Gene Structure Prediction and Alternative Splicing Analysis Using Genomically Aligned ESTs (original) (raw)
Abstract
With the availability of a nearly complete sequence of the human genome, aligning expressed sequence tags (EST) to the genomic sequence has become a practical and powerful strategy for gene prediction. Elucidating gene structure is a complex problem requiring the identification of splice junctions, gene boundaries, and alternative splicing variants. We have developed a software tool, Transcript Assembly Program (TAP), to delineate gene structures using genomically aligned EST sequences. TAP assembles the joint gene structure of the entire genomic region from individual splice junction pairs, using a novel algorithm that uses the EST-encoded connectivity and redundancy information to sort out the complex alternative splicing patterns. A method called polyadenylation site scan (PASS) has been developed to detect poly-A sites in the genome. TAP uses these predictions to identify gene boundaries by segmenting the joint gene structure at polyadenylated terminal exons. Reconstructing 1007 known transcripts, TAP scored a sensitivity (Sn) of 60% and a specificity (Sp) of 92% at the exon level. The gene boundary identification process was found to be accurate 78% of the time. TAP also reports alternative splicing patterns in EST alignments. An analysis of alternative splicing in 1124 genic regions suggested that more than half of human genes undergo alternative splicing. Surprisingly, we saw an absolute majority of the detected alternative splicing events affect the coding region. Furthermore, the evolutionary conservation of alternative splicing between human and mouse was analyzed using an EST-based approach. (See http://stl.wustl.edu/∼zkan/TAP/)
Deciphering the human genome is no less a challenge than the sequencing effort itself. A primary task in genome annotation is to elucidate the locations and structures of protein-coding genes. Over the last decade, computational gene finders have made significant advances toward accomplishing this goal. Recent evaluation studies (Claverie 1997; Reese et al. 2000) estimate that nearly all of the coding regions in anonymous genomic sequences can be identified. However, available prediction tools still have difficulty defining gene boundaries and predicting complete gene structures.
Expressed sequence tags (ESTs), which are single sequencing reads from cDNA clones, provide a tremendous resource for gene identification. As of February 10, 2001, the dbEST database has nearly 3.2 million human ESTs and continues to grow rapidly. Several software tools have used the EST resource to predict genes by aligning ESTs to the genomic sequence (Kulp et al. 1996; Xu et al. 1997; Jiang and Jacob 1998). However, EST-based gene inference still suffers from low specificity (Jiang and Jacob 1998; Reese et al. 2000). Sorting out the complex and often self-conflicting patterns of genomic EST alignment to predict the correct gene structure is a difficult problem. First, EST coverage of the gene is partial and some genes lack EST coverage altogether. In addition, EST resources are plagued by problems such as poor sequence quality, chimerism, and vector or intronic contamination (Wolfsberg and Landsman 1997). The prevalence of alternative splicing variants further compounds the difficulty. Even when all splice sites are correctly defined, it may be difficult to determine which combinations of splice sites are present in a full-length transcript. As a result, most gene finders do not take alternative splicing into consideration.
Alternative splicing of pre-mRNA serves versatile regulatory functions in controlling major developmental decisions and fine-tuning of gene function (Lopez 1998). Two recent studies estimate that 35%–38% of human genes undergo alternative splicing (Mironov et al. 1999; Brett et al. 2000). Hence, there is a vast “hidden” transcriptome that remains poorly characterized. Because ESTs are derived from genes expressed in a myriad of tissues and developmental processes, EST-based prediction would be an ideal approach to discover and delineate these alternative-splicing variants. A number of studies have relied on EST self-clustering to assemble alternative transcripts (Burke et al. 1998; Mironov et al. 1999). However, because of the error-prone nature of ESTs, the accuracy of EST self-clustering is problematic (Bouck et al. 1999).
Gene boundary determination is also an unsolved problem for EST-based and statistical gene finders (Claverie 1997; Reese et al. 2000). 5′ EST alignments frequently spread along the transcript because of varying degrees of cDNA truncation. 3′ EST alignments may also be scattered because of internal priming (Hillier et al. 1996). Moreover, genes on opposite strands often overlap at the 3′ UTR (untranslated region) (Tsai et al. 1994; Burke et al. 1998). Labeling errors and clone inversions can make ESTs from these reverse-strand genes difficult to distinguish. As a result, entirely EST-based methods are not expected to effectively identify gene boundaries.
We have developed a software tool, Transcript Assembly Program or TAP, that infers the predominant gene structure and reports alternative splicing events using genomic EST alignments. The gene structure is assembled from individual splice junction pairs using connectivity information encoded in the ESTs. A method called PASS (polyadenylation site scan) is used to infer poly-A sites from 3′ EST clusters. The gene boundaries are identified using the poly-A site predictions. We evaluated the accuracy of TAP by reconstructing 1007 functionally cloned and multiexon genes using ESTs from dbEST. The program scored a specificity of 92% at the exon level and 78% precision in defining gene boundaries. We also used TAP to conduct an analysis of alternative splicing in 1124 genic regions. By taking EST coverage into account, we estimated that over 55% of human genes undergo alternative splicing. Furthermore, 11% of the detected alternative splicing patterns were found to be conserved in mouse ESTs.
RESULTS
Overview
TAP uses EST sequence data to predict gene structures in anonymous genomic sequences. The test set we used consists of 1124 functionally cloned and genomically mapped human transcripts derived from the RefSeq database (Maglott et al. 2000). The transcript reconstruction process consists of the following three steps:
- Alignment construction. The goal of the first step is to obtain a set of native EST alignments to the genomic template. After the genomic locus for a RefSeq transcript is identified, the genomic sequence containing the genic region and up to 20 kb extensions at both ends is extracted. This genomic template is searched against dbEST using WU-BLASTN (Gish 1996–2000) and sequences of high-scoring EST hits are aligned to the genomic template using sim4 (Florea et al. 1998).
- Gene structure prediction. In the second step, genomic EST alignments with near-identity are used to infer the exon/intron structures. Although TAP can predict genes simultaneously on both strands, for simplicity we herein describe the gene prediction results with respect to the plus strand for each RefSeq gene. First, the splice pair, donor, and acceptor splice junctions that define the boundaries of an intron, are inferred from segmentation patterns in EST alignments and screened according to splice site patterns. The test set contains 1007 multiexon genes with 8879 pairs of known splice junctions. TAP correctly identified 5111 known splice pairs, yielding a sensitivity of 58%. Separately, PASS scans the genomic sequence for poly-A sites by clustering 3′ ESTs. For 290 RefSeq sequences with known poly-A sites, PASS scored a 84.5% sensitivity. Second, mutually exclusive splicing patterns are resolved by selecting the “predominant” splice pairs, according to EST coverage, and a joint gene structure for the entire genomic region is assembled from individual splice pairs. The EST-based connectivity between two adjacent splice pairs is examined to define exons and to delineate gaps in EST coverage. Finally, the gene boundaries are defined by segmenting the joint gene structure into individual genes at inferred intergenic regions (Fig. 1).
- Evaluation. The predicted gene structure are compared with the known gene structures to evaluate the accuracy of TAP. (A Web-based interface to TAP and the reconstruction results for 1124 RefSeq genes are available at http://stl.wustl.edu/∼zkan/TAP/.)
Figure 1.

Gene boundary identification. TAP uses a computer-generated graphic plot to illustrate the predicted gene structures. Shown here is the prediction on the genomic template of FANCG gene (NM_004629, Fanconi anemia, complementation group G). The blocks in the top level of each window represent exons in the reference gene structure. The blocks in the second level represent the predicted exons. Predicted poly-A sites are labeled by vertical lines. The region between two exons is either a splice junction pair (line) or a gap. Genes are colored differently according to inferred gene boundaries. In this plot, a boundary happens to be defined in each gap that follows a polyadenylated exon.
Gene Boundary Identification
In this study, we used genomic templates consisting of a 20-kb extension at both ends of the known genic region. According to TAP predictions, there is an average of 1.85 genes per template. For each template, TAP identifies gene boundaries by first assembling a single intron/exon structure containing coverage gaps and then dividing up this joint gene structure at gaps based on identifying features indicative of an intergenic gap, such as predicted poly-A sites. We evaluated the accuracy of this approach by comparing the inferred gene boundaries with known gene boundaries (Table 1). An extension error is counted if a predicted gene contains one or more exons located entirely outside the known boundaries. A split error is counted when a known genic region is divided into more than one gene. A total of 846 TAP reconstructions were evaluated as they consisted of at least one splice pair. We found 147 extension errors and 40 split errors, leaving 662 correct boundary predictions. Hence, TAP was correct 78% of the time in terms of identifying gene boundaries. For comparison, Genscan (Burge and Karlin 1997) was applied to the same set of genomic sequences and evaluated under the same criteria. It made 441 extension errors and 58 split errors, yielding a specificity of 44%.
Table 1.
Evaluation of Gene Boundary Identification
| 5′ Extension | 3′ Extension | Extension | Split | Correct | Accuracy | |
|---|---|---|---|---|---|---|
| Genscan | 303 | 203 | 441 | 58 | 375 | 44% |
| TAP | 89 | 66 | 147 | 40 | 662 | 78% |
Evaluation of Transcript Reconstruction
We also evaluated the performance of TAP in terms of reconstructing the gene structures for 1007 multi-exon genes. With an average of 10 exons per gene, this dataset spans a broad range of gene structure complexity in human genes. The predicted gene structures were compared with the known gene structures to calculate sensitivity (Sn) and specificity (Sp) measures at the exon and gene levels (Table 2). First, predicted gene structures within the known gene boundaries were evaluated. At the exon level, TAP scored an Sn of 0.60 and an Sp of 0.92; Genscan scored an Sn of 0.8 and an Sp of 0.81. The sensitivity of TAP is dependent on EST coverage, so it is likely to increase as available EST resources continue to grow. The fact that TAP had a lower sensitivity than Genscan is consistent with the observation that ESTs provide incomplete coverage of the transcripts. On the other hand, it is reasonable that TAP had a higher specificity than Genscan, because TAP is based on empirical evidence whereas Genscan tackles the difficult problem of ab initio gene prediction. At the gene level, TAP made predictions for 872 genes (Sn > 0) and Genscan made predictions for 979 genes. There were 611 accurate TAP gene structure predictions with 100% specificity and 362 accurate Genscan predictions. At the exon level, 272 TAP predictions had perfect sensitivity and specificity, whereas only 166 Genscan predictions were complete and accurate. Both programs did a better job of reconstructing simpler genes. On average, the genes successfully reconstructed by TAP had 6.8 exons per gene and those completely and accurately predicted by Genscan had 6.1 exons per gene.
Table 2.
Evaluation of Transcript Reconstruction
| Exon level | Gene level | |||||
|---|---|---|---|---|---|---|
| Sn | Sp | ME | WE | Sn | Sp | |
| TAP | 0.60 | 0.92 | 0.36 | 0.01 | 0.27 | 0.31 |
| Genscan | 0.80 | 0.81 | 0.15 | 0.09 | 0.165 | 0.17 |
| TAP1 | 0.58 | 0.87 | 0.37 | 0.07 | 0.23 | 0.27 |
| Genscan1 | 0.72 | 0.66 | 0.18 | 0.26 | 0.125 | 0.13 |
We also evaluated gene predictions using inferred boundaries. In this case, an exon prediction is considered correct only if it matches a known exon and is assigned into the correct genic region. Hence, a gene-splitting error reduces sensitivity, whereas an extension error reduces specificity. Overall, the sensitivity of TAP decreased from 0.60 to 0.58, and its specificity decreased from 0.92 to 0.88; the sensitivity of Genscan decreased from 0.80 to 0.72, and its specificity decreased from 0.81 to 0.66. The drop in the prediction performance of a program reflects the extent to which it miscalls gene boundaries.
Alternative Splicing Analysis
TAP identifies alternative splicing patterns of a gene by comparing predicted splice pairs with the known gene structure. EST-inferred splice pairs not found in the known gene structure are considered “alternative.” We do not have sufficient evidence to assert that all alternative splicing events found in EST alignments are biologically meaningful. However, detection is a necessary first step toward understanding those events that are truly important. We have found 669 alternative splice pairs in 365 of the 1007 multiexon genes, and 575 pairs were mutually exclusive with known splicing patterns, meaning that two splicing patterns have different but overlapping genomic coordinates. Mutually exclusive splicing patterns can only come from alternatively spliced transcript forms. The other 94 were novel splice pairs inserted into known exons.
An analysis of the distribution of alternative splicing events in different functional regions of the transcript revealed several distinctive features. First, the majority of alternative splicing events affected the coding regions (CDS). In 311 genes, 540 (81%) alternative splice pairs overlapped with the coding region (Table 3). A total of 639 known splice pairs were affected by alternative splicing, and 564 (88%) of them were located within the coding region. Thus, it appears that most of the alternative splicing events would alter the protein products if translation were successful. However, alternative splicing seemed to be more frequent in the untranslated region than in the coding region. Only 6.7% of known splice pairs in the CDS were affected by alternative splicing, whereas 15% of 5′ UTR and 14.6% of 3′ UTR splice pairs were affected. Intriguingly, we observed that alternative splicing often skips the known 5′ or 3′ terminal by inserting a new intron or by expanding an existing intron. In 62 genes, 75 alternative introns were found to skip the 5′ terminals, and 50 alternative introns in 47 genes were found to skip the 3′ terminals. In addition, a sizable fraction of novel splice pairs skipped the start or stop codons. Out of 94 novel splice pairs, eight spliced out the region between 5′ UTR and CDS, and 17 spliced out the region between CDS and 3′ UTR.
Table 3.
Regional Distribution of Alternative Splicing Patterns
| Region | REF | REFalt | REFest | REFalt, est | ALT | NEW |
|---|---|---|---|---|---|---|
| 5′ UTR | 335 | 50 | 135 | 25 | 61 | 38 |
| 5′ UTR and CDS | N/A | N/A | N/A | N/A | 20 | 8 |
| CDS | 8373 | 564 | 4895 | 451 | 447 | 13 |
| CDS and 3′ UTR | N/A | N/A | N/A | N/A | 23 | 17 |
| 3′ UTR | 171 | 25 | 81 | 17 | 22 | 18 |
| Total | 8879 | 639 | 5111 | 493 | 573 | 94 |
The conservation of splicing patterns between human and mouse was examined by BLAST searching sequence probes, each specifically representing a splice junction pair, against mouse ESTs. A probe was made by joining two 50-nucleotide exonic sequences that flank the donor and acceptor splice sites, respectively. A splice pair is said to be “conserved” if the BLASTN search produces an HSP alignment that exceeds 70% identity and spans the midpoint of the sequence probe. Out of 8879 known splice pairs, 4347 (49%) were found to be conserved, whereas only 73 (11%) of the 669 alternative splice pairs were conserved. If two mutually exclusive splice pairs both have matches in mouse ESTs, it suggests that the underlying alternative splicing event is conserved across species and, therefore, functionally significant. We have found 575 alternative splice pairs mutually exclusive with known splice pairs, and both patterns were conserved in 23 cases (Fig. 3).
Figure 3.

An example of conserved alternative splicing pattern. Shown here is a conserved exon skipping event at the 3′ end of RPN2 gene (NM_002951). The reference gene structure is displayed in the top level. The alternative splice pairs predicted from human ESTs, 76969–78662 and 78709–81563, are shown in the second level. We found that both the reference and alternative patterns were conserved. The mouse ESTs were aligned to the human genomic template using sim4. Each aligned block has ± 88% identity. The graphic plot was modified to illustrate these alignments. In the third level, the alignment of EST AI154341 shows the same pattern as the reference gene structure. In the bottom level, the alignment of EST AA038525 displays the alternative splicing pattern.
The frequency of alternative splicing in 1124 known genes was estimated by calculating the proportion of genes that were alternatively spliced. A total of 374 genes (33%), of which 365 were multiexon and nine were single exon, were found to have alternative splicing patterns. Genes expressed at lower levels are more likely to have alternative splicing variants that are poorly represented in the EST collection. We found that the frequency was indeed higher in genes with more EST hits (Fig. 4). Of the 575 genes with 40 or more EST hits, 49% were alternatively spliced. The frequency stabilized around 55% as the threshold was raised from 80 to 280 EST hits. This trend suggests that an alternative splicing frequency around 33% is likely an underestimate because of insufficient EST coverage, and that over 55% of human genes might undergo alternative splicing. Interestingly, the frequency of alternative splicing seemed to decrease below 50% for genes with more than 300 EST hits, indicating that highly expressed genes may be less likely to be alternatively spliced.
Figure 4.

Correlation of alternative splicing frequency with EST coverage. The frequency of alternative splicing was measured by the proportion of sequences that were alternatively spliced. A threshold on minimum EST coverage was imposed to select a subset of sequences. As the threshold was raised from zero to 340, the fraction of sequences (bar, left axis) that met the requirement was decreasing. The alternative splicing frequency (line, right axis) increased from 33% to 55% at lower EST coverage, and stabilized at roughly 55% at higher EST coverage.
DISCUSSION
We have developed an EST-based gene finder (TAP) for predicting the predominant gene structures and alternative splicing patterns in anonymous genomic sequences. By reconstructing 1007 multiexon RefSeq genes, we have shown that TAP is superior to existing gene finders with respect to accuracy in defining exons and gene boundaries. It scored a specificity of 92% at the exon level, and nearly a third of the reconstructions were both complete and accurate at the gene level. Furthermore, TAP achieved 78% accuracy in identifying gene boundaries. We also used TAP to conduct a large-scale analysis of alternative splicing in human genes. In 1007 multiexon genes, 669 alternative splice junction pairs were found. The majority of these alternative-splicing events affected the coding regions. A novel strategy was used to assess human-mouse conservation of alternative splicing patterns and revealed that 11% of the detected alternative splice pairs were conserved in mouse ESTs. Of 1124 RefSeq genes, 374 (33%) were found to contain alternative splicing patterns. Taking EST coverage into account, we estimated that over 55% of human genes undergo alternative splicing.
Analysis at the Splice Pair Level
We believe that alternative splicing is a major factor that negatively affects the specificity of TAP as shown by an analysis at the splice pair level. TAP assembles each gene structure from a selected set of EST-inferred splice pairs. When there are mutually exclusive splicing patterns, the algorithm selects the “predominant” splice pair that receives more EST coverage than other “alternative” splice pairs. Comparing 1007 known gene structures with all EST-inferred splice pairs, we found 575 alternative splice pairs mutually exclusive with known splicing patterns, but only 410 cases in which the known splice pair was detected in EST alignments. Hence, the remaining 165 alternative splice pairs had to be incorporated into the predicted gene structures because of a lack of predominant counterpart, and eventually counted as false positives in evaluation. However, we found our method to be fairly effective at selecting the known splicing patterns when mutually exclusive splicing patterns were both present in EST alignments. On average, each detected known splice pair was found in 13 EST alignments, whereas each alternative splice pair was only found in three ESTs. The known splice pair received more EST coverage than its alternative counterpart in 332 out of 410 cases, or 81% of the time. Only in 27 cases was the alternative splicing pattern more highly expressed.
Sim4 is an efficient and accurate tool for aligning EST sequences to the genome. However, because of poor sequence quality and the large volume of EST data, we found that a substantial number of inferred splice sites deviated slightly from the correct sites. To filter out these alignment errors, TAP requires that an accepted splice pair either contains the canonical GT..AG splice site pattern or is found in more than two EST alignments. An evaluation at the splice pair level suggested that this filtering step was crucial to making accurate gene predictions. During the reconstruction of 1007 multiexon transcripts, TAP filtered out a total of 1472 putative splice pairs. A comparison with the reference gene structures showed that 1054 were slight variations of known splice pairs. Nonetheless, the filtering step only results in a tiny loss in sensitivity as 95% of these splice pairs were still correctly predicted.
Partially spliced mRNAs often give rise to intronic EST sequences (Wolfberg and Landsman 1997). In 1007 multi-exon genes, we have found 141 intron-retaining events in which a known intronic region is completely covered by EST alignments. Only four of these introns (< 3%) appeared to be conserved in mouse ESTs. We also observed that the frequency of intron retention increased linearly with increasing EST coverage. This was taken as further evidence that intron retention was mostly caused by contamination. Curiously, 41 (31%) retained introns were 3′ terminal introns, but only four were 5′ terminal introns. One possible explanation is that 3′ end of a transcript received the most redundant EST coverage and, therefore, gave rise to more irregular ESTs. To be cautious, TAP did not report intron retaining as alternative splicing pattern.
Issues in Gene Prediction
TAP partitions EST alignments into strand-specific sets to predict genes on both strands (Fig. 5). ESTs are first classified according to labels and alignment directions (Jiang and Jacob 1998). As strand misclassification may occur because of incorrect labeling or clone inversion, EST alignments are partitioned again following the prediction of splice pairs by examining the strand specificity of splice site patterns. EST alignments that span splice junctions reveal their strand origins, whereas EST alignments that do not span splice junctions contribute little to the predicted gene structure, which is based on an assembly of splice junction pairs. Genes on opposite strands sometimes overlap in the 3′ UTRs. When misclassified, ESTs from the 3′ UTR of the reverse gene could result in false extension of the gene boundary. TAP deals with this type of cross-strand contamination by defining the 3′ gene boundary at a predicted poly-A site. To make a positive prediction, PASS requires either a minimum of four 3′ ESTs that cluster at the 3′ ends, or one EST containing a polyadenylation motif near the 3′ end. The misclassified ESTs, with their 3′ ends flipped to the 5′, can only meet these requirements by coincidence.
Figure 5.

Gene prediction on both strands. This is a graphic illustration of gene predictions in the genomic template of NM_005155, palmitoyl-protein thioesterase 2 (PPT2). Reference gene structure is shown in the first level of each window. Gene predictions on the plus strand are plotted in the second level and gene predictions on the minus strand are plotted in the bottom level. The transcriptional direction of a gene is also indicated by the arrow shape of its terminal exon. Only poly-A sites on the plus strand are shown in vertical lines. The middle levels are used to display alternative splicing patterns that are inferred by comparing predicted splice junction pairs with the reference gene structure. Note that the predicted gene structure of NM_005155 consists of extensions to the reference gene structure at both ends. The second predicted gene on the plus strand overlaps with the 3′ UTR of a gene on the opposite strand, but its 3′ boundary is not extended.
TAP predicts both multiexon and single-exon genes. However, EST-based single-exon gene prediction is likely to have low sensitivity and specificity for the following reasons. First, intronless genes are known to have lower expression levels than genes with introns (Hamer and Leder 1979). In addition, many factors such as aforementioned cross-strand contamination, processed pseudogenes (Vanin 1985), and spurious transcription (Liang et al. 2000) can give rise to false single-exon gene predictions. TAP predicts a single-exon gene by extending its poly-A site, which is not detected unless a minimum number of 3′ ESTs are clustered. This measure is intended to reduce the false positive rate, but it also reduces sensitivity. When tested on 117 single-exon RefSeq genes, TAP yielded an Sn of 0.33 at the nucleotide level, whereas Genscan scored an Sn of 0.77. Predictions were made (Sn > 0) for 56 genes, only 48% of the total. For these genes, TAP scored an Sn of 0.69, and Genscan scored an Sn of 0.70. It is worth noting that 36 reference sequences were histone genes, most of which are not polyadenylated and therefore cannot be identified by EST-based methods.
Evaluation of gene boundary prediction is a complicated issue. Transcripts often have alternative terminals because of alternative transcriptional initiation or differential polyadenylation. In addition, functionally cloned transcripts are not necessarily full length (Gautheret et al. 1998; Kan et al. 2000). It is also impractical to define the exact 5′ terminals of genes using an EST-based approach because ESTs are end-sequenced from cDNA clones often truncated at the 5′ end. The primary aim of our gene boundary identification process is, therefore, not to define the exact gene terminals, but to partition EST alignments into the correct genic regions. Hence, our evaluation criteria do not require the correct boundary predictions to exactly match the known boundaries. The predicted boundaries for a gene are considered to be correct as long as the known genic region is not divided up or joined with exons entirely outside of the known boundaries. Even these criteria may be too stringent. An extension beyond the known boundaries does not necessarily constitute a prediction error, but could be a part of the gene missing in the published sequence. For example, TAP has predicted 63 5′ extensions and 27 3′ exon extensions connected to the known genic region through EST-confirmed splicing events.
The Hidden Transcriptome
Recent studies suggest that the human genome codes for fewer than twice the number of genes in the genome of Caenorhabditis elegans or Drosophila melanogaster, and raise an intriguing hypothesis that alternative splicing accounts for much of the molecular complexity in vertebrates (Crollius et al. 2000; Ewing and Green 2000). Our estimate that more than half of human genes undergo alternative splicing appears to support this argument. In addition, frequent alternative splicing may partially explain the drastic difference in the estimated numbers of human genes, ranging from 30,000 to 120,000 (Crollius et al. 2000; Ewing and Green 2000; Liang et al. 2000). The gene index analysis could be counting the number of unique transcripts because they used strict clustering rules that partitioned alternative transcripts into separate clusters, whereas other studies were more focused on the number of genic regions. The huge excess in the number of transcripts over the number of genic regions suggests a vast realm of alternative transcripts.
There is growing interest in gene prediction based on cross-species sequence comparison. Using preselected sets of orthologous gene pairs, several studies have shown that comparing human and mouse genomic sequences is highly effective in predicting gene structures (Batzoglou et al. 2000; Bafna and Huson 2000). However, conservation analysis based on mouse ESTs showed that the conservation rate was 49% for reference splice pairs, but only 11% for alternative splice pairs, suggesting that a substantial fraction of alternative splicing events may be species-specific and, therefore, undetectable through genome comparison. Moreover, genome comparison is unable to predict certain types of alternative splicing patterns such as exon skipping. Thus, it appears that the primary purpose of gene prediction based on genome comparison is to delineate the evolutionarily conserved gene structures. Even if the genomic sequence of mouse becomes available, the challenge of gene prediction in term of elucidating alternative splicing variants will remain. To that end, EST-based methods such as TAP can provide a powerful means of detection and characterization.
METHODS
Construction of Datasets and Genomic EST Alignments
The genomic contig dataset consists of 2861 Homo sapiens contigs and 541,943,911 bases of finished sequences retrieved from the Genome Contigs Database dated February 2000 (Rouchka and States 1999). A total of 1,521,800 H. sapiens EST sequences and 916,528 Mus musculus sequences were derived from the dbEST database (release 021800). We retrieved 6616 H. sapiens mRNA sequences from the NCBI RefSeq database (Maglott et al. 2000) dated February 2000. These mRNA sequences were searched against the contig database using WU-BLASTN 2.0 (Gish 1996–2000). The high scoring sequences were aligned to the matching contig sequences using sim4 1.4 (Florea et al. 1998). If the overall percent identity of an alignment was above 99%, the genomic locus was assumed to be found. This process produced 1124 sequences that made up the test set. 1007 of these were multi-exon and 117 were single exon. For each gene, the genomic template including the genic region, and up to 20 kb extensions at both ends were extracted and set to the same orientation as the known mRNA. Repetitive elements in these genomic sequences were masked using RepeatBlaster (Bedell et al. 2000), an accelerated RepeatMasker (Smit and Green 1996). The masked genomic templates were searched against the human ESTs. After removing residual poly-A sequences, high scoring ESTs were aligned to the template using sim4. Only EST alignments with greater than 92% identity were used. Poorly aligned terminal regions were also trimmed from the alignments.
Prediction of Splice Junction Pairs and Poly-A Sites
Each gap between two adjacent exon segments in an EST alignment delineates a possible splice pair: a pair of genomic sequence coordinates that denote the boundaries of an intron. An EST alignment that gives rise to the splice pair is referred to as a carrier. The number of EST carriers, also called EST coverage, is proportional to the expression level of the underlying transcript. Splice pairs with more EST coverage are predicted with more confidence. A splice pair prediction is accepted if it has the consensus GT.AG splice pattern or more than two EST carriers.
TAP partitions EST alignments into strand-specific sets. First, strand origins are determined from EST labels and alignment directions. 5′ ESTs aligned to the genome in the plus direction and 3′ ESTs aligned in the minus direction are considered to belong to the plus strand. 5′ ESTs aligned in the minus direction and 3′ ESTs ligned in the plus direction are partitioned onto the minus strand. For each strand, splice junction pairs and poly-A sites are inferred using its specific set of EST alignments. EST alignments are further partitioned according to strand specificity of splice patterns following the prediction of splice pairs. For a plus-strand EST, if its splice pairs only carry a splice site pattern that is the reverse complement of GT.AG, it is reassigned to the minus strand. Likewise, minus-strand ESTs carrying GT.AG patterns are reassigned to the plus strand.
We used PASS to define the poly-A sites in the genome using 3′ EST sequences (Kan et al. 2000). EST alignments that terminate within 20 nucleotides of each other are clustered. For each cluster, all 3′ ends are sorted and the center position is taken to represent a potential poly-A site. On the genomic template, a 30-nucleotide region upstream of a possible site is searched for canonical poly-A patterns, AATAAA or ATTAAA (Gautheret et al. 1998). Twenty nucleotides of downstream sequence are searched for an A-rich region, defined as windows of 10 nucleotides containing eight or more As. All possible sites are scored. By default, the score is the natural log of the cluster size. The presence of poly-A signal confers a two-point addition and the presence of an A-rich region results in a two-point penalty. The threshold value used is 1.1, requiring a minimum cluster size of four for a positive identification when none of the other factors is present.
Gene Structure Assembly
A joint gene structure that could include multiple genes in the genomic region is assembled from accepted splice pairs. A biological gene structure consists of either introns or exons, but an EST-inferred gene structure may contain a region for which there is no EST coverage and no information about the biological gene structure, which is referred to as a coverage gap. TAP delineates the boundaries of these coverage gaps by examining the EST-based connectivity between adjacent splice pairs.
All inferred splice pairs are sorted by their 5′ coordinates. The connectivity between two adjacent splice pairs is classified into four types: conflicting, contiguous, transitive, and gapped (Fig. 6A). A conflicting connection arises when two splice pairs i and j overlap with one another but have different coordinates, and are given a conflict score A(i,j) = −∞. The connection is contiguous when one or more EST alignments carry two splice pairs i and j in adjacent positions, indicating a complete exon. This type of connection is scored by B(i,j), set to the number of ESTs that carry introns i and j contiguously. When no EST spans the entire exon, the program examines if there is overlapping EST coverage between two splice pairs. If there is overlapping coverage, the connection is transitive, and the transitive score C(i,j) is set to 1. Otherwise, the connection is gapped, and C(i,j) is set to 0. A matrix M is constructed to record the connectivity relationships between splice pairs (Fig. 6B). The value M(i,j) for the connection between splice pair i and j is the sum of A(i,j), B(i,j), and C(i,j).
Figure 6.

Gene structure assembly. (A) Shown here is a hypothetical gene structure (block) and genomic EST alignments (line). There are five inferred splice pairs. Splice pairs 1 and 2 are transitively connected. Splice pairs 2 and 4 are contiguously connected. Both splice pairs 2 and 4 are mutually exclusive with splice pair 3. There is a coverage gap between splice pair 5 and the 3′ end. (B) The connectivity matrix for assembling this gene structure. The nodes include the 5′ beginning (BEG) of the EST alignments, the 3′ end (END) and five predicted splice pairs. The numerical value in cell M(i,j) is determined from the EST-encoded connectivity between the ith and jth nodes. For instance, two EST alignments link splice pairs 2 and 4, so M(2,4) = 2. (C) Two alternative gene structures inferred from two different traces through the matrix. The higher scoring trace gives rise to the predominant gene structure.
The filled connectivity matrix is traced to assemble the gene structure, represented by a path through the matrix M in the 5′ to 3′ direction (Fig. 6C). At each elongation step from splice pair i to j, the tracing process selects the downstream splice pair(s) with the maximum M(i,j) score. This rule means that contiguous connections take precedence over transitive connections, which in turn take precedence over gapped connections. Mutually exclusive splice pairs are not allowed in the same path. Alternative paths are generated by branching the path when multiple downstream splice pairs have the maximum connectivity score. Hence, if the algorithm cannot determine the predominant connection among mutually exclusive splicing patterns, it recursively traces all of these connections. For each path, the individual M(i,j) scores are summed up to yield a cumulative connectivity score, taken as a heuristic measure of expression level for the underlying transcript. Finally, the algorithm selects an optimal path with the maximum cumulative connectivity score.
Gene Boundary Determination
The assembly process yields an optimal path of splice pairs, which is then converted into a joint gene structure spanning the entire template. A genomic region is defined as an exon if it is enclosed between two splice pairs connected contiguously or transitively. The gapped region in between two splice pairs is defined by setting boundaries for the flanking exons. The 3′ boundary of the upstream intron defines the 5′ end of the upstream exon, and the 5′ boundary of the downstream intron defines the 3′ end of the downstream exon. The upstream exon is extended toward the 3′ direction, and the downstream exon is extended toward the 5′ direction. This extension process examines all EST alignments and successively extends the boundary whenever an exon segment overlaps with the existing boundary and indicates an extension in the right direction. Only one coverage gap is defined in between two splice pairs. As a result, a gapped region could contain singleton EST alignments that sometimes represent single-exon genes. To identify single-exon genes, TAP searches for poly-A sites within coverage gaps, extends them toward upstream to define exon segments, and inserts them into the joint gene structure. This process sets the 3′ boundary of the exon segment at the poly-A site and extends the 5′ boundary using the successive extension strategy based on overlapping EST alignments. The terminals of the joint gene structure are defined by the outermost EST alignment ends on the genomic template. The EST-based connectivity between a terminal and its adjacent splice pair is examined using the same approach as above to define the terminal exon.
The joint gene structure is assembled regardless of gene boundaries and could incorporate multiple genes. The boundaries of each gene are determined by identifying intergenic gaps surrounding the gene. A gap in the joint gene structure can be intragenic or intergenic. An intergenic gap is invariably preceded by a terminal exon containing one or more poly-A sites. In addition, the distal poly-A site tends to be distant from the next poly-A site downstream. Terminal exons are also longer than internal exons in general. Each gap in the joint gene structure is examined as a possible intergenic gap. A score is determined by the following formula:
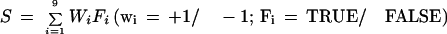
Fi is a feature that either evaluates to true or false. Wi is the weight assigned to each feature. We used six positively weighted features that are characteristic of an intergenic gap. F1 stands for the presence of a poly-A site. F2 stands for the presence of multiple poly-A sites. F3 stands for long distance (distance >5000 nucleotides) between the distal poly-A site and the immediately downstream site. F4 stands for long gap (length >20,000 nucleotides). F5 stands for long exon (length >600 nucleotides). F6 stands for the presence of multiexon genes on the opposite strand. We use three negatively weighted features. F7 stands for short gap (length <500 nucleotides). F8 stands for short exon (length <150 nucleotides). F9 stands for short distance (distance <1000 nucleotides). All weights had an absolute value of 1. The threshold values used were empirically determined. A gap was determined to be intergenic if it received a positive score. Finally, the joint gene structure is segmented in the predicted intergenic gaps into individual genes, setting the gene boundaries at either the terminal of the joint gene structure or the boundary of a coverage gap. A postprocessing step defines the 3′ gene terminal at the distal poly-A site in the terminal exon.
Evaluation of Transcript Reconstruction
The reference gene structures were obtained by aligning RefSeq sequences to the genomic templates. Splice pair predictions and gene structure predictions were evaluated separately. First, all predicted splice pairs located within the known gene boundaries were examined. A predicted splice pair is correct if it has an exact match with a reference splice pair, meaning that they have the same boundary positions. A splice pair has an approximate match when both of its boundary positions differ by <10 nucleotides from a pair of known intron boundaries. We found only 35 approximately matched splice pairs in total. If a predicted splice pair does not have any match, it is considered an alternative splice pair.
The standard evaluation methods (Burset and Guigo 1996; Reese et al. 2000) were used to evaluate gene structure predictions. All predicted exons within the known gene boundaries were evaluated, whereas exonic regions beyond the boundaries were ignored. We require that a correctly predicted internal exon must have both sides exactly matching a reference exon. The initial exon only needs to match the 3′ side, and the terminal exon needs to match the 5′ side. A total of 476 gapped exons (containing internal gaps) and 195 exon fragments (bounded by gaps) were excluded from evaluation. At the exon level, Sn is the proportion of reference exons correctly predicted. Sp is the proportion of predicted exons that are correct. ME (missed exon) is the proportion of reference exons that are completely missed. WE (wrong exon) is the proportion of predicted exons that do not overlap with any reference exons. In addition, the evaluation was performed using the inferred gene boundaries. In that case, the predicted gene structure with the greatest overlap with the known genic region was evaluated. If a known exon is predicted but assigned to another gene structure, it is considered a missed exon. Likewise, any predicted exons outside the known boundaries are considered to be wrong exons. At the gene level, Sn represents the proportion of complete gene structures that are accurately reconstructed at the exon level. Sp represents the proportion of predicted gene structures that are completely accurate.
Figure 2.

Prediction of alternative splicing patterns. This plot shows both the predicted gene structure and alternative splicing patterns for D6S52E gene (NM_004639, HLA-B associated transcript-3). The reference gene structure is displayed in the first level. The predicted gene structure is shown in the second level. TAP detected nine alternative splice pairs by comparing with the reference gene structure. Sorted by the start coordinates, these are 20211–20855 (AW408054.1), 23432–23880 (AL046298.1), 23954–24805 (AL046298.1), 27064–33110 (AW182608.1), 27684–28653 (AL041773.1), 28105–28216 (AW380963.1), 28323–28436 (AW380963.1), 31988–32327 (AI024684), and 32434–33407 (AI024684). For each splice pair, one of the EST carriers is denoted in parentheses.
Acknowledgments
We thank Mr. Jarret Glasscock for providing us with the early release of Eugene database. This work was supported in part by grants from the National Institutes of Health (R01-HG01391), and the Department of Energy (DE-FG02-94ER61910).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Corresponding author.
E-MAIL states@ibc.wustl.edu; FAX (314) 362-0234.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.155001.
REFERENCES
- Bafna V, Huson DH. The conserved exon method for gene finding. Intell Syst Mol Biol. 2000;8:3–12. [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander E. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell JA, Korf I, Gish W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. doi: 10.1093/bioinformatics/16.11.1040. [DOI] [PubMed] [Google Scholar]
- Bouck J, Yu W, Gibbs R, Worley K. Comparison of gene indexing databases. Trends Genet. 1999;15:159–161. doi: 10.1016/s0168-9525(99)01709-6. [DOI] [PubMed] [Google Scholar]
- Brett D, Hanke J, Lehmann G, Hasse S, Delbruck S, Krueger S, Reich J, Bork P. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 2000;47:83–86. doi: 10.1016/s0014-5793(00)01581-7. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Burke J, Wang H, Hide W, Davison DB. Alternative gene form discovery and candidate gene selection from gene indexing projects. Genome Res. 1998;8:276–290. doi: 10.1101/gr.8.3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burset M, Guigo R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–367. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- Claverie JM. Computational methods for the identification of genes in vertebrate genomic sequences. Hum Mol Genet. 1997;6:1735–1744. doi: 10.1093/hmg/6.10.1735. [DOI] [PubMed] [Google Scholar]
- Crollius HR, Jaillon O, Bernot A, Basilva C, Bouneau L, Fischer C, Fizames C, Wincker P, Brottier P, Quetier F, et al. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat Genet. 2000;25:235–238. doi: 10.1038/76118. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Analysis of expressed sequence tags indicate 35,000 human genes. Nat Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D, Poirot O, Lopez F, Audic S, Claverie JM. Alternate polyadenylation in human mRNAs: A large-scale analysis by EST clustering. Genome Res. 1998;8:524–530. doi: 10.1101/gr.8.5.524. [DOI] [PubMed] [Google Scholar]
- Gish, W. 1996–2000. WU-BLAST 2.0. http://blast.wustl.edu/
- Hamer D, Leder P. Splicing and formation of stable RNA. Cell. 1979;18:1299–1302. doi: 10.1016/0092-8674(79)90240-x. [DOI] [PubMed] [Google Scholar]
- Hillier L, Lennon G, Becker M, Bonaldo MF, Chiapelli B, Chisoe S, Dietrich N, Dubuque T, Favello A, Gish W, et al. Generation and analysis of 280,000 human expressed sequences tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Jiang J, Jacob HJ. EbEST: An automated tool using expressed sequence tags to delineate gene structure. Genome Res. 1998;8:268–275. doi: 10.1101/gr.8.3.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Gish W, Rouchka E, Glasscock J, States D. UTR reconstruction and analysis using genomically aligned EST sequences. Intell Syst Mol Biol. 2000;8:218–227. [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden markov model for the recognition of human genes in DNA. Intell Syst Mol Biol. 1996;4:134–142. [PubMed] [Google Scholar]
- Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25:239–240. doi: 10.1038/76126. [DOI] [PubMed] [Google Scholar]
- Lopez AJ. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu Rev Genet. 1998;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Fickett JW, Gelfand MS. Frequent alternative splicing of human genes. Genome Res. 1999;9:1288–1293. doi: 10.1101/gr.9.12.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouchka, E.C. and States, D.J. 1999. Assembly and analysis of extended genomic contig regions. Technical report WUCS-99-10. http://stl.wustl.edu/contigs/
- Smit, A.F.A. and Green, P. 1996. RepeatMasker. http://ftp.genome.washington.edu/RM/RepeatMasker.html
- Tsai JY, Namin-Gonzales ML, Silver LM. False association of human ESTs. Nat Genet. 1994;2:321–322. doi: 10.1038/ng1294-321. [DOI] [PubMed] [Google Scholar]
- Vanin EF. Processed pseudogenes: Characteristics and evolution. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, Landsman D. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 1997;25:1626–1632. doi: 10.1093/nar/25.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Mural RJ, Uberbacher EC. Inferring gene structures in genomic sequences using pattern recognition and expressed sequence tags. Intell Syst Mol Biol. 1997;5:344–353. [PubMed] [Google Scholar]