High-Throughput Genotyping with Single Nucleotide Polymorphisms (original) (raw)
Abstract
To make large-scale association studies a reality, automated high-throughput methods for genotyping with single-nucleotide polymorphisms (SNPs) are needed. We describe PCR conditions that permit the use of the TaqMan or 5′ nuclease allelic discrimination assay for typing large numbers of individuals with any SNP and computational methods that allow genotypes to be assigned automatically. To demonstrate the utility of these methods, we typed >1600 individuals for a G-to-T transversion that results in a glutamate-to-aspartate substitution at position 298 in the endothelial nitric oxide synthase gene, and a G/C polymorphism (newly identified in our laboratory) in intron 8 of the 11–β hydroxylase gene. The genotyping method is accurate—we estimate an error rate of fewer than 1 in 2000 genotypes, rapid—with five 96-well PCR machines, one fluorescent reader, and no automated pipetting, over one thousand genotypes can be generated by one person in one day, and flexible—a new SNP can be tested for association in less than one week. Indeed, large-scale genotyping has been accomplished for 23 other SNPs in 13 different genes using this method. In addition, we identified three “pseudo-SNPs” (WIAF1161, WIAF2566, and WIAF335) that are probably a result of duplication.
Single-nucleotide polymorphisms, or SNPs, have become prominent in human genetics, and their popularity can be attributed to several reasons. The failure of linkage analysis to identify, in a convincing way, loci for complex diseases has led to interest in large-scale association studies for mapping genes for complex traits (Risch and Merikangas 1996; Collins et al. 1997). Because SNPs are the most abundant, accessible class of polymorphisms present in the human genome, the use of as many as one million SNPs scattered across the human genome was envisioned for such studies. Substantial effort is being devoted by the SNP Consortium toward developing a more modest SNP map comprising 300,000 markers (Masood 1999). The second reason for the current popularity of SNPs is the potential ease with which they can be used for genotyping. In contrast to the tri- and tetranucleotide markers used in linkage analysis, SNPs, because they are usually biallelic, are more amenable to automated detection. This difference could result in considerable savings in the cost and time required for genotyping.
Our group, the Stanford, Asia, Pacific Program for Hypertension and Insulin Resistance (SAPPHIRe), is devoted to identifying susceptibility genes for essential hypertension, a complex trait, in populations of Chinese and Japanese origin. To this end, we have begun a systematic survey of candidate genes that might be involved in regulating blood pressure. Our approach is to identify SNPs in such genes and test these for association in a large population comprised of subjects that have very high or low-normal blood pressure. The general validity of association results needs to be determined by testing the same polymorphism in different populations. The Family Blood Pressure Program (FBPP) of the National Heart, Lung, and Blood Institute, of which SAPPHIRe is a member, was established with this objective in mind. Because members of the FBPP have recruited subjects of diverse origins—from Asian samples like ours, as well as from Caucasian, Hispanic, and African-American populations from the United States—a positive association result in one population can be quickly tested in another. Thus, the FBPP in general, and SAPPHIRe in particular, needed methods to rapidly test different SNPs in a large number of subjects (>4000). We therefore focused on developing tools that will facilitate high-throughput genotyping using SNPs.
Here we have examined the suitability of the 5′ nuclease allelic discrimination or TaqMan assay (Livak et al. 1995) for high-throughput genotyping. In this method, the region flanking the polymorphism, typically 100 base pairs, is amplified in the presence of two probes each specific for one or the other allele. Probes have a fluor, called “reporter,” at the 5′ end but do not fluoresce when free in solution because they have a “quencher” at the 3′ end that absorbs fluorescence from the reporter. During PCR, the Taq polymerase encounters a probe specifically base-paired with its target and unwinds it. The polymerase cleaves the partially unwound probe and liberates the reporter fluor from the quencher, thereby increasing net fluorescence. The presence of two probes, each labeled with a different fluor, allows one to detect both alleles in a single tube. Moreover, because probes are included in the PCR, genotypes are determined without any post-PCR processing, a feature that is unavailable with most other genotyping methods (for a recent review, see Landegren et al. 1998).
We describe PCR conditions that facilitate accurate and rapid genotyping of large numbers of individuals with large numbers of SNPs and computational methods that permit one to automate the allele-calling procedure—a key requisite for any high-throughput genotyping method. To demonstrate the utility of these methods, we typed >1600 subjects for a SNP in the endothelial nitric oxide synthase gene and another in the 11–β-hydroxylase gene. In addition, we uncovered three “pseudo-SNPs” that appear to be the result of adjacent duplications.
RESULTS
High-Throughput Genotyping with the TaqMan Assay
We used the TaqMan assay to type 1699 individuals for two unrelated SNP markers on different chromosomes: One is a G/T transversion that results in a glutamate-to-aspartate substitution at position 298 in the endothelial nitric oxide synthase gene (eNOSE298D; Miyamoto et al. 1998), and the other is a C/G transversion in intron 8 (newly identified in our laboratory) of the 11–β hydroxylase gene (CYP11B15). DNA samples and a mixture containing buffer, probes, primers, and polymerase were distributed in 96-well plates and fluorescence in each was measured prior to PCR. Following PCR in a standard 96-well machine, fluorescence was measured again for each sample. Post-PCR data from all the plates were imported into a statistical software package and fluorescence from the two reporters was plotted.
As shown in Figure 1A, for the eNOSE298D SNP, there were no obvious outliers, but samples from one PCR plate formed a separate group. Comparison of mean pre-PCR fluorescence values for both dyes from this plate with those from other plates revealed that there was significantly less fluorescence from the reporter dyes in all wells of this plate. Because the magnitude of post-PCR fluorescence was proportional to that prior to PCR, we adjusted post-PCR fluorescence for this plate accordingly. _k_-means clustering was then used to automatically classify samples into four groups—the three genotypes and a “no DNA” control group (Fig. 1B).
Figure 1.

Genotyping results for the E298D SNP in the endothelial nitric oxide synthase gene. Data for 1699 genotypes are shown. (A) Raw data from the TaqMan PCR. (B) Data corrected for variation in pre-PCR fluorescence and _k_-means clustering. Fluorescence for the E allele is plotted along the _X_-axis (the dye is “FAM”) and for the D allele, labeled with the fluor “VIC”, along the _Y_-axis. “Rn” is fluorescence from the reporter dye divided by that from the passive reference dye. (Red squares) Samples homozygous for the E allele; (blue squares) samples homozygous for the D allele; (green squares) E/D heterozygotes; (black squares) “no DNA” controls or samples that failed to amplify. Arrows indicate samples with a very low conditional probability of belonging to that particular cluster.
The particular _k_-means clustering algorithm used for assigning genotypes in this study is based on nearest-centroid sorting (Anderberg 1973; Sharma 1996). In this method, data are classified into a predetermined number of groups or clusters; a case is assigned to the cluster with the smallest distance between the case and the center of the cluster (centroid). Cluster centers are not known in advance but are iteratively estimated from the data. As can be seen (Fig. 1B), the classification is good with no overlap between different genotype clusters. A small number of samples (15) failed to amplify, and the algorithm correctly placed these in the no DNA control group.
The results for the CYP11B15 SNP are shown in Figure 2. As can be seen from the raw data (Fig. 2A), there were no outliers and, unlike in the eNOSE298D case, all the plates yielded similar post-PCR fluorescence values. The output of the _k_-means clustering is plotted in Figure 2B. For this particular SNP, only four samples failed to amplify robustly and these were classified appropriately in the no DNA control group. For both SNPs, assay failure was probably due to pipetting error because these samples gave robust genotypes for other SNPs.
Figure 2.

Genotyping results for the CYP11B15 SNP. Data for 1699 genotypes are shown. (A) Raw data from the TaqMan PCR. (B) Output of the _k_-means clustering. Fluorescence values for the C and G alleles are plotted along the X and Y axes, respectively. (Red squares) C/C homozygotes; (green squares) G/C heterozygotes; (blue squares) G/G homozygotes; (black squares) “no DNA” controls or failed PCRs. Arrows indicate samples with a very low conditional probability of belonging to that particular cluster.
Over twenty different SNPs (Table 1) have been used for large-scale genotyping using this method and yield results that are similar to those presented here (see www.genome.org for these data).
Table 1.
Other SNPs Used in Large-Scale Genotyping
| Gene | # of SNPs | Source |
|---|---|---|
| Urea transporter | 5 | This lab* |
| Mineralocorticoid receptor | 2 | This lab* |
| CD36 | 1 | This lab* |
| Angiotensinogen 2 receptor 2 | 2 | This lab* |
| Arginine Vasopressin receptor | 2 | This lab* |
| Protein tyrosine phosphatase-1 | 1 | This lab* |
| Aldosterone synthase | 2 | Halushka et al. 1999 and this lab |
| Kallikrein | 1 | Halushka et al. 1999 |
| Apolipoprotein C4 | 1 | Halushka et al. 1999 |
| β3 adrenergic receptor | 1 | Walston et al. 1995 |
| Intercellular adhesion molecule-1 | 1 | Halushka et al. 1999 |
| Alpha adducin | 1 | Halushka et al. 1999 |
| β2 adrenergic receptor | 3 | McGraw et al. 1998; Reibsaus et al. 1993 |
| Total | 23 |
To monitor the quality of genotyping and allele-calling procedure, >200 samples, of which 56 were blind, were typed in duplicate for both SNPs; there were no discordancies. Based on similar genotyping data for SNPs listed in Table 1, we estimate the error-rate to be <1 in 2000 genotypes. Further, for the CYP11B15 SNP, for 17 samples we compared TaqMan genotypes to those obtained by direct sequencing. This set of samples included nine C/C homozygotes, seven C/G heterozygotes, and one G/G homozygote; again, there were no discrepancies.
Detection of Pseudo-SNPs
In the course of developing TaqMan assays for a genome-wide map of SNPs, we encountered three SNPs (WIAF1161, WIAF2566, and WIAF335) that were apparently not polymorphic—30 unrelated individuals tested were heterozygous (see Fig. 3, Genomic DNA). A trivial possibility is that the probes used in the TaqMan assay failed to distinguish between the two alleles. As can be seen in Figure 3 (Synthetic templates), this is not the case. Synthetic templates carrying one or the other allele were constructed by annealing the appropriate oligonucleotides and “filling in” the resulting partial duplexes. For all three SNPs, the two synthetic alleles can be distinguished from each other by the same probes and primers used to type genomic DNA. Furthermore, artificial heterozygotes made by mixing the two synthetic alleles can be easily distinguished from the two homozygotes.
Figure 3.

Pseudo-SNPs. (Left) Genomic DNA from 25–30 individuals was typed for the indicated SNPs. The axes are as in Figs. 1 and 2. Green dots are inferred to be heterozygotes; homozygotes are conspicuously absent. (Right) Synthetic templates bearing one (red dots) or the other allele (blue dots) were typed in duplicate using the same probes and primers used at left. Heterozygotes made by mixing equal amounts of homozygotes are shown in green.
We hypothesized, therefore, that duplications that differ from one another at a single nucleotide, and thus render all individuals heterozygous, had been misinterpreted as SNPs. If this were true, and the duplications were scattered across the genome, then the two alleles would be expected to segregate among cell lines used in radiation-hybrid mapping. If, on the other hand, the duplications were in close proximity, then they would be expected to “cosegregate” in the low-resolution mapping panel used here. The results presented in Table 2 show that this latter hypothesis appears to be correct. We typed the Genebridge 4 radiation-hybrid mapping panel for the presence or absence of these three pseudo-SNPs and for three others that were polymorphic. For all three pseudo-SNPs, all cell-lines tested harbor both copies of the duplication or neither. In contrast, for markers WIAF2065, 2042, 896, and 610 that are indeed polymorphic, the two alleles “segregate” among the radiation-hybrid cell lines—cell lines carry one or the other allele. In the cases of WIAF896, 610, and 2065 a handful of cell lines are “heterozygous” and bear both alleles.
Table 2.
Radiation Hybrid Analysis
| SNP | Allele 1, but not Allele 2, positivea | Allele 2, but not Allele 1, positivea | Allele 1 and Allele 2 positivea | Polymorphicb |
|---|---|---|---|---|
| WIAF2065 | 17 | 13 | 2 | yes |
| WIASF896 | 22 | 22 | 5 | yes |
| WIAF610 | 15 | 9 | 2 | yes |
| WIAF2042 | 11 | 15 | 0 | yes |
| WIAF1161 | 0 | 0 | 35 | no |
| WIAF2566 | 0 | 0 | 28 | no |
| WIAF335 | 0 | 0 | 38 | no |
DISCUSSION
High-Throughput Genotyping with SNPs
For large-scale association studies to become a reality, high-throughput genotyping methods that are accurate and flexible and use uniform conditions for typing different SNPs will be required. Several methods are currently available that offer the promise of accurate high-throughput genotyping. These include the TaqMan assay (Livak et al. 1995), oligonucleotide-ligation assays or OLAs (Tobe et al. 1996), minisequencing (Chen and Kwok 1997; Pastinen et al. 1997), molecular beacons (Tyagi et al. 1998), dye-labeled oligonucleotide ligation (Chen et al. 1998), chips (Hacia et al. 1998; Wang et al. 1998), mass spectrometry (Ross et al. 1998), and the invader assay (Mein et al. 2000). These methods, as presently employed, rely on a PCR step to increase the concentration of a segment of DNA sequence carrying the SNP; they diverge in their method of detecting alleles following this amplification step.
TaqMan and molecular beacons, because they incorporate allele-specific probes in the PCR, combine the amplification and detection steps and require no post-PCR processing for determining genotypes—for each reaction, fluorescence is merely measured after PCR and genotypes are inferred based on these values. The other methods, in contrast, require significant post-PCR processing. For instance, in the chip-based method used by Wang et al. (1998), amplified products are purified to remove nucleotides, enzymes, primers, etc. These purified samples are then hybridized for 15 h to oligonucleotides arrayed on chips; after several additional washing and developing steps, genotypes are determined. In some assays, such as the invader, after amplification, separate reactions are performed to distinguish the two alleles (Mein et al. 2000). These separate reactions could potentially lead to errors because if one reaction fails and the other succeeds, then a heterozygote could be misinterpreted as a homozygote. Although such artifacts can be controlled and many of the post-PCR steps automated, we believe that methods that do not require processing of amplified products are more suited to accurate and high-throughput genotyping. We have, therefore, focused our efforts on one such method—the 5′ nuclease allelic discrimination assay or TaqMan. Here we describe tools that make the method accurate, rapid, and flexible, and each of these points is considered below.
We estimate the error-rate to be <1 in 2000 genotypes. Two factors contributed to achieving this high level of accuracy: uniform buffer conditions and automated assignment of genotypes. We ensured uniform buffer conditions by using a single large batch of master-mix (i.e., buffer, nucleotides, polymerase, probes, and primers mixture) for typing all of the samples for a given SNP. We have found that small differences in buffer conditions, such as might result from errors pipetting into individual wells or plates, cause variation in post-PCR fluorescence values. It turned out to be the general case that applying the correction factor derived from the relative pre-PCR fluorescence of the several plates being compared (exemplified in Fig. 1A,B) deals well with this problem. Thereafter, we could pool data across different plates and thereby fully automate the procedure for assigning genotypes. Genotypes are assigned automatically using _k_-means clustering. This method of allele-calling eliminates human bias and allows one to assign a “quality score” to each genotype. This score is the probability that a particular sample falls within a genotype class given its fluorescence values for each reporter dye. If fluorescence values within a cluster are approximately normally distributed, then calculating this probability is straightforward—it is simply the probability of observing a certain value given a bivariate normal density (see Methods). As shown in Figures 1B and 2B, a few samples (three and two, respectively) have a low probability of belonging to the assigned cluster using this criterion. To our knowledge, this is the first time that _k_-means clustering coupled with a quality score has been used in assigning genotypes at SNPs.
The particular _k_-means clustering algorithm implemented here is not foolproof. Egregious outliers with very high or very low fluorescence values, which can result, for example, from contaminants in the DNA, defeat the clustering algorithm and result in classification that is obviously wrong. We note, however, that cursory visual examination of a plot of the data can usually identify these outliers.
The conditions for PCR used here—900 nM each primer, 250 nM each probe, and an annealing/extension temperature of 62°C—are, with minor modifications, generally applicable. One parameter, the annealing/extension temperature needs to be optimized for each new SNP. We generally test two temperatures, 62°C and 64°C, for a new SNP. With improvements in programs that calculate melting temperatures, we expect that even this step can be eliminated. Over 20 different SNPs (Table 1) have been used for large-scale genotyping under these conditions, and yield results that are similar to those presented here. These uniform conditions make the assay flexible and enable one to accommodate a new SNP easily. With five 96-well PCR machines, one fluorescent reader and no automated pipetting, >1000 genotypes can be generated by one person in one day. Thus, in our hands, once a new SNP is identified, a large-scale association study with 2000 samples can be performed in less than a week. With more PCR machines and automated pipetting stations, we expect that the throughput could be increased by at least an order of magnitude.
The genotyping routine described here is suitable for typing large numbers of individuals for selected SNPs in tens or even hundreds of candidate genes. However, there are two impediments to the use of TaqMan, as implemented here, for whole-genome association studies: the amount of DNA used in the PCR and the cost per genotype. If 30 ng of DNA are used for typing each SNP, then a genome scan with 300,000 SNPs will require a minimum of 9 mg of DNA, an amount far in excess of that available from typical blood samples obtained at clinics. We estimate that the cost of reagents per TaqMan genotype is ∼$1.50; at this price, a whole-genome scan for 2000 individuals with 300,000 SNPs will cost almost one billion US dollars. Clearly, the amount of DNA and the cost per genotype will need to be reduced significantly for whole-genome association studies to become practical.
For the TaqMan method we can envision at least two improvements. First, the PCR can be miniaturized, perhaps to nanoliter volumes (25μL reactions were used in this study), thereby decreasing the cost of reagents and conserving precious DNA samples. A thousand-fold reduction in volume would bring the assay into a scale that might be both feasible and affordable. Second, with improvements in TaqMan chemistry (e.g., probes with higher specificity) it should be possible to genotype pools of DNA, as opposed to individual samples as was done here, thus further conserving reagents and effort. An alternative method to determining allele frequencies in pools of DNA is to run the TaqMan assay in real time (Germer et al. 2000). However, for high-throughput genotyping, this approach would require a large number of expensive fluorescent readers. A limitation of the pooling method is that individual genotypes would not be provided, thus complicating analyses of haplotypes.
To conclude, we believe that the TaqMan assay is technically adequate for fully automated genotyping of SNPs on a scale required for genome-wide association studies, provided only that further miniaturization to a nanoliter scale is carried out. We have used the current implementation to genotype ∼1700 individuals for tens of markers and have found the method to be robust in daily use. To our knowledge, this is the first time that a method for typing SNPs has been assessed on such a large scale. Furthermore, the automated allele-calling procedure we have implemented should be generally applicable to any fluorescence-based genotyping method.
METHODS
Genotyping
The PrimerExpress program (Perkin-Elmer, Applied Biosystems Division) was used to design probes and primers. For the eNOSE289D SNP the approximate melting temperatures of probes and primers were 67°C and 61°C, respectively. For the CYP11B15 SNP, probes and primers were calculated to have melting temperatures of ∼70°C and 62°C, respectively. The sequences of the primers and probes used in this study are given in Table 3. Each 25 μL PCR contained 30 ng of genomic DNA, 900 nM primers, 250 nM probes, and 12.5 μL of TaqMan Universal PCR master mix (Perkin-Elmer, Applied Biosystems Division), which is a solution containing buffer, Uracil-N-glycosylase, deoxyribonucleotides, uridine, passive reference dye (ROX), and TaqGold DNA polymerase. We found that, for authentic SNPs, over 90% of the TaqMan assays we tested under these conditions give usable genotypes without further optimization of the assay. We have also found that as little as 100 nM probes can be used in the PCR reaction, with results that are comparable to those presented here. For consistently poolable results (see below), it was found necessary to type all subjects for a given SNP with a single lot of PCR master mix. Amplification was done under the following conditions: 50°C, 2 min; 95°C, 10 min; followed by 40 cycles of 94°C, 15 sec and 62°C, 1 min in a Perkin-Elmer 9600 thermocycler. Fluorescence in each well was measured before and after PCR using a ABI 7700 machine (Perkin Elmer, Applied Biosystems Division). Patient population has been described previously (Ranade et al. 2000).
Table 3.
Probes and Primers
| SNP | Probesa | Primersb |
|---|---|---|
| eNOSE298D | CCCCAGATGAGCCCCCAGAACT | CGGTCGCTTCGACGTGCT |
| CCCCAGATGATCCCCCAGAACTC | CCAGTCAATCCCTTTGGTGCT | |
| CYP11B15 | TCTCCCAGTACCCGCTCTGCCCA | TACACAGCCTCAACCTGGCC |
| TCTCCCAGTACCGGCTCTGCCCA | GGCCTGCATGTTTCCTGGA | |
| WIAF2065 | AACAAGCTTTCCACTCCCACTTCCCT | CAAGCATTTGTCTTAATTTACAGACATTAA |
| ACAAGCTTTCCGCTCCCACTTCC | GTGGATGAAGAGGTTGAGGTGATA | |
| WIAF896 | CCAAAATTCCGAGTCCCTTGGCT | TCTGCCTATAGCTTGGATATCTTAATCTCT |
| CAGCCAAAATTCCTAGTCCCTTGGCT | CTGAGAGGTTTTGGAATGACTTGAA | |
| WIAF610 | TCTGGGTATGTGACATGCCTGCTCC | ACTCTGAATTATGGCAGTAGGCAAA |
| TGGGTATGTGACGTGCCTGCTCC | CTTGAGAGAACTCATCATTTAAAAATGTGT | |
| WIAF2042 | TCTATCTCTAAGGGGAGTCTCAAAACCCCA | TGAGCACATTTCTTGGGTCTGTT |
| TCTATCTCTAAGGGCAGTCTCAAAACCCCA | ||
| WIAF1161 | CATTATGTGGACTGAACCGACTTTTCTAAAGC | ATGAGCATTAGCTACTTTTCAGAATTGA |
| CATTATGTGGACTGAATCGACTTTTCTAAAGCT | TGTTGCAAAAGGAAAGAAAAGCTT | |
| C | ||
| WIAF2566 | TGTGCATAGTATCCATTAGTTTTTCATGATCCA | ACTCAGAAGGGTCATGGGTGA |
| TGTGCATAGTATCCAGTAGTTTTTCATGATCCA | GCTTCTAGGGTATCCATCTCCCTAGTA | |
| WIAF335 | TGTTAACAATTTTGGGCAGCCGAACT | GGATGTTGCATAAATTCAGGTTCTTT |
| TGTTAACAATTTTTGGCAGCCGAACTC | ATGTGCGTTTGTAGACAGCATCA |
Statistical Analyses
Normalized fluorescence values (Rn), defined as the amount of fluorescence from each reporter dye divided by that from the reference dye, were imported into a statistical software package (SPSS version 6.1). To correct for pipetting errors, fluorescence values were measured prior to the PCR. In principle, prior to PCR all wells of all of the plates should have equal amounts of fluorescence from either reporter or reference dyes. In practice, however, because of variation in pipetting, these amounts tend to vary and eventually cause predictable variation in post-PCR fluorescence values. To account for these pre-PCR differences, mean fluorescence values prior to PCR for each reporter dye were calculated for each plate. If the mean value of a particular plate is significantly different from the others, as judged by a non-parametric Wilcoxon signed-ranks test, then post-PCR values for that particular plate are adjusted accordingly. For this study, only one plate of samples needed to be adjusted for the eNOSE298D SNP (see Fig. 1).
_k_-means clustering was used to classify data into four groups. In this method of partitioning the data, cases are assigned to a predetermined number of groups. In this case, the number of groups is the number of genotype classes—three—and a no DNA control group. Squared Euclidean distances were used in the clustering, and cluster centers were estimated iteratively from the data. Fifty iterations were permitted, but clustering was terminated after only four iterations because there was no change in cluster centers.
If one assumes that the distribution of fluorescence values within a cluster is approximately bivariate normal, then the conditional probability (Pi(x)) that a sample with particular Rn values (x) falls within a particular cluster or genotype class is given by the formula:
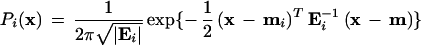
where Ei is the covariance matrix for the i th cluster, |Ei| is the determinant of the covariance matrix, mi is the mean vector for the i th cluster, and T denotes the transpose of the vector. If the normality assumption is invalid, then the distribution can be modeled as a mixture of more than one normal distribution, and the conditional probability calculated based on this new distribution. Bayesian posterior probabilities that take into account prior probabilities can also be calculated using this framework (Sharma 1996), probably with little added benefit, however.
Radiation Hybrid Analysis
The Genebridge 4 panel (Research Genetics) was used to analyze pseudo-SNPs. The primers and probes used to detect each SNP are listed in Table 3. PCR conditions were as described above, except that 25 ng of DNA from each radiation-hybrid cell line was used and PCR was done using an ABI 7700 machine. Following PCR, fluorescence values were read on the same machine and each cell line was scored for the presence or absence of signal from either reporter.
For SNPs WIAF1161, WIAF2566, and WIAF335, synthetic templates bearing one or the other allele were constructed as follows. The “top” oligonucleotide (∼40 nucleotides) was annealed to two “bottom” oligonucleotides (∼70 nucleotides), which differed from each other at the single polymorphic nucleotide. Annealing was carried out in a 100 μL volume containing 2 μM of each oligonucleotide, 10 mM Tris at pH 7.5, 6 mM MgCl2, 50 mM NaCl, and 5.76 mM β-mercaptoethanol. After denaturing the oligonucleotides at 95°C for 1 min, the solution was slowly cooled to room temperature. Five units of Klenow polymerase were added and the reaction was incubated at room temperature for 20 min. The reaction was stopped by adding EDTA to a final concentration 10 mM and heating to 70°C for 20 min. One or two μL of a 10−3 or 2 × 10−3 dilution of this solution was used in the TaqMan PCR.
Acknowledgments
We thank subjects for participating in this study. Ken Livak and Mike Lucero of Perkin-Elmer, Applied Biosytems Division, helped greatly with the TaqMan assays. This paper is written on behalf of members of the Stanford, Asia, and Pacific program for Hypertension and Insulin resistance (SAPPHIRe). We thank Susan Old, Steve Mockrin, and Cashell Jaquish of the National Heart, Lung, and Blood Instittute (NHLBI) for helpful discussions. This work is funded by a grant from the Family Blood Pressure Program of the NHLBI, National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Article published on-line before print: Genome Res., 10.1101/gr. 157801.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.157801.
REFERENCES
- Anderberg MR. Cluster analysis for applications. New York: Academic Press; 1973. [Google Scholar]
- Chen X, Kwok PY. Template-directed dye-terminator incorporation (TDI) assay: A homogeneous DNA diagnostic method based on fluorescence resonance energy transfer. Nucleic Acids Res. 1997;25:347–353. doi: 10.1093/nar/25.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chex X, Livak KJ, Kwok PY. A homogeneous ligase-mediated DNA diagnostic test. Genome Res. 1998;8:549–556. doi: 10.1101/gr.8.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Chakravarti A. Variations on a theme: Cataloguing human DNA sequence variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Hacia JG, Sun B, Hunt N, Edgermon K, Mosbrook D, Robbins C, Fodor SP, Tagle DA, Collins FS. Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res. 1998;8:1245–1258. doi: 10.1101/gr.8.12.1245. [DOI] [PubMed] [Google Scholar]
- Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nature Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- Germer S, Holland MJ, Higuchi R. High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res. 2000;10:258–266. doi: 10.1101/gr.10.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landegren U, Nilsson M, Kwok PY. Reading bits of genetics information: Methods for single-nucleotide polymorphism analysis. Genome Res. 1998;8:769–776. doi: 10.1101/gr.8.8.769. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Marmaro J, Todd JA. Towards fully automated genome-wide polymorphism screening. Nature Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- Lyamichev V, Mast AL, Hall JG, Prudent JR, Kaiser MW, Takova T, Kwiatkowski RW, Sander TJ, de Arruda M, Arco DA, et al. Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes. Nature Biotechnol. 1999;17:292–296. doi: 10.1038/7044. [DOI] [PubMed] [Google Scholar]
- Masood E. …As consortium plans free SNP map of human genome. Nature. 1999;398:545–546. doi: 10.1038/19126. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Forbes SL, Kramer LA, Liggett SB. Polymorphisms of the 5′ leader cistron of the human β2-adrenergic receptor regulate receptor expression. J Clin Invest. 1998;102:1927–1932. doi: 10.1172/JCI4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mein CA, Barratt BJ, Dunn MG, Siegmund T, Smith AN, Esposito L, Nutland S, Stevens HE, Wilson AJ, Phillips MS, et al. Evaluation of single nucleotide polymorphism typing with invader on PCR amplicons and its automation. Genome Res. 2000;10:330–343. doi: 10.1101/gr.10.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Saito Y, Kajiyama N, Yoshimura M, Shimasaki Y, Nakayama M, Kamitani S, Harada M, Ishikawa M, Kuwahara K, et al. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension. 1998;32:3–8. doi: 10.1161/01.hyp.32.1.3. [DOI] [PubMed] [Google Scholar]
- Pastinen T, Kurg A, Metspalu A, Peltonen L, Syvanen AC. Minisequencing: A specific tool for DNA analysis and diagnostics on oligonucleotide arrays. Genome Res. 1997;7:606–614. doi: 10.1101/gr.7.6.606. [DOI] [PubMed] [Google Scholar]
- Ranade K, Hsuing AC, Wu KD, Chang MS, Chen YT, Hebert J, Chen YI, Olshen R, Curb D, Dzau V, Botstein D, Cox D, Risch N. Lack of evidence for an association between alpha-adducin and blood-pressure regulation in Asian populations. Amer J Hyper. 2000;13:704–709. doi: 10.1016/s0895-7061(00)00238-7. [DOI] [PubMed] [Google Scholar]
- Reihsaus E, Innis M, MacIntyre N, Liggett SB. Mutations in the gene encoding for the b2– adrenergic receptor in normal and asthmatic subjects. Amer J Respir Cell Mol Biol. 1993;3:334–339. doi: 10.1165/ajrcmb/8.3.334. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;13:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Ross P, Hall L, Smirnov I, Haff L. High level multiplex genotyping by MALDI-TOF mass spectrometry. Nature Biotechnol. 1998;16:1347–1351. doi: 10.1038/4328. [DOI] [PubMed] [Google Scholar]
- Sharma S. Applied Multivariate Techniques. New York: John Wiley & Sons; 1996. [Google Scholar]
- Tobe VO, Taylor SL, Nickerson DA. Single-well genotyping of diallelic sequence variations by a two-color ELISA-based oligonucleotide ligation assay. Nucleic Acids Res. 1996;24:3728–3732. doi: 10.1093/nar/24.19.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination. Nature Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- Walston J, Silver K, Bogardus C, Knowler WC, Celi FS, Austin S, Manning B, Strosberg AD, Stern MP, Raben N, et al. Time of onset of non-insulin-dependent diabetes mellitus and genetic variation in the β3-adrenergic-receptor gene. N Engl J Med. 1995;333:343–347. doi: 10.1056/NEJM199508103330603. [DOI] [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao A, Berno CJ, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]