 and had no effect on the residual Ca2+ influx measured in Trpc3−/− cells (Fig. 2A-D). We also analyzed the effect of Pyr3 on the receptor-evoked Ca2+ signaling in SMG cells stimulated with physiological and pathological agonist concentrations. Pyr3 reduced the sustained increase in Ca2+ observed at 0.3 or 100 μM carbachol by about 50%.
Fig. 2. Deletion of Trpc3 and Pyr3 similarly inhibit SOC-mediated Ca2+ influx.

Fura2-loaded pancreatic (A, B) or SMG (C, D) acini from wild-type (A, C) or _Trpc3_−/− mice (B, D) were used to measure Ca2+ release in response to inhibition of the SERCA pumps with 25 μM CPA in Ca2+-free solution and Ca2+ influx in response to Ca2+ re-addition. Dark traces are controls and gray traces are cells treated with 3 μM Pyr3. * denotes P<0.05 or better with n=6–8 experiments with acini from 3 mice of each phenotype. NS denote not significant. In (E) GMS acini were stimulated with 0.3 (upper traces) or 100 μM carbachol (lower traces) and treated with or without 3 μM Pyr3, as indicated (gray lower trace). The upper traces show example of individual cells. The lower traces are averages of 4–5 experiments in cells stimulated in Ca2+-free solution to measure Ca2+ release and then Ca2+ was re-added to measure influx.
Together, the results in Figs. 1 and 2 provide strong evidence that Pyr3 is selective Trpc3 channel inhibitor and can be used to assay the roles of Trpc3 in cell function and dysfunction.
Acute inhibition of Trpc3 reduces the severity of pancreatitis associated with activation of SOCs
Trpc3-mediated Ca2+ influx has a prominent role in mediating the tissue damage seen in a model of acute pancreatitis15. Towards searching for a drug for the treatment of acute pancreatitis we asked whether inhibition of Trpc3 by Pyr3 can similarly protect the pancreas. The results of the multiple assays below indicate that this is the case. In initial control experiments to test for toxicity, mice were injected hourly up to six times with 0.1 μg/g body weight Pyr3 and isolated acini were treated with 3–10 μM Pyr3 up to 1 hr. No obvious effects on appearance of the pancreas or microscopic appearance of acinar cells in treated mice or isolated acini were observed. In addition, no obvious change in appearance or behavior of the mice was noted. This is in agreement with the previous study, in which no toxic effects of Pyr3 were found upon infusing mice with Pyr3 for up to two weeks28. Hence, the mice appear to tolerate well acute and chronic exposure to Pyr3 with no apparent toxicity.
We then used the cerulein model of acute pancreatitis to determine the possible protecting effect of Pyr3. Injecting Pyr3 at 0.1 μg/g body weight during the four hourly cerulean injections reduced plasma amylase by about 50% (Fig. 3A) and reduced pancreatic edema to the level measured in control mice (Fig. 3B). On the cellular level, 3 μM Pyr3 prevented the intracellular trypsin activation caused by stimulation with supra-maximal concentration of CCK8 (Fig. 3C). Finally, Pyr3 prevented the pathological endocytic vacuoles formation (Fig. S1) that is commonly observed in acini stimulated with supramaximal agonist concentration29.
Fig. 3. Inhibition of Trpc3 by Pyr3 ameliorates CCK8-induced acute pancreatitis.

Mice were treated with or without 0.1 μg/g body weight Pyr3 and injected with saline (controls) of cerulein to induce acute pancreatitis. (A) shows the mean±s.e.m of plasma amylase content in 3–6 mice under each condition. (B) shows typical images of damaged tissue under the various treatments and the columns are the mean±s.e.m of at least 5 images from each of the 3–6 mice used in each condition. In (C) isolated pancreatic acini were stimulated for 1 hr with 10 nM CCK8 in the presence or absence of 3 μM Pyr3 and incubated for 20 min with fluorescence trypsin substrate. The Fig. shows example images and the mean±s.e.m of 6 experiments.
Trypsin activation and vacuoles formation requires mistargeting of secretory granules to the lysosomes30. This can be assayed in vivo by measuring overlap between granular and lysosomal markers31. Fig. 4A shows that induction of pancreatitis with cerulean resulted in 70% overlap between amylase and the lysosomal marker LAMP2. Pyr3 reduced the overlap to the level observed in control mice. Acute pancreatitis is associated with induction of autophagy and inhibition of autophagy protects against acute pancreatitis31, 32. We assayed autophagy by measuring autophagosomes formation that can be traced by the autophagy marker LC315, 31, 32. Fig. 4B shows that inhibition of Trpc3 with Pyr3 inhibited induction of autophagy observed in acute pancreatitis. To support the in vivo studies by in vitro measurements, we determined the accumulation of LC3-II in isolated acini from the SMG and the pancreas of wild-type and Trpc3−/− mice. Stimulation with supramaximal agonist concentration markedly increased the LC3-II/LC3-I ratio in wild-type acini, which was inhibited by Pyr3. No significant increase was observed in Trpc3−/− stimulated acini (n=3).
Fig. 4. Effect of Trpc3 deletion and inhibition of Trpc3 activity by Pyr3 on granules mistargeting and induction of autophagy.

In (A), tissue sections were obtained from mice treated with or without Pyr3 and with and without cerulein to induce acute pancreatitis. The sections were co-stained with Lamp2 and the secretory granules marker Amylase. Overlap of Lamp2 with Amylase was determined by MataMorph and the columns show the mean±s.e.m. In (B), tissue sections from mice treated with or without Pyr3 and with and without cerulein, were stained with the autophagy marker LC3. The number of autophagosomes was determined by MataMorph and the columns show the mean±s.e.m of the indicated number of experiments. In (C), isolated SMG and pancreatic acini from wild-type (upper) or Trpc3−/− mice (lower) treated with or without 3 μM Pyr3 were stimulated with 1 mM carbachol or 10 nM CCK8, respectively, for 30 min and extracts were used to analyze accumulation of LC3-II. The columns are the mean±s.e.m (n=3) for acini from wild-type mice. In acini from 3 Trpc3−/− mice LC3 did not change.
Trpc3-mediated Ca2+ influx is required salivary glands physiological function
To extend the usefulness of Pyr3 as a protector against cell damage in the GI tract we asked whether it affects the function and dysfunction of the salivary submandibular gland (SMG). For this, we first tested if Trpc3 has a role in salivary secretion in vivo. Fig. 5 provides the first evidence that Trpc3 function is required for the function of the salivary glands. Salivation was induced by a single intraperitoneal injection of pilocarpine to activate the M3 receptors. Deletion of Trpc3 in mice reduced salivation by about 50%. Notably, intraperitoneal injection of the mice with Pyr3 for 1 hr prior to stimulation of salivation inhibited salivation to the same extent as deletion of Trpc3. Previous study reported the contribution of Trpc1 to SOC-mediated Ca2+ influx and salivary secretion27. Ca2+ influx by TRPC1 and TRPC3 are likely related since TRPC1 and TRPC3 heteromultimerize33, 34 to form a Ca2+ influx pathway that is regulated by STIM133. Hence, deletion of Trpc1 and of Trpc3 in mice likely targets the same pathway.
Fig. 5. Effect of Trpc3 deletion and inhibition of Trpc3 activity by Pyr3 on salivary secretion in vivo.

Anesthetized wild-type (■, ▲) injected with saline (■) or 0.1 μg/g body weight Pyr3 (▲) and Trpc3−/− mice (●) were used to measure cumulative salivary secretion in response to injection of pilocarpine, as detailed in Methods. The results are the mean±s.e.m of 4–7 mice under each condition. * denotes P< 0.05 or better relative to secretion by untreated wild-type mice.
Genetic deletion and pharmacological inhibition of Trpc3 similarly reduce the severity of Ca2+ influx-dependent cell damage
Aberrant increase in [Ca2+]i likely plays a role in salivary glands damage observed in Sjögren's syndrome, medications and other inflammatory conditions9–12. Therefore, for the purpose of the present work we developed a Ca2+-dependent cell damage model in SMG acini using supramaximal stimulation of isolated acini. In preliminary experiments, we tested several incubation times between 15–120 min and carbachol concentrations between 0.1–1 mM while measuring release of glucose 6-phosphate dehydrogenase (G6PD) and in the presence and absence of extracellular Ca2+ to asses the Ca2+-dependent damage. Massive cell damage was observed by stimulating isolated acini with 1 mM carbachol for 30–40 min at 37 °C in the presence of external Ca2+. Longer stimulation period resulted in massive irreversible cell damage and removal of external Ca2+ prevented the cell damage due to 30 min stimulation with carbachol. Therefore, we selected 30 min stimulation at 37 °C with 1 mM carbachol.
First, we tested the effect of Trpc3 deletion and of Pyr3 on induction of autophagy (Fig. 3 and Fig. 6A-C) and mistargeting of secretory granules to lysosomes (Fig. 6D-F) in SMG acini. Freshly isolated acini incubated at 37 °C for 30 min in the absence of receptor stimulation had minimal LC3-II and autophagosomes and the LC3-II level and the number of autophagosomes was minimally reduced by deletion of Trpc3 or by Pyr3. Stimulation with 1 mM carbachol for 30 min increased LC3-II and autophagosomes formation about four fold. Deletion of Trpc3 and inhibition of Trpc3 with Pyr3 markedly inhibited the carbachol-induced increase in LC3-II/LC3-I ration and autophagosomes formation (Figs. 3, 6A-C). Fig. 6D shows that 30 min of superamaximal stimulation with carbachol resulted in significant mistargeting of SMG acini secretory granules to the lysosomes, a phenomenon well established in pancreatitis and discovered here for the SMG acini. Deletion of Trpc3 and Pyr3 similarly prevented granules mistargeting, while treating Trpc3−/− cells with Pyr3 had no further effect.
Fig. 6. Deletion of Trpc3 and inhibition of Trpc3 activity by Pyr3 inhibit induction of autophagy and granules mistargeting in SMG acini.

Acini obtained from wild-type (A, D) or Trpc3−/− mice (B, E) and treated with PBS (control) or 3 μM Pyr3 were left unstimulated (upper images) or were stimulated with the supramaximal concentration of 1 mM (Car). The acini were fixed and stained for the autophagy marker for LC3 (A, B) or co-stained for the late endosomal/lysosomal marker LAMP2 and the secretory granules marker Amylase (D, E). The number of LC3 particles was counted with Metamorph and the mean±s.e.m of the fold increase measured in 8 experiments with acini from wild-type mice and 5 experiments with acini from Trpc3−/− mice is shown in (C). The % LAMP2/Amylase overlap was determined with MetaMorph and the mean±s.e.m of the overlap measured in 7 experiments with acini from wild-type mice and 5 experiments with acini from Trpc3−/− mice is shown in (F). The P values are listed in the figures.
Cell damage can be assayed by endocytic pathological vacuole formation that can be followed with TRD 29. Fig. S1B shows that stimulation of SMG acini with 1 mM carbachol caused massive vacuolization that was reduced by Pyr3. Another established parameter of cell stress is activation of the ER unfolding protein response that can be followed by phosphorylation and activation of the ER kinase PERK35. Fig. 7A shows that supramaximal stimulation of wild-type SMG acini with carbachol and pancreatic acini with CCK8 increased PERK phosphorylation, which was inhibited by Pyr3. Fig. 7B shows that deletion of Trpc3 inhibited PERK phosphorylation and treatment with Pyr3 of the Trpc3−/− acini had no further effect. We also assayed cell damage directly by measuring G6PD release. Fig. 7C shows that supramaximal stimulation of pancreatic and SMG acini resulted in the release of 25–30% of total G6PD and the release was reduced by more than 50% by inhibition of Trpc3 with Pyr3.
Fig. 7. Deletion of Trpc3 and inhibition of Trpc3 activity by Pyr3 inhibit PERK phosphorylation and cell damage.
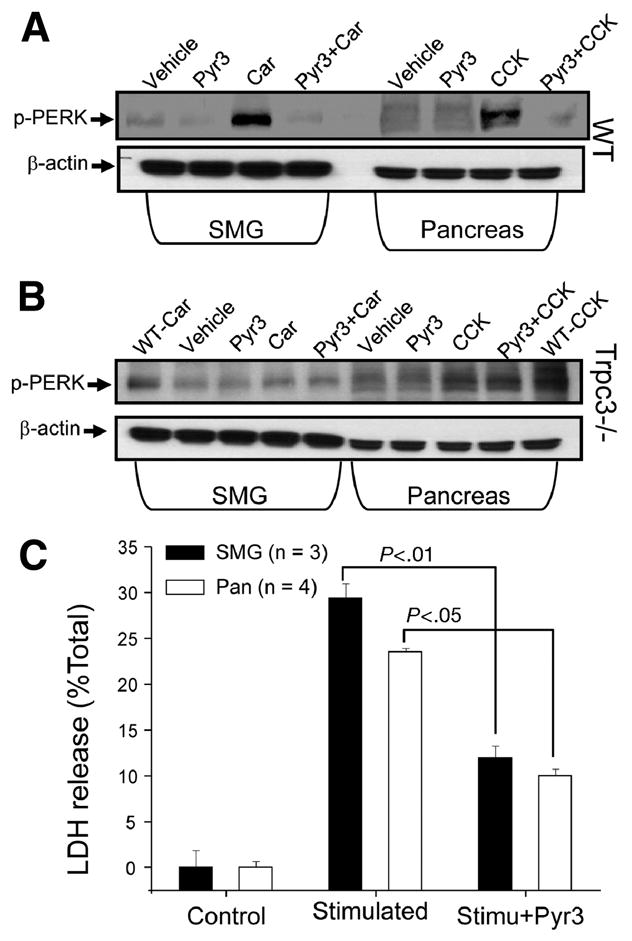
SMG and pancreatic acini obtained from wild-type (A) or Trpc3−/− mice (B) were treated with PBS (vehicle) or 3 μM Pyr3, as indicate. Part of the SMG acini were stimulated with 1 mM carbachol (Car) for 30 min and part of the pancreatic acini were stimulated with 10 nM CCK8 (CCK) for 30 min. Lysates prepared from the acini were analyzed for phosphor-PERK (p-PERK). Analysis of beta;-actin provides the loading controls. In (B) the first and last lanes are positive controls obtained with acini from wild-type mice. In (C) the supernatants of SMG and pancreatic acini treated with or without 3 μM Pyr3 and stimulated with 1 mM carbachol or 10 nM CCK8 for 30 min, respectively, were collected and used to measure released G6PD. G6PD release is expressed as % of total G6PD and is given as the mean±s.e.m of 3 and 4 experiments with SMG and pancreatic acini, respectively. The P values are listed in the Fig.
Cell damage causes excessive lipid hydrolysis and mistargeting of sphingolipids to increase cellular ceramide content36, 37. Most commonly, ceramide accumulates at the plasma membrane of damaged cells36. However, Fig. S2A shows the unique accumulation of ceramide in the secretory granules area of damaged pancreatic acini, possibly due to activation of lipases in the secretory granules. On the other hand, the images in Fig. S2B show that ceramide accumulated mostly at the plasma membrane of damaged SMG acini. Fig. S2 also shows that supramaximal stimulation increased ceramide level in both acini and the increase was prevented by inhibition of Trpc3 with Pyr3.
Conclusions
The present study examined the potential use of a specific Trpc3 inhibitor in the treatment of the autoimmune inflammatory diseases acute pancreatitis and Sjögren's syndrome. The first novel finding of the present study is the importance of Trpc3 for salivary secretion. Salivary secretion is a Ca2+-mediated process18 and Ca2+ influx is essential to sustain the secretion. Salivary glands express several TRPC channels, including TRPC1, TRPC3 and TRPC620. Deletion of Trpc3 in mice reduced salivary secretion (present work) to the same extent as deletion of Trpc127. This likely reflects the heteromultimerization of these TRPC channels and their mutual gating by STIM133. Hence, the present findings further highlight the importance of TRPC channels in exocrine cell function.
The contribution of TRPC channels to SOC activity continues to be debated with variable findings depending on cell type and the method used to evaluate the SOC function of the TRPC channels26, 38. We note that deletion of Trpc3 and inhibition of Trpc3 by Pyr3 reduced Ca2+ influx by about 50%, whether activated by receptor stimulation or by passive store depletion to activate Ca2+ influx by the SOC channels. Similarly, about 50% reduction in Ca2+ influx was observed by deletion of Trpc1 in salivary gland cells27 and pancreatic acini39. These findings indicate that Trpc3 and Trpc1 contribute to SOC activity in both pancreatic and salivary gland cells. Moreover, the residual 50% of the receptor-evoked and SOC-dependent Ca2+ influx must be mediated by a pathway that is independent of Trpc3 and Trpc1, suggesting the existence of at least two independent Ca2+ influx pathway in pancreatic and salivary gland cells. The residual Ca2+ influx can be mediated by another TRPC channel, like Trpc6 or by one of the Orai channels21, 26. An unresolved question at this stage is whether the different Ca2+ influx pathways mediate different cellular functions. If such is found, it will not be unprecedented since acinar cells show receptor-specific Ca2+ signaling due to differential coupling to RGS proteins14, 40, selective generation of Ca2+ releasing second messengers41 and regulation of IP3Rs42, 43.
The use of the Trpc3−/− mice allowed us to show that Pyr3 is indeed a highly selective Trpc3 inhibitor. The lack of a suitable model for studying mechanism of salivary gland cells damage, lead us to develop such a cell model. The model is based on inducing a receptor-mediated sustained increase in cytoplasmic Ca2+. We used this model since Ca2+ likely plays a role in acute salivary gland damage caused by medications and local release of cytokines and oxidative mediators, and sustained Ca2+ increase disrupts many cellular activities as it is a nodal point in many diseases. The salivary glands cell damage and the cerulein-induced acute pancreatitis models were used to show the Trpc3-mediated Ca2+ influx plays an essential role in cell damage. Notably, pharmacological inhibition of Trpc3 with Pyr3 protected the pancreas and salivary gland cells to the same extent as deletion of Trpc3. Since chronic28 and acute (present work) application Pyr3 to the mice appears to be tolerated well with no apparent toxicity, Pyr3 should be considered as a promising lead compound for the treatment of the glandular inflammatory diseases.
It is quite significant that Pyr3 is effective in preventing cell damage when given to the mice acutely by intraperitunial injection since it can be used when the diseases are in progress and patients are at highest risk, as is the case in acute pancreatitis. Targeted delivery of Pyr3 should increase its efficacy and use as a drug for the treatment of Ca2+ influx and TRPC3-associated diseases.
Supplementary Material
01
02
Acknowledgments
Grant Support: This work was supported by the Intramural Research Program of the NIH NIDCR/DIR to S.M and Z01-ES-101684 to L.B. and by the Science Research Program of the National Research Foundation of Korea (NRF), the Ministry of Education, Science and Technology grants 2011-0001167 and 2010-0000315 to DM.S.
Abbreviations
Pyr3
pyrazole3
SOC
store-operated Ca2+ influx channels
SMG
submandibular glands
TRPC3
transient receptor potential (canonical) isoform 3 channel
[Ca2+]i
free cytoplasmic Ca2+
ER
endoplasmic reticulum
CPA
cyclopiazonic acid (SERCA inhibitor)
SERCA
sarcoplasmic/endoplasmic Ca2+ ATPase pump
PERK
PKR-like ER kinase
Footnotes
All authors declare no conflict of interests
Authors contributions:
Min Seuk Kim, acquisition and analysis of data
Kyu Pil Lee, acquisition and analysis of data
Dongki Yang, acquisition and analysis of data
Dong Min Shin, drafting the manuscript
Joel Abramowitz, produced the KO mice
Shigeki Kiyonaka, synthesized pyrazole 3
Lutz Birnbaumer, produced the KO mice, drafting the manuscript
Yasuo Mori, synthesized pyrazole 3, drafting the manuscript
Shmuel Muallem, directing the study, drafting the manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakamura T, Lipton SA. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: possible pharmacological strategies. Cell Calcium. 47:190–7. doi: 10.1016/j.ceca.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naidoo N. ER and aging-Protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–9. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukherjee R, Criddle DN, Gukovskaya A, Pandol S, Petersen OH, Sutton R. Mitochondrial injury in pancreatitis. Cell Calcium. 2008;44:14–23. doi: 10.1016/j.ceca.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen OH, Tepikin AV, Gerasimenko JV, Gerasimenko OV, Sutton R, Criddle DN. Fatty acids, alcohol and fatty acid ethyl esters: toxic Ca2+ signal generation and pancreatitis. Cell Calcium. 2009;45:634–42. doi: 10.1016/j.ceca.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Lee MG, Muallem S. Pancreatitis: the neglected duct. Gut. 2008;57:1037–9. doi: 10.1136/gut.2008.150961. [DOI] [PubMed] [Google Scholar]
- 7.Reina S, Orman B, Anaya JM, Sterin-Borda L, Borda E. Cholinoreceptor autoantibodies in Sjogren syndrome. J Dent Res. 2007;86:832–6. doi: 10.1177/154405910708600905. [DOI] [PubMed] [Google Scholar]
- 8.Kruger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol. 2000;157:43–50. doi: 10.1016/S0002-9440(10)64515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roescher N, Tak PP, Illei GG. Cytokines in Sjogren's syndrome: potential therapeutic targets. Ann Rheum Dis. 2010;69:945–8. doi: 10.1136/ard.2009.115378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perl A. Emerging new pathways of pathogenesis and targets for treatment in systemic lupus erythematosus and Sjogren's syndrome. Curr Opin Rheumatol. 2009;21:443–7. doi: 10.1097/BOR.0b013e32832efe6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordmark G, Alm GV, Ronnblom L. Mechanisms of Disease: primary Sjogren's syndrome and the type I interferon system. Nat Clin Pract Rheumatol. 2006;2:262–9. doi: 10.1038/ncprheum0173. [DOI] [PubMed] [Google Scholar]
- 12.Ramos-Casals M, Tzioufas AG, Stone JH, Siso A, Bosch X. Treatment of primary Sjogren syndrome: a systematic review. JAMA. 304:452–60. doi: 10.1001/jama.2010.1014. [DOI] [PubMed] [Google Scholar]
- 13.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–20. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Kiselyov K, Wang X, Shin DM, Zang W, Muallem S. Calcium signaling complexes in microdomains of polarized secretory cells. Cell Calcium. 2006;40:451–9. doi: 10.1016/j.ceca.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology. 2009;137:1509–17. doi: 10.1053/j.gastro.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 17.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 18.Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–69. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- 19.Sutton R, Petersen OH, Pandol SJ. Pancreatitis and calcium signalling: report of an international workshop. Pancreas. 2008;36:e1–14. doi: 10.1097/MPA.0b013e3181675010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambudkar IS, Bandyopadhyay BC, Liu X, Lockwich TP, Paria B, Ong HL. Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium. 2006;40:495–504. doi: 10.1016/j.ceca.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the CRAC channel. Cell Calcium. 2007;42:133–44. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, Tepikin AV. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol. 2009;19:1648–53. doi: 10.1016/j.cub.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Ong HL, Pani B, Johnson K, Swaim WB, Singh B, Ambudkar I. Effect of cell swelling on ER/PM junctional interactions and channel assembly involved in SOCE. Cell Calcium. 47:491–9. doi: 10.1016/j.ceca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 25.Naidoo N. Cellular stress/the unfolded protein response: relevance to sleep and sleep disorders. Sleep Med Rev. 2009;13:195–204. doi: 10.1016/j.smrv.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee KP, Yuan JP, Hong JH, So I, Worley PF, Muallem S. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 584:2022–7. doi: 10.1016/j.febslet.2009.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proc Natl Acad Sci U S A. 2007;104:17542–7. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherwood MW, Prior IA, Voronina SG, Barrow SL, Woodsmith JD, Gerasimenko OV, Petersen OH, Tepikin AV. Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc Natl Acad Sci U S A. 2007;104:5674–9. doi: 10.1073/pnas.0700951104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–69. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 31.Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–55. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S, Okumura Y, Takahashi E, Kido H, Araki K, Baba H, Mizushima N, Yamamura K. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181:1065–72. doi: 10.1083/jcb.200712156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–45. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem. 2005;280:21600–6. doi: 10.1074/jbc.C400492200. [DOI] [PubMed] [Google Scholar]
- 35.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–61. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vazquez P, Blazquez C, Torres S, Garcia S, Nowak J, Fimia GM, Piacentini M, Cecconi F, Pandolfi PP, Gonzalez-Feria L, Iovanna JL, Guzman M, Boya P, Velasco G. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–72. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Cholesterol and sphingolipids in alcohol-induced liver injury. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S9–15. doi: 10.1111/j.1440-1746.2007.05280.x. [DOI] [PubMed] [Google Scholar]
- 38.Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- 39.Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S. Polarized but Differential Localization and Recruitment of STIM1, Orai1 and TRPC Channels in Secretory Cells. Traffic. 2011;12:232–45. doi: 10.1111/j.1600-0854.2010.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, Zeng W, Popov S, Berman DM, Davignon I, Yu K, Yowe D, Offermanns S, Muallem S, Wilkie TM. RGS proteins determine signaling specificity of Gq-coupled receptors. J Biol Chem. 1999;274:3549–56. doi: 10.1074/jbc.274.6.3549. [DOI] [PubMed] [Google Scholar]
- 41.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol. 2008;70:273–99. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 42.Bruce JI, Straub SV, Yule DI. Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium. 2003;34:431–44. doi: 10.1016/s0143-4160(03)00150-7. [DOI] [PubMed] [Google Scholar]
- 43.Yule DI, Betzenhauser MJ, Joseph SK. Linking structure to function: Recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium. 2010;47:469–79. doi: 10.1016/j.ceca.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02