SGP-1: Prediction and Validation of Homologous Genes Based on Sequence Alignments (original) (raw)
Abstract
Conventional methods of gene prediction rely on the recognition of DNA-sequence signals, the coding potential or the comparison of a genomic sequence with a cDNA, EST, or protein database. Reasons for limited accuracy in many circumstances are species-specific training and the incompleteness of reference databases. Lately, comparative genome analysis has attracted increasing attention. Several analysis tools that are based on human/mouse comparisons are already available. Here, we present a program for the prediction of protein-coding genes, termed SGP-1 (Syntenic Gene Prediction), which is based on the similarity of homologous genomic sequences. In contrast to most existing tools, the accuracy of SGP-1 depends little on species-specific properties such as codon usage or the nucleotide distribution. SGP-1 may therefore be applied to nonstandard model organisms in vertebrates as well as in plants, without the need for extensive parameter training. In addition to predicting genes in large-scale genomic sequences, the program may be useful to validate gene structure annotations from databases. To this end, SGP-1 output also contains comparisons between predicted and annotated gene structures in HTML format. The program can be accessed via a Web server at http://soft.ice.mpg.de/sgp-1. The source code, written in ANSI C, is available on request from the authors.
Given homologous genomic sequences from two species, their local alignment usually shows a patchwork pattern of conserved and less conserved segments. Generally, coding sequences tend to be more conserved than noncoding sequences. Highly conserved fragments may sometimes also be attributed to gene regulatory (Hardison et al. 1997) or other DNA elements, such as clade-specific repeats. Although the level of sequence similarity depends strongly on the evolutionary distance of the compared species, recent studies (Roest Crollius et al. 2000; Wiehe et al. 2000; R. Guigó, L. Duret, and T. Wiehe, unpubl.) suggest that homology-based gene prediction can be very reliable over a fairly wide spectrum of species and evolutionary divergence times. Gene prediction has received considerable attention from computer scientists and biologists during the past decade (for reviews, see Burge and Karlin 1998; Claverie 1998). These efforts have led to considerable progress, but the problem is still far from a satisfactory solution (Guigó et al. 2000). Current methods can be roughly grouped into two main categories, ab initio and homology-based methods. The former methods recognize signals or compositional features in a single input sequence by pattern-matching, probabilistic, or statistical methods. An example is Genscan (Burge and Karlin 1997). The homology-based methods use external information such as comparison of the query sequence with protein, EST, or cDNA databases. Examples are BLASTX (Altschul et al. 1990) and the more sophisticated spliced alignment algorithms Procrustes (Gelfand et al. 1996) or GeneWise (www.sanger.ac.uk/Software/Wise2). Lately, gene prediction tools have become available that infer gene structures from alignments of anonymous genomic sequences and the resulting pattern of conserved segments. For instance, ExoFish (Roest Crollius et al. 2000) predicts human exons by comparison with a database of random sequences from Tetraodon nigroviridis. Bafna and Huson (2000) and Batzoglou et al. (2000) have developed programs for gene prediction by pairwise comparison of human and mouse homologous sequences. SGP-1 is a similar gene prediction tool, but it is not species specific. It is designed for large-scale genomic sequences, such as complete bacterial artificial chromosomes (BACs), of vertebrates and plants.
Today we are in a situation where analysis programs need to cope with low sequence quality of published draft genomes. Whereas classical tools are sensitive to sequence quality, similarity-based tools are much less so, because similarity levels are less affected by sequencing or assembly errors. Rather, such errors may even be rectified with the help of sequence comparison programs.
For gene prediction in SGP-1, two lines of reasoning are combined (Fig. 1). First, a pairwise local alignment is computed. This may be done either on a DNA level (e.g., with BLASTN or SIM96 [Huang and Miller 1991]) or on an amino acid level (e.g., with TBLASTX [Altschul et al. 1990]). The evolutionary distance of the compared sequences is an important criterion to choose between the methods. Generally, we obtained better results with DNA-based alignments for closely related sequences and with amino acid-based alignments for distantly related sequences (Wiehe et al. 2000). If computation speed is a main concern, a BLAST-like alignment is preferable over a dynamic programming algorithm such as SIM96. In any case, a postprocessing step to reduce noise may be applied to the resulting local alignment. Second, for both sequences we generate separate lists of potential exons, termed precandidates. A subroutine called filter retains only those precandidates that are compatible with the alignment. Subsequently, exons are rescored and then assembled into a gene model.
Figure 1.

Flowchart of SGP-1.
Because the two tasks, computation of the alignment and gene prediction, are separated from each other, SGP-1 can work with the (possibly reformatted) output of an arbitrary alignment program. Furthermore, run time of the program can be significantly reduced, if a precomputed alignment is provided as input.
We applied SGP-1 to several sets of homologous sequence pairs from vertebrates and from plants. Unlike ProGen (Novichkov et al. 2000) or ExoFish (Roest Crollius et al. 2000), SGP-1 emphasizes similarity and is therefore particularly suited for well-conserved genes or sufficiently closely related species. For instance, good results can be obtained for species that are evolutionarily at least as close as Homo sapiens and Gallus gallus (R. Guigó, L. Duret, and T. Wiehe, unpubl.). Finally, we show how SGP-1 may be useful for verifying gene structure annotations.
RESULTS
Algorithm
Gene prediction with SGP-1 proceeds in two separate steps, calculation of a pairwise alignment and processing of sequence and alignment files. This modularity makes the tool very fast and versatile: given a suitable format-conversion tool, SGP-1 may be combined with any pairwise alignment program. We successfully ran SGP-1 on alignments produced by SIM96 (Huang and Miller 1991), BLASTN, TBLASTX (Altschul et al. 1990), BLASTZ (W. Miller, pers. comm.), and MUMMER (Delcher et al. 1999). Given two sequences and their alignment as input, the program calls subroutines for (1) alignment postprocessing, (2) generating exon precandidates, (3) filtering, (4) rescoring, and (5) gene assembly and output. The subroutine with the highest time complexity is filter (see Methods). A very rough bound for its run time is given by O(nm), where n and m are the lengths of the input query sequences. This is due to the fact that the size of the two exon precandidate lists depends linearly on n and m, respectively, but pairs of precandidates, one from each list, have to be processed. For all other subroutines the time requirement is subquadratic. Memory space is dynamically allocated in all subroutines. An upper bound for the required memory space is also given by O(nm), because lists with pairs of exon precandidates have to be stored and handled. However, absolute running times and space requirements also depend on sequence properties such as the level of similarity. For instance, gene prediction with SGP-1 for two homologous sequences from the human and mouse HOX regions (176 kb and 214 kb, respectively) took 10.5 sec CPU time on a Linux PC (RedHat, distribution 7.0) with a 400 MHz Pentium II processor and 256 Mb RAM. Memory size was sufficient for the program to run without swapping. In detail, the time requirements for the individual subroutines (1) to (5) were 0.3 sec, 2.6 sec, 4.5 sec, 1.9 sec, and 1.2 sec, respectively. In contrast, calculation of the pairwise alignment with BLASTZ, a heuristic alignment method (W. Miller, pers. comm.), for these two sequences took 89 sec. To take advantage of the modularity, the Web server provides the possibility of uploading precomputed alignment files.
Evaluation of Test Sets
To measure gene-prediction accuracy of SGP-1, we generated several test sets from human/rodent (S1, S2) and plant (T1) homologous sequences. S1 is the set originally used by Batzoglou et al. (2000) as a test set for Rosetta. S2 is a set of large homologous chromosomal fragments that contains multiple genes in both species. Accuracy is measured in terms of sensitivity and specificity (Burset and Guigó 1996; see Methods). The results for SGP-1 on set S1 of single genes are comparable to those of Genscan and Rosetta (Table 1), Genscan being slightly inferior and Rosetta being slightly superior to SGP-1 on nucleotide level accuracy. Data set S1 contains several exons with nonstandard splice sites, which are not detected in the current version of SGP-1. This explains the low sensitivity on exon level (SN) of SGP-1 compared with Rosetta in set S1. Similarity-based programs tend to be more accurate than conventional methods for large-scale sequences with multiple genes (Guigó et al. 2000; Wiehe et al. 2000). This property was also found when we evaluated test set S2. Because the divergence between two species may considerably vary along their genomes (Fig. 2), measures have to be taken to cope with different levels of conservation. For less conserved sequences, SGP-1 performs better if it is based on an amino acid alignment; for highly conserved sequences it performs better if it is based on a DNA alignment. More generally, this pattern was found both for vertebrate and for plant sequences. On set T1, the performance of SGP-1 on nucleotide level was better when based on a DNA alignment; on exon level the performance was better when based on an amino acid alignment (Table 1). In both cases, SGP-1 performed better than Genscan, which was in particular due to a higher specificity.
Table 1.
Evaluation of Gene Prediction Accuracy
| Test set | Nucleotide level | Exon level | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn | Sp | AC | SN | SP | (SN + SP)/2 | ME | WE | |
| Set S1 (human/rodent single genes) | ||||||||
| SGP SIM1 | 0.94 | 0.96 | 0.94 | 0.70 | 0.76 | 0.73 | 0.12 | 0.04 |
| SGP TBLASTX1 | 0.87 | 0.96 | 0.90 | 0.64 | 0.73 | 0.68 | 0.15 | 0.03 |
| Rosetta2 | 0.95 | 0.97 | 0.96 | 0.84 | 0.84 | 0.84 | 0.04 | 0.05 |
| Genscan | 0.97 | 0.89 | 0.92 | 0.79 | 0.74 | 0.76 | 0.06 | 0.13 |
| Set S2 (human/rodent multiple genes) | ||||||||
| ERCC2 | ||||||||
| SGP SIM | 0.91 | 0.96 | 0.93 | 0.72 | 0.80 | 0.76 | 0.15 | 0.06 |
| SGP TBLASTX | 0.89 | 0.97 | 0.92 | 0.69 | 0.84 | 0.77 | 0.18 | 0.00 |
| Genscan | 0.99 | 0.71 | 0.83 | 0.79 | 0.70 | 0.74 | 0.05 | 0.16 |
| MHC | ||||||||
| SGP SIM | 0.79 | 0.99 | 0.88 | 0.60 | 0.80 | 0.70 | 0.28 | 0.02 |
| SGP TBLASTX | 0.67 | 1.00 | 0.83 | 0.45 | 0.69 | 0.57 | 0.38 | 0.00 |
| Genscan | 0.93 | 0.73 | 0.82 | 0.72 | 0.58 | 0.65 | 0.14 | 0.31 |
| HOX | ||||||||
| SGP SIM | 0.86 | 0.86 | 0.85 | 0.62 | 0.50 | 0.56 | 0.10 | 0.23 |
| SGP TBLASTX | 0.51 | 0.91 | 0.69 | 0.24 | 0.33 | 0.29 | 0.33 | 0.07 |
| Genscan | 0.94 | 0.77 | 0.85 | 0.67 | 0.30 | 0.44 | 0.05 | 0.47 |
| MeCP2 | ||||||||
| SGP SIM | 0.99 | 0.93 | 0.96 | 0.92 | 0.89 | 0.91 | 0.08 | 0.11 |
| SGP TBLASTX | 0.72 | 0.85 | 0.78 | 0.46 | 0.50 | 0.48 | 0.19 | 0.21 |
| Genscan | 0.97 | 0.73 | 0.83 | 0.80 | 0.73 | 0.76 | 0.07 | 0.21 |
| Set T1 (plant single genes) | ||||||||
| SGP SIM | 0.93 | 0.99 | 0.94 | 0.57 | 0.63 | 0.60 | 0.12 | 0.00 |
| SGP TBLASTX | 0.88 | 0.97 | 0.91 | 0.64 | 0.76 | 0.70 | 0.18 | 0.00 |
| Genscan | 0.92 | 0.90 | 0.87 | 0.61 | 0.57 | 0.59 | 0.09 | 0.15 |
Figure 2.

Sequence similarity between three homologous human/mouse genomic regions, which diverged at different evolutionary rates. (a) ERCC2 locus (Accession nos. L47234; L47235). (b) MHC-II region (Accession nos. X87344; AF100956, AF027865). (c) HOX cluster (Accession nos. AC009336; L1084). Abscissa: sequence position along the human sequence. Ordinate: sequence position along the mouse sequence (upper panels) and identity of locally aligned fragments (middle panels). For better illustration, the lower panels show a sliding-window-plot of the identity. The position of exons is indicated by grey vertical lines. Note that the HOX cluster is highly conserved also in intronic and intergenic parts of the sequence.
When comparing duplicated regions within a single species, the same dependence of prediction accuracy on conservation levels is observed. Genome duplication is particularly common among plants. For example, the most recent large-scale duplication events in Arabidopsis thaliana are estimated to have occurred between 50 and 100 Myr B.P. (Vision et al. 2000). In such cases, similarity-based gene prediction can be useful to detect genes even if a homologous sequence of a second species is not available. For an example, we tested a pair of duplicated segments residing on chromosomes 3 and 5 in Arabidopsis thaliana. Sensitivity and specificity results are Sn = 0.87 and Sp = 0.84 (nucleotide level), and SN = 0.62 and SP = 0.65 (exon level), respectively. Comparing a pair of BACs from Oryza sativa and Zea mays that contain orthologous genes for AdHI (rice and maize) and the paralogous AdHII gene (rice), SGP-1 detects these three genes with Sn = 0.94 and Sp = 0.98 on nucleotide level, and SN = 0.65 and SP = 0.72 on exon level. Finally, we also applied—without specific training—SGP-1 to the complete chloroplast genomes of Oryza sativa and Zea mays. Sensitivity and specificity on nucleotide level are both 0.80. For comparison, Genscan with standard settings for nuclear genes in Zea mays yielded sensitivity and specificity of 0.01 and 0.38, respectively, for the chloroplast genome of Zea mays. Not surprisingly, both programs do very poorly in terms of exon level accuracy (0.01). However, it would be a relatively simple task to provide SGP-1 with a splice-site profile that is adequate for organellar instead of nuclear genes and to enhance the exon level accuracy.
Codon Bias Versus Splice-Site Conservation
Codon bias can vary to a large extent among species within the same taxonomic group, and even among genes within the same species (Sharp et al. 1995). On the other hand, the average splice-site profile, that is, the nucleotide distribution around splice sites, appears to be more conserved. In particular, this is true for human/rodent comparisons. Codon bias is on average much lower in mice and rats than in humans, which is perhaps due to the different rates of neutral evolution in the two lineages: an acceleration in the rodent lineage and a slow-down in the primate lineage (Britten 1986, Li et al. 1987). We calculated codon bias (Peden 1997) individually for the human and rodent genes in set (S1) of single human/rodent genes and then determined the difference in codon bias for each pair of homologous genes. Applying a two-tailed _t_-test (level α = 0.01) to the differences, we rejected the null hypothesis that the difference is zero (p = 4.5×10–6, Fig. 3).
Figure 3.

Distributions of score differences in data set S1. Shown is the distribution for the scores of acceptors (a) and donors (d) and for the codon bias (c). Codon bias was calculated with CodonW (Peden, 1997) separately for each pair of homologous coding sequences in set S1. To bring the numerical values for a,c, and d on the same scale, we divided the numbers obtained by the respective sample standard deviation σi, i = a,d,c. Based on a two-tailed Student's _t_-test, the hypothesis that the mean of the distribution is zero is not rejected for acceptors nor for donors. However, it is rejected for codon bias (P = 4.5×10–6).
Similarly, we calculated the difference of the scores of homologous human and rodent acceptor and donor sites. The distribution of both the acceptor and donor score differences is more symmetrical around zero than is the distribution of the codon bias difference (Fig. 3). In fact, the null hypothesis of the difference being zero cannot be rejected in a two-tailed _t_-test (level α = 0.05).
Application to Gene Structure Validation
Annotations of gene structures submitted to sequence databases such as GenBank (www.ncbi.nlm.nih.gov), EMBL (www.ebi.ac.uk), or DDBJ (www.ddbj.nig.ac.jp) can sometimes be erroneous. SGP-1 provides an option to compare a CDS (coding sequence) annotation with the gene prediction result. This feature may be helpful for cross-checking and validating annotations because discrepancies between the given annotation and the prediction are highlighted. To that end, annotated and predicted exons are written (in GFF [General Feature; http://www.sanger.ac.uk/Software/formats/GFF] format) into an HTML file that can be viewed with any Web browser. Such potential annotation errors may include sequencing errors, wrongly annotated start or stop codons, or wrongly annotated splice sites. Figure 4 shows a discrepancy between GenBank annotation and prediction in the mouse preproinsulin gene II (Accession Number X04724). Donor site of exon 1 and acceptor site of exon 2 are wrongly annotated. As a consequence, the inferred intron phase would differ from that of the homologous human intron.
Figure 4.

Example of potential annotation errors. Comparison of GenBank annotation (CDS field) and SGP-1 prediction for human and mouse insulin genes (accession nos. M10039 and X04724). From left to right, the fields are identifier (1), source (2), feature (3), sequence positions of beginning and end of a coding exon (4 and 5), score (6), strand (7), reading frame (8), grouping (9), and sequence (10). Discrepancies between annotation and prediction are marked by black circles around murine donor and acceptor positions and reading frame for exon 2. Capital letters indicate the coding sequence.
DISCUSSION
Genome analysis has entered a stage in which comparative methods play an increasingly important role, not only for computational gene finding but also for determining gene regulatory regions and delineating gene function. Various programs (Bafna and Huson 2000; Batzoglou et al. 2000; Roest Crollius et al. 2000; Novichkov 2000) have already been published or are under development. Here, we present a method that is based on DNA or amino acid pairwise alignments to predict coding regions and exon–intron structure of multiple genes, and to validate gene-structure annotations. One of the shortcomings of traditional gene prediction tools has been that they are extremely species specific and that their accuracy may drop dramatically when they are applied to species for which they have not been trained. In contrast, comparative gene prediction may rely exclusively or primarily on the pattern of conservation between a pair of species, exploiting the fact that functional (which here means amino acid coding) parts of the genome are generally more conserved than nonfunctional parts. Therefore, such programs should be more versatile and perform well across a wide spectrum of species, no matter whether bacterial, animal, or plant genomes are compared. In practice, however, there is probably no single tool that works equally well regardless of the evolutionary divergence between the compared sequences. Underprediction and overprediction, depending on the evolutionary distance, are common problems. Furthermore, prediction accuracy is sensitive not only to the choice of an appropriate species pair, but may also vary considerably along the genome within a particular species pair. SGP-1 is designed for comparative analysis in evolutionarily closely related species such as Homo sapiens and Mus musculus, Arabidopsis thaliana and Brassica oleracea, Caenorhabditis elegans and Caenorhabditis briggsae, or more closely related species. A central strategy of SGP-1 is to rely as little as possible on species-specific DNA characteristics, such as nucleotide composition, isochore distribution, codon bias, or repetitive elements. Therefore, the precandidate exons (see Methods) do not receive scores that depend on the coding potential or codon usage. Rather, scoring at the initial step relies exclusively on splice-site quality. Splice profiles are generally less variable within a taxonomic group than is codon usage. SGP-1 is an alignment-based method. Ideally, the alignment is computed with dynamic programming such as that implemented in SIM96, and which guarantees an optimal alignment to be found. Often, however, the time requirement is prohibitive for such a method to be applicable. The current Web-server version of SGP-1 provides alternative alignment options: BLASTN, TBLASTX, or the possibility to upload a precomputed alignment. SGP-1 relies on local rather than global alignments. It is well known (Doolittle 1990) that local alignments are more appropriate to identify short regions of similarity that may be embedded in regions of high dissimilarity—as is the case with coding regions embedded in large intragenic stretches. With a global alignment program, such short conserved stretches may only be detected if the gap penalties are extremely well adapted to the problem, which would pose a severe restriction on program versatility. The generally accepted strategy to individually anchor highly conserved, but possibly short, stretches is to produce a set of suboptimal local alignments rather than a single, global alignment. Furthermore, global alignments necessarily yield a colinear similarity pattern. Therefore, particularly in the absence of colinearity, two sequences may sensibly be compared only in terms of a local alignment. The currently distributed version of SGP-1 is designed for nuclear eukaryotic DNA sequences as input. A parameter file, which is easily accessible by the user and which describes splice profiles and/or genetic code, needs to be edited to treat nonnuclear DNA.
When comparing SGP-1 with other, not similarity based, gene finders, one of the most remarkable features is the generally much higher specificity. SGP-1 also performs well in large-scale genomic sequences. In particular, in problem zones, such as unusually large introns, SGP-1 may be superior to other gene-finding programs: the prediction of SGP-1 of the Human MeCP2 gene structure is correct around intron 2 (size 60 kb), whereas Genscan returns a number of false-positive results in this region. We compared SGP-1 with other similarity-based gene finders such as Rosetta and ProGen (see Table 1). ProGen uses an amino acid alignment rather than a DNA alignment. Its strength is in detecting more distant relationships, such as seen when, for example, Human and Fugu sequences are compared. Rosetta is primarily designed for human/rodent comparisons (Batzoglou et al. 2000). ExoFish (Roest Crollius et al. 2000) compares a human query sequence with a sequence database of the pufferfish T. nigroviridis; it is designed for gene prediction in humans, not in arbitrary species. Prediction accuracy of SGP-1 does not depend on the availability of ESTs or CDSs or the completeness of EST or CDS databases. Given two homologous genomic sequences, SGP-1 is expected to be superior to programs that rely on extrinsic information, and spliced alignment programs of the first generation, such as Procrustes (Gelfand et al. 1996), which were designed for a particular species. Even if homologous BACs of two or more species are not available, gene prediction by homology may still yield reliable results. We applied SGP-1 to a self-aligned 340-kb genomic BAC of Oryza sativa (Accession Number AF172282). The rice AdH region is known (Tarchini 2000) to have undergone several micro duplication events. In principle, duplicated genes may be identified by homology-based programs. Again, accuracy depends on the time when the duplication event occurred and on the speed of divergence. Comparing the rice AdH (AdHI and AdHII) genes with that in Arabidopsis thaliana and assuming that the split between Dicotyledons and Monocotyledons occurred about 100 Myr ago, we estimate the duplication event between AdHI and AdHII in rice at about 44 Myr B.P. SGP-1 correctly predicts the gene structures of AdHI and AdHII, except for the terminal exon. More generally, members of a gene family may be identified by comparing a single gene, or even only a CDS, with an entire chromosome or genome of the same or a related organism. The question of whether two genes are an orthologous or paralogous pair is per se irrelevant for gene identification by similarity. What matters, however, is the time and speed of their divergence. In addition to local duplications, extant organisms carry traces of a history of genome or chromosome duplications. This is particularly common in plants that may have undergone several rounds of genome duplication. This fact can be usefully applied to homology-based gene prediction by aligning two chromosomes of a single organism. For example, chromosome 3 and 5 of Arabidopsis thaliana contain syntenic regions over large parts of the chromosomes (Blanc et al. 2000; Vision et al. 2000). Applying SGP-1 to two 230-kb regions (Fig. 5) in the two chromosomes, related genes and gene families are identified. Clearly, unique genes will be missed by such an approach. Therefore, values of prediction accuracy, in particular sensitivity, for SGP-1, or any other homology-based program, are not very informative in such a region. This is aggravated in the preceding example by the fact that most of the annotated genes in this region are not experimentally confirmed but are only computer predicted.
Figure 5.

(Left panel) Alignment of two 230-kb regions on chromosomes 3 (abszissa) and 5 (ordinate) of Arabidopsis thaliana. (Right panel) CDS exon structure (filled boxes and grey bands) of a gene family with two copies on chromosome 5 and one copy on chromosome 3.
In the future, we will see an increasing need not only for computerized prediction of gene structures, but also of regulatory regions in particular, and for reliable statements about inferred gene function in the absence of experimental validation. Comparative genome analysis will undoubtedly play an important role in accomplishing these tasks.
METHODS
Algorithm
Sequence Alignment
SGP-1 requires a pairwise local alignment of two genomic sequences, such as produced by SIM96 (Huang and Miller 1991), MUMMER (Delcher et al. 1999), BLASTN, or TBLASTX (Altschul et al. 1990). Because alignment calculation is computationally intensive, this task can be skipped if a precomputed alignment is available. Thus, in addition to two sequence files, an alignment file (the admissible formats are described in soft. ice.mpg.de/sgp-1/man) can be uploaded to the Web server.
Given the alignment, it is postprocessed to select high-scoring segments. The user may choose among three options. The first generates a monotonic set of aligned segments such that all segments are disjoint and that the sum of their alignment scores is maximized (Myers and Miller 1995). This option is adequate if the two sequences are colinear in the sense that they are inversion-free and that gene order is preserved. The second option generates a set of disjoint but not necessarily monotonic segments. This option is adequate for sequences that are duplication free but that may contain inversions or translocations. The third option does nothing and hands all segments from the original alignment to the next subroutine. This option is adequate for sequences that contain duplications. Time complexity and space requirement to perform the first or second option are subquadratic and depend only on the number of aligned blocks given as input.
Generating Precandidates
Input DNA sequences are scanned for patterns such as start codons and stop codons and splice sites. The patterns are represented in a tree-like data structure, known as keyword tree (Aho and Corasick 1975; Gusfield 1997). Thus, the input sequence is scanned for multiple patterns in a single pass. Furthermore, it is easy to achieve a significant acceleration of the scanning process because at each position the cursor is maximally advanced (i.e., from one occurrence of a pattern to the next), and intermediate sequence positions are skipped whenever possible. The required computation time scales linearly with the length of the input sequence (Gusfield 1997). Each pattern has an associated likelihood profile (for an example, see http://soft.ice.mpg.de/sgp-1/example/profile)that is derived from the nucleotide distribution in a reference set. Hence, to each pattern hit a score can be assigned by evaluating the likelihood profile for the sequence segment under consideration. Evaluation at any given position takes a constant amount of time and does not increase time complexity. Scanning of an input sequence results in a list of so-called precandidate exons. A precandidate exon is a sequence stretch with a well-defined reading frame, without internal stop codon, which is delimited at each end by an admissible pattern: acceptor, donor, start codon, or stop codon. The score of a precandidate is the sum of the scores of both flanking patterns as given by their likelihood profiles. The profiles are based on reference sets of annotated and validated exons, extracted from GenBank (release 117). Currently, SGP-1 provides two sets of profiles, one for vertebrates and one for crucifers. When running the program, the user can select a profile via a command line switch. It should match the origin of the input sequence.
Because stop codons are distributed roughly uniformly along a DNA sequence, exon precandidates cannot become arbitrarily long. Therefore, the required memory space to store all precandidates scales linearly with the length of the input sequence.
Filtering
The subroutine filter checks whether begin and end positions of any pair of precandidates are contained in the postprocessed alignment. If there is a discrepancy, a pair is discarded. Optionally, the filter can be relaxed to allow for an offset between alignment and exon precandidate. There are two parameters: x, the number of base pairs by which locally aligned segments are extended, and d, the maximal distance (in bp) by which the ends of two paired precandidates may be separated (Fig. 6). The parameter values can be selected by the user via a command line switch. Computation time depends on parameter settings. For the general case one has as upper limit
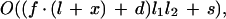
where _l_1 and _l_2 are the sizes of the precandidate lists, f is the number of aligned segments, l is their average length, and s is the total sequence length. An even coarser upper bound for time complexity is given by O(nm), where n and m are the input sequence lengths.
Figure 6.

Relaxed filtering of precandidates. (a) A blunt end, but complete coverage by the alignment. (b) A blunt end and partial coverage by the alignment. Setting parameters d and/or x to a value >0 retains precandidates with unaligned splice sites.
Rescoring
The output of filter consists of pairs of precandidates, where each one is uniquely characterized by its position, strand label (“+” or “−”) and reading frame. Hence, translation is unambiguous and for each pair of amino acid sequences a similarity score can be computed, for example, by a dynamic programming method similar to the Needleman and Wunsch (1970) algorithm. In addition to the splice-site score, each precandidate receives a second score: its similarity score. The score depends on the amino acid substitution matrix. The user may select from a list of matrices via a command line switch, with BLOSUM80 being the default. Time and space requirements for the module rescore are essentially linear in the number of pairs to be rescored. The final score attached to each candidate is a weighted combination of the splice-site quality and amino acid similarity. We found a ratio of amino acid to splice site weight of 4:1 to be optimal on our training set (see following). We use this value as default; other weights can be selected on the command line. The output of rescore consists of two lists of exon candidates, one list for each query sequence. Precandidates have now turned into candidates.
Gene Assembly
Assembly is performed independently for both species. Here, we use the method described by Guigó (1998). It is based on one-dimensional chaining and runs in linear time (O[_k_], k the number of candidate exons). The assembly program attempts to build complete gene models consisting of either a single exon or one initial exon, an arbitrary number of internal exons, and one terminal exon. Multiple genes, on either strand, may be assembled.
Evaluating Gene Prediction Accuracy
Prediction accuracy is measured by the quantities Sn (“sensitivity”) and Sp (“specificity”), as defined by Burset and Guigó (1996). On the level of nucleotides, sensitivity is
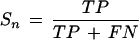
and specificity

where TP (true positives) is the number of coding nucleotides predicted as coding, FP (false positives) is the number of noncoding nucleotides incorrectly predicted as coding, and FN (false negatives) is the number of coding nucleotides that are predicted as noncoding. Similarly, on the level of complete exons one defines

and
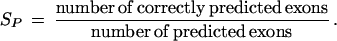
The quantity approximate correlation, AC, has been introduced to summarize sensitivity and specificity in a single measure. It is defined as
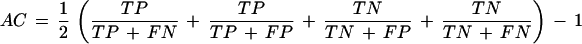
On exon level, the average (SN + SP)/2 is used instead. Furthermore, the quantities ME (number of exons that are missing in the prediction) and WE (number of incorrectly assigned exons) are recorded.
Output and Visualization of Results
The program returns an ASCII file with predicted genes in GFF format. The file also contains the amino acid sequence (in FASTA format) of the predicted proteins. Optionally, gene prediction results may be visualized via an annotated two-dimensional dotplot (Abril et al. 1999). The Web server contains a switch to produce such a graphical output (see Fig. 2). It is in PostScript or PDF format and can be saved to a local file. Furthermore, an HTML file can be generated. It contains a list of the DNA sequences of predicted and annotated exons. Special features, such as splice sites or start or stop codons, are highlighted along the sequence (see Fig. 4).
Test Sets
Gene prediction accuracy is evaluated on several test sets. A problem with available test sets is that they often contain only sequences with single genes. However, the analysis of megabase-sized draft sequences is a routine task in many laboratories, and gene finders need to perform well also with large-scale sequences that may include multiple genes on both strands.
Human/Rodent
A set (S1) of 116 homologous human/rodent single gene sequences was kindly provided by S. Batzoglou. A further human/rodent test set of 57 pairs was compiled by N. Jarborg and is available at www.sanger.ac.uk/Software/Alfresco/mmhs.shtml. Because the two sets are not disjoint, we generated a disjoint subset from the latter, comprising 39 homologous sequence pairs, which we then used as a training set to optimize program parameters. Set S2 consists of four homologous human/rodent pairs of partially unfinished BACs. They include the human and mouse MHC-II (accession numbers X87344; AF100956, AF027865), ERCC2 (accession numbers L47234; L47235) and MeCP2 regions (accession numbers AF030876, Z47046, Z47066; AF121351), and the HOX cluster (accession numbers AC009336; L1084). Parameter optimization was done manually on a low-dimensional discrete grid. Parameters to be tuned were c, the lower score cutoff for exon precandidates; d, the radius of fuzziness; x, the alignment extension (both in module filter); the weight w of splice site- versus similarity-score (module rescore); and s, a value by which the entire distribution of candidate scores is shifted (module assembly, Guigó 1999).
Plants
A set (T1) of 20 homologous nuclear gene pairs of Brassicaceae was obtained from U. Göbel (pers. comm.). In each pair, one species is Arabidopsis thaliana and the other is Brassica oleracea or Brassica napus. This set was generated by first searching SWISS-PROT (release 39.0) for the taxon name Brassicaceae. Sequences were then clustered into species. Each possible pair of sequences from different species was globally aligned (GAP in GCG package [1999]). Pairs with a minimum protein identity of 30% were considered further and their respective DNA entry was extracted from GenBank, release 117. If the GenBank entry contained the keyword “gene” or “complete cds” in the DEFINITION line, the pair was retained; otherwise it was discarded. The remaining entries were manually checked for complete annotation. We also analyzed (set T2) several BACs of Arabidopsis thaliana (AC002291, AC009465, AC009177; AC007123, AF007271), Oryza sativa (AF172282), and Zea mays (AF123535), which are known to contain several sets of duplicated genes. Finally, we applied the program to the complete chloroplast genomes of Oryza sativa (NC_001320) and Zea mays (NC_001666).
Implementation
The program is written in ANSI C. The source is available under the general GNU license agreement from the authors on request. Furthermore, a Web server is accessible at http://soft.ice.mpg.de/sgp-1. Memory requirement depends on the size of the sequences to be analyzed and on the chosen options. Running the program on a Linux PC with a single Pentium II processor, we found 256 Mbyte RAM generally to be sufficient for analyzing sequences in the range of up to 200 kb.
Acknowledgments
We thank two anonymous reviewers for valuable comments, and Bernhard Haubold, Webb Miller, and Matthias Platzer for stimulating discussions. We are grateful to Ulrike Göbel who helped to compile a set of homologous plant genes and to René Kiessig for support in CGI-programming. Laurent Duret provided unpublished material on orthologous vertebrate genes. This work has been supported by the Max-Planck Gesellschaft (Germany), by Proyecto del Plan Nacional de I+D, BIO98–0443-C02–01, Beca de Formación de Personal Investigador, FP95–3881 7943 from the Ministero de Educación y Ciencia (Spain), and by a TMR grant (ERBFMGECT950062) from the European Community.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL twiehe@ice.mpg.de; FAX 49-3641-64-3668.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.177401.
REFERENCES
- Abril J, Wiehe T, Guigó R. APLOT: 2D-Visualization of genome annotations. 1999. http://www1.imim.es/software/gfftools/APLOT.html http://www1.imim.es/software/gfftools/APLOT.html. [Google Scholar]
- Aho A, Corasick M. Efficient string matching: An aid to bibliographic search. Comm ACM. 1975;18:333–340. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bafna V, Huson DH. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. Menlo Park, CA: AAAI Press; 2000. The conserved exon method for gene finding; pp. 3–12. [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Meserov J, Berger B, Lander ES. Human and mouse gene structure comparative analysis and applications to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Barakat A, Guyot R, Cooke R, Delseny M. Extensive duplication and reshuffling in the Arabidopsis genome. Plant Cell. 2000;12:1093–1102. doi: 10.1105/tpc.12.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Rates of DNA sequence evolution differ between taxonomic groups. Science. 1986;231:1393–1398. doi: 10.1126/science.3082006. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Burset M, Guigó R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–357. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- Claverie JM. Computational methods for exon detection. Mol Biotechnol. 1998;10:27–48. doi: 10.1007/BF02745861. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. Alignment of whole genomes. Nucleic Acids Res. 1999;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF. Searching through sequence databases. In: Doolittle RF, editor. Methods in enzymology. Molecular evolution: Computer analysis of protein and nucleic acid sequences. Vol. 183. New York: Academic Press; 1990. pp. 99–110. [Google Scholar]
- GCG. The Wisconsin Package (Version 10). Madison, WI: Wisconsin Genetics Computer Group; 1999. [Google Scholar]
- Gelfand MS, Mironov AA, Pevzner PA. Gene recognition via spliced alignment. Proc Natl Acad Sci. 1996;93:9061–9066. doi: 10.1073/pnas.93.17.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigó R. Assembling genes from predicted exons in linear time with dynamic programming. J Comp Biol. 1998;5:681–702. doi: 10.1089/cmb.1998.5.681. [DOI] [PubMed] [Google Scholar]
- Guigó R. DNA composition, codon usage and exon prediction. In: Bishop M, editor. Genetic databases. New York: Academic Press; 1999. pp. 53–80. [Google Scholar]
- Guigó R, Agarwal P, Abril JF, Burset M, Fickett JW. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 2000;10:1631–1642. doi: 10.1101/gr.122800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield D. Algorithms on strings, trees, and sequences. Cambridge: Cambridge University Press; 1997. [Google Scholar]
- Hardison R, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;7:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
- Li W, Tanimura M, Sharp P. An evaluation of the molecular clock hypothesis using mammalian DNA sequences. J Mol Evol. 1987;25:330–342. doi: 10.1007/BF02603118. [DOI] [PubMed] [Google Scholar]
- Myers E, Miller W. Proceedings of the Sixth Annual ACM-SIAM Symposium. New York: ACM; 1995. Chaining multiple-alignment fragments in sub-quadratic time; pp. 38–47. [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequences of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Novichkov P, Gelfand MS, Mironov AA. Prediction of the exon-intron structure by comparison of genomic sequences. Mol Biol (Mosk) 2000;34:230–236. [PubMed] [Google Scholar]
- Peden J. CodonW. Correspondence Analysis of Codon Usage. 1997. www.molbiol.ox.ac.uk/cu www.molbiol.ox.ac.uk/cu. [Google Scholar]
- Roest Crollius H, Jaillon O, Bernot A, Dasilva C, Bouneau L, Fischer C, Fiyames C, Wincker P, Brottier P, Quetier F, et al. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat Genet. 2000;25:235–238. doi: 10.1038/76118. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Averof M, Lloyd AT, Matassi G, Peden JF. DNA sequence evolution: The sounds of silence. Philos Trans R Soc Lond B Biol Sci. 1995;349:241–247. doi: 10.1098/rstb.1995.0108. [DOI] [PubMed] [Google Scholar]
- Tarchini R, Biddle P, Wineland R, Tingey S, Rafalski A. The complete sequence of 340kb of DNA around the rice AdH1-AdH2 region reveals interrupted colinearity with maize chromosome 4. Plant Cell. 2000;12:381–391. doi: 10.1105/tpc.12.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science. 2000;290:2114–2117. doi: 10.1126/science.290.5499.2114. [DOI] [PubMed] [Google Scholar]
- Wiehe T, Guigó R, Miller W. Genome sequence comparisons: Hurdles in the fast lane to functional genomics. Briefings in Bioinformatics. 2000;1:381–388. doi: 10.1093/bib/1.4.381. [DOI] [PubMed] [Google Scholar]