Adaptor Protein Complex 4 Deficiency Causes Severe Autosomal-Recessive Intellectual Disability, Progressive Spastic Paraplegia, Shy Character, and Short Stature (original) (raw)
Abstract
Intellectual disability inherited in an autosomal-recessive fashion represents an important fraction of severe cognitive-dysfunction disorders. Yet, the extreme heterogeneity of these conditions markedly hampers gene identification. Here, we report on eight affected individuals who were from three consanguineous families and presented with severe intellectual disability, absent speech, shy character, stereotypic laughter, muscular hypotonia that progressed to spastic paraplegia, microcephaly, foot deformity, decreased muscle mass of the lower limbs, inability to walk, and growth retardation. Using a combination of autozygosity mapping and either Sanger sequencing of candidate genes or next-generation exome sequencing, we identified one mutation in each of three genes encoding adaptor protein complex 4 (AP4) subunits: a nonsense mutation in AP4S1 (NM_007077.3: c.124C>T, p.Arg42∗), a frameshift mutation in AP4B1 (NM_006594.2: c.487_488insTAT, p.Glu163_Ser739delinsVal), and a splice mutation in AP4E1 (NM_007347.3: c.542+1_542+4delGTAA, r.421_542del, p.Glu181Glyfs∗20). Adaptor protein complexes (AP1-4) are ubiquitously expressed, evolutionarily conserved heterotetrameric complexes that mediate different types of vesicle formation and the selection of cargo molecules for inclusion into these vesicles. Interestingly, two mutations affecting AP4M1 and AP4E1 have recently been found to cause cerebral palsy associated with severe intellectual disability. Combined with previous observations, these results support the hypothesis that AP4-complex-mediated trafficking plays a crucial role in brain development and functioning and demonstrate the existence of a clinically recognizable syndrome due to deficiency of the AP4 complex.
Main Text
With a worldwide prevalence of around 2%, early-onset cognitive impairment, commonly referred to as intellectual disability (ID), is the most frequent cause of severe disability and a leading socioeconomic healthcare problem in Western countries.1 For the last two decades, remarkable progress has been made in the elucidation of ID conditions. About 30% percent of severe ID cases have been ascribed to chromosomal imbalances.2 Defects in X-linked genes account for about 10% of male ID cases.3 Yet despite these recent advances, the cause of ID remains unexplained in the majority of cases, and this leaves families without accurate diagnosis or genetic counseling. In particular, very little is known about the autosomal-recessive forms of ID (ARID). The broad genetic heterogeneity of ARID precludes any possibility of pooling families, and the scarcity of large pedigrees suitable for linkage analyses have hitherto hampered identification of the genes responsible for most of these cases. This problem has been successfully circumvented by the use of autozygosity mapping in large consanguineous families; in nonspecific ARID ten genes have been identified so far.1,4–12 Nevertheless, mutations in each of these genes only account for one or very few families, suggesting that many genes remain to be identified.
Here, we present linkage analyses, mutation discovery, and clinical characterization of a recognizable ARID syndrome in eight affected individuals from three consanguineous families.
Family ID01 is a sibship of three affected and two healthy siblings born to healthy parents, who are second cousins of Israeli–Arab descent (Figure 1A and Table 1). Pregnancy and delivery were unremarkable in all three affected cases. At birth all three siblings presented with microcephaly and muscular hypotonia, which later developed to hypertonia. At the time of examination, a clinical assessment showed hyperreflexia, spastic paraplegia, and an inability to walk unaided. All affected individuals revealed a severe cognitive deficit, marked speech delay, and adaptive impairment. Furthermore, they presented with microcephaly, a high palate, mildly remarkable facial gestalt with a wide nasal bridge, short stature, hyperlaxity, genu recurvatum, pes planus, and a waddling gait. All three affected individuals had stereotypic laughter and markedly shy character. None of the patients had seizures, vision or hearing impairments, or any anomalies of inner organs (Table 1).
Figure 1.
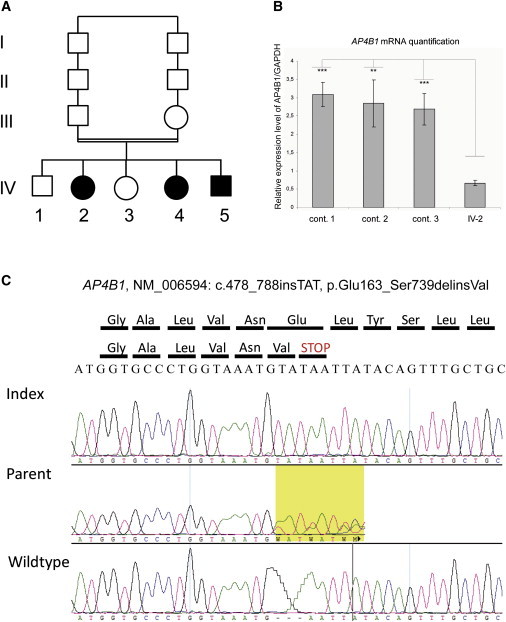
Genetic Analysis of Family ID01
(A) Pedigree of the family.
(B) Electrophoregrams illustrating the c.487_488insTAT, p.Glu163_Ser739delinsVal variant in exon 5 of AP4B1. Data are shown for homozygous affected individuals, heterozygous healthy parents, and homozygous wild-type healthy siblings.
(C) Quantitative RT-PCR analysis of AP4B1 mRNA. AP4B1 expression in fibroblast cells from three controls and from patient IV-2.
Data are normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Means ± standard deviation are given (n = 3 independent experiments). ∗∗∗p value of < 0.01 (Student's test) for the difference of expression. ∗∗p value of < 0.05 (Student's test) for the difference of expression.
Table 1.
Clinical Findings in Patients with Mutation in Distinct AP4 Subunits
| Family | This Report | Moreno-De-Luca et al.32 | Verkerk et al.31 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID01 | MR061 | MR071 | |||||||||||||
| Patient | IV-2 | IV-4 | IV-5 | V-14 | V-28 | VI-3 | III-5 | III-11 | IV-4 | IV-5 | IV-1 | IV-3 | IV-4 | IV-5 | IV-6 |
| AP4 subunit disrupted | B1 | B1 | B1 | S1 | S1 | S1 | E1 | E1 | E1 | E1 | M1 | M1 | M1 | M1 | M1 |
| Sex | F | F | M | M | F | M | M | F | F | M | F | M | F | M | M |
| Age at evaluation (years) | 23 | 15 | 11 | 22 | 20 | 18 | 11 | 6 | 23 | 22 | 24 | 23 | 22 | 1.5 | 21 |
| Severe ID | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Normal speech | + | − | − | − | − | − | − | − | − | − | − | − | − | NA | − |
| Stereotypic laughter | + | + | + | + | + | + | − | + | + | + | + | + | + | NA | + |
| Shy character | + | + | − | + | + | + | − | − | NA | NA | NA | NA | NA | NA | NA |
| Neonatal hypotonia | + | + | + | + | NA | + | + | + | + | + | + | + | + | + | + |
| Progressing to hypertonia | + | + | + | + | + | + | + | + | + | + | + | + | + | NA | + |
| Hyperreflexia | + | + | + | + | NA | + | NA | NA | + | + | + | + | + | NA | + |
| Babinski sign | + | − | + | NA | NA | NA | NA | NA | + | + | + | + | + | NA | + |
| Spasticity | + | + | + | + | − | + | + | + | + | + | + | + | + | NA | + |
| Drooling | − | − | + | + | + | + | − | − | + | + | + | + | + | NA | + |
| Walk independently (years) | 2.5 | 2.5 | 2.5 | 2 | 2 | 2.5 | − | − | − | − | − | − | − | − | − |
| Ambulation | wheelchair | + | wheelchair | − | − | − | crawling | crawling | − | − | − | − | − | NA | − |
| Foot deformity | − | − | − | + | + | + | + | + | NA | NA | − | + | − | − | − |
| Head circumference | −2 SD | −2.5 SD | −3 SD | −1 SD | −4 SD | −2 SD | −3 SD | −4 SD | −3 SD | −3 SD | −1 SD | 0 SD | −2 SD | NA | −2.5 SD |
| Height (cm) | ↓ | ↓ | ↓ | 145 | 130 | 140 | 125 | 105 | NA | NA | NA | NA | NA | NA | NA |
| Epilepsy | − | − | − | − | − | − | − | + | + | + | − | − | − | − | − |
| Sphincter control | − | − | − | + | + | + | − | − | − | − | − | − | − | NA | − |
| Eye evaluation | normal | normal | normal | normal | normal | amblyopia | normal | normal | normal | normal | NA | normal | NA | NA | POD |
| Hearing evaluation | normal | normal | normal | normal | normal | normal | normal | normal | normal | normal | NA | NA | NA | NA | NA |
| Overweight | + | + | normal | normal | normal | normal | normal | normal | NA | NA | NA | NA | NA | NA | NA |
Family MR061 is composed of three nuclear families from middle Syria; the families have one affected child each: the index, V-14, his nephew VI-3, and his cousin once removed V-28 (Figure 2). Pregnancy, delivery, and neonatal period were unremarkable. Affected individuals sat at age one and walked at age two to four but lost their ability to walk six to 24 months later. At the time of examination, clinical assessment showed muscular hypertonia, especially of lower limbs; contractures; talipes equinovarus; weak and decreased muscle mass of the shanks; and a clinical suspicion of peripheral neuropathy. All affected individuals revealed a severe cognitive deficit and absent speech and could only express basic needs (i.e., thirst, hunger, and strangury). Further, they presented with microcephaly (VI-3 and V-28); mild facial dysmorphisms, including a prominent and bulbous nose, a wide mouth, and coarse features; short stature; and mild spasticity in flection of upper limbs, which could be used only for simple tasks (e.g., holding a bottle of water). Similar to the former family, all affected individuals were markedly shy, amicable, and calm and kept smiling or laughing for no obvious reason but did not have laughter bursts. None of the patients had seizures, a hearing impairment, or any anomalies of inner organs (Table 1). The nephew of the index, VI-3, has an unclassified vision impairment. In addition, V-9 and VI-7 presented with moderate intellectual disability, could walk and speak, and did not have growth retardation and thus presented clinically with a different form of disability.
Figure 2.
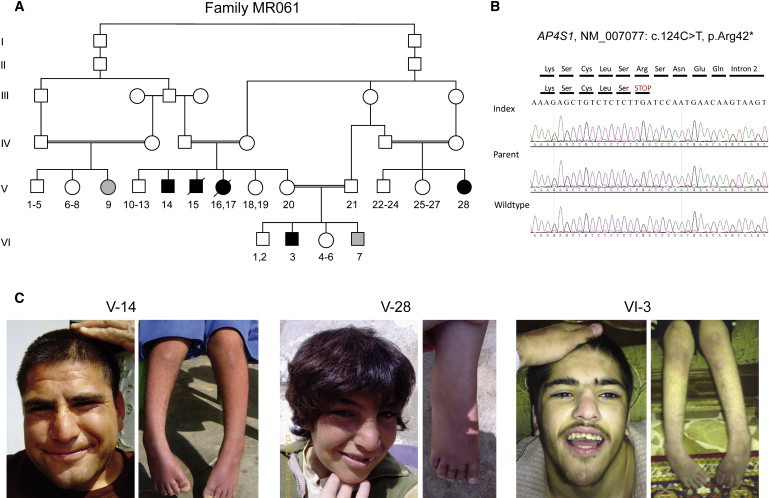
Genetic Analysis of Family MR061
(A) Pedigree of family; arrows indicate index. Family MR061 is large with multiple affected individuals with variable phenotypes. Grey symbols denote individuals in whom clinical presentation is markedly different and who have no AP4S1 mutation (heterogeneity within the family).
(B) Electropherograms illustrating the mutation in exon 2 of AP4S1.
(C) Facial appearance of affected individuals with discreet remarkable facial gestalt, including a prominent and bulbous nose, a wide mouth, and coarse features and photographs of lower limbs with foot deformity and decreased muscle mass of the shanks.
Family MR071 is composed of two nuclear families who are from the north of Syria and had one affected child each: the index, III-5, and his cousin, III-11 (Figures 3A and 3B). The pregnancy and birth were unremarkable in both cases. The parents reported muscular hypotonia in the neonatal period; this later developed to muscular hypertonia, especially of the lower limbs. At the time of examination, clinical assessment revealed contractures; talipes equinovarus; and decreased muscle mass of the shanks; together these findings resembled peripheral neuropathy. All affected individuals presented with a severe cognitive deficit and absent speech. Furthermore, they had microcephaly; short stature; and a mildly remarkable facial gestalt that included a prominent and bulbous nose, a wide mouth, and coarse features. Individual III-11 had epilepsy. Both patients showed a shy, amicable, and calm character. They smiled or laughed for no obvious reason but did not have laughter bursts. None of the patients had vision or hearing impairments or any anomalies of inner organs (Table 1). In this family one other cousin, III-1, presented clinically with a different form of disability. She had mild ID, could walk and speak, and did not have growth retardation.
Figure 3.
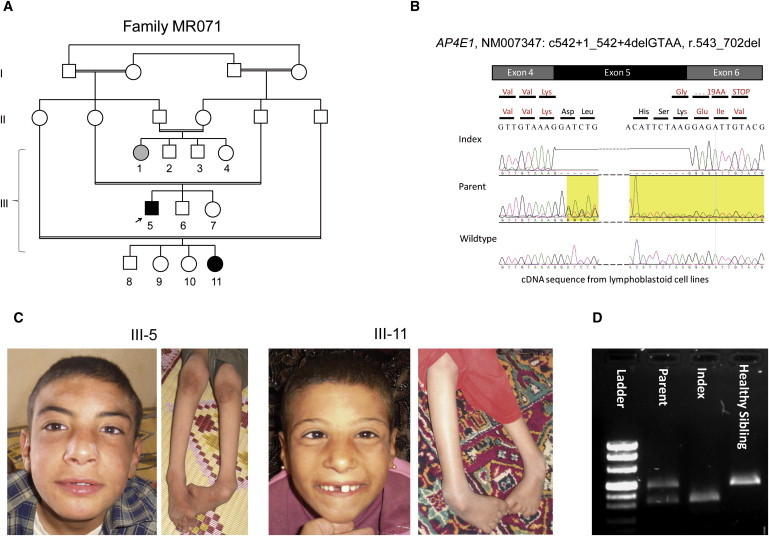
Genetic Analysis of Family MR071
(A) Pedigree of family MR071.
(B) Representative sequence traces from cDNA showing skipping of exon 5.
(C) Facial appearance of affected individuals includes discreet remarkable facial gestalt with prominent and bulbous nose, wide mouth, and coarse features. Also shown are photographs of the lower limbs with foot deformity and decreased muscle mass of the shanks.
(D) RT-PCR products of mRNA from homozygous affected individuals, heterozygous healthy parents, and homozygous wild-type healthy siblings; the expected size from the normal AP4E1 allele (512 bp) as well as a smaller band corresponding to aberrant splicing of the mutated allele with skipping of exon 5 (389 bp) is shown.
Chromosome analysis and metabolic screening (including plasma amino acid, lactate, carnitine and urinary oligosaccharides, mucopolysaccharidoses, and organic acids) as well as biochemical screening for GM1 Gangliosidosis, Tay Sachs, and Krabbe diseases were all normal. No brain magnetic resonance imaging was available for any of the patients.
All procedures followed in this study were approved by the local ethical committee at the contributing Universities and proper informed consent was obtained. Additional consent was obtained from parents of affected persons whose photographs are presented in this work. Blood samples were collected from all affected and most unaffected siblings and parents. Blood lymphoblasts (families MR061 and MR071) and skin fibroblasts (patient IV-2 of family ID01) were cultured. Genomic DNA was extracted by standard methods and analyzed with the Affymetrix GeneChip Mapping 250K array (family ID01) or 6.0 array (families MR061 and MR071) (Affymetrix, Santa Clara, CA, USA). Analysis did not reveal pathogenic deletions or duplications. Mendelian segregation was calculated with PedCheck software and was confirmed in all instances.13–15 We performed linkage analysis with ALLEGRO under an autosomal-recessive mode of inheritance with 99% penetrance and a disease allele frequency of 0.001 by using the EasyLinkage interface software.14–16 Multipoint linkage analysis resulted in significant linkage peaks at 1p13.2-q21.2 for family ID01, at 14q11-q12 for MR061,17 and at 15q21.1-q25.1 for MR071. Further genotype and haplotype analyses confirmed homozygosity by descent and defined critical intervals of 34 Mb, 9.1 Mb, and 32.9 Mb, respectively (Table S1, available online). Individuals from families MR061 and MR071, who presented with a clinically distinct form of ID, neither showed linkage to the above-mentioned regions nor, in the case of individuals V-9 and VI-7, showed a common locus. This means that in families MR061 and MR071 three and two different ID disorders exist, respectively.
After prioritizing genes according to their expression in the brain and their putative role in the central nervous system, we screened eight genes mapping to 1p13.1-q21.2 in family ID01 by using direct sequencing of all coding exons and exon-intron junctions. We identified a 3 bp homozygous insertion (NM_006594.2: c.487_488insTAT) within exon 5 of AP4B1 (encoding the β subunit of the adaptor complex 4 [MIM 607245]) (Figure 1B). This variant cosegregated with the disease and was not present in NCBI dbSNP (build 131) or detected in any of 796 control chromosomes, including 160 chromosomes from individuals of Israeli-Arab origin. This frameshift mutation (p.Glu163_Ser739delinsVal) was predicted to cause premature termination of translation. Quantitative real-time PCR with the ΔCT method showed significantly less AP4B1 transcript in patient skin fibroblasts than in three controls (p = 0.0084–0.013 with Student's test, Figure 1C). These results suggest that the primary effect of this frameshift mutation is nonsense-mediated decay.
DNA from individual V-14 of family MR061 was enriched with the SureSelect Human All Exon Kit, which targets approximately 38 Mb, that is 1.22%, of the human genome (Agilent, Santa Clara, Ca, USA). Sequencing was carried out on an Illumina Genome Analyzer IIx (Illumina, San Diego, CA, USA) as 54 bp or 76 bp paired-end runs. Image analysis and base calling were performed with the Genome Analyzer Pipeline version 1.5 with default parameters. We performed read alignment with BWA (v 0.5.8) by using the default parameters18 with the human genome assembly hg19 (GRCh37) as reference. Single-nucleotide variants and small insertions and deletions (indels) were detected with SAMtools (v 0.1.7).19 Variant annotation was performed with custom Perl scripts integrating data from dbSNP (v131) and the UCSC Genome Browser knownGene track. Additionally, we compared variants to 80 sequenced exomes in an in-house database to identify further common variants that are not present in dbSNP. We captured 26,037 variants; 6,655 variants were coding and homozygous, 345 of those were not annotated, and 139 of those were nonsynonymous. Only two variants were located in the linked region on chromosome 14: a missense variant in the last exon of SLC22A17 (NM_020372.2: c.1429G>A, p.Val477Met) and a nonsense variant in the first coding exon of AP4S1 (NM_007077.3: c.124C>T, p.Arg42∗). In silico analysis with MutationTaster20 and PolyPhen21 indicated a high probability for a pathogenic effect for both variants. The variants were also absent in 740 Syrian control chromosomes and cosegregated with the affected status within the family. Both SLC22A17 (highly expressed in the brain and belonging to the organic cation transporter family [MIM 611461]) and AP4S1 (encoding the small subunit of the adaptor complex 4, MIM [607243]) are thus good a priori candidates for ID. Because the clinical presentation of affected persons with mutations in the different AP4 subunits shows high similarity (Table 1), we assume that the AP4S1 mutation, which truncates the protein at its very beginning, is the main determinant of the phenotype in this family, although we cannot exclude an additional effect of the SLC22A17 or other variant in the linked region.
Finally, we tested homozygosity at the four loci coding for the subunits B1, E1, S1, and M1 of the AP4 complex by genotyping cohorts of 22 French and 62 Syrian17 ARID families with Affymetrix GeneChip Mapping 6.0, Affymetrix GeneChip 250K (Affymetrix, Santa Clara, CA, USA) or Illumina 610K arrays (Illumina, San Diego, CA, USA). One Syrian family (MR071) showed linkage to chromosome 15 containing AP4E1. Direct sequencing of this gene (encoding the ɛ subunit of the adaptor complex 4 [MIM 607244]) identified a homozygous splice-donor site mutation in intron 5 (NM_007347.3, c.542+1_542+4delGTAA) (Figure 3). This variant cosegregated with the phenotype, was not present in dbSNP build 131, and was absent in 740 control chromosomes from healthy individuals of Syrian descent. This mutation was predicted to abolish the intron 5 splice-donor site. Consistently, analysis of AP4E1 transcript from patient lymphoblastoid cell lines revealed skipping of exon 5 (Figure 3). This deletion was predicted to cause a frameshift and a premature termination of translation in exon 6 (NM_007347.3: r.421_542del, p.Glu181Glyfs∗20). In an additional Syrian family, an individual who had a homozygous 11 bp deletion in intron 11 of AP4E1 (c.1346+44_1346+54delGCAGTGACTTT) was identified. In silico analysis indicated that this deletion is not pathogenic and it was present in 20% of 320 control chromosomes from healthy individuals of Syrian descent. No additional, not annotated variants were identified.
Adaptor protein complexes (AP1, AP2, AP3, and AP4) play a key role in signal-mediated trafficking of integral membrane proteins. They mediate different types of vesicle formation and the selection of cargo molecules for inclusion into these vesicles. These evolutionarily conserved heterotetrameric complexes share a common structural organization and are composed of four subunits: two large subunits or adaptins (∼100 kD; α-ɛ/β1-4), one medium (∼50 kD; μ1-4), and one small subunit (∼17 kD; σ1-4).22 AP1-3 complexes are widely distributed among eukaryotes from yeast to humans. By contrast, the AP4 complex is absent in organisms such as Saccharomyces cerevisiae, Caenorhabditis elegans, and Drosophila melanogaster.23 Moreover, although AP1, AP2, and AP3 complexes have been shown to be associated with clathrin vesicles, the AP4 complex functions independently of clathrin and is preferentially linked to membranes regulated by the small GTPase ARF, another coat-vesicle protein.24–26
As shown in Figure S1, expression analysis of _AP4B1, AP4E1,_and AP4S1 transcript levels by quantitative RT-PCR in various adult and fetal tissues revealed ubiquitous expression in all fetal and adult brain structures tested. It has been suggested that the AP4 complex is involved in various sorting processes. In HeLa cells, the μ4 subunit interacts with the cytoplasmic tyrosine motif of lysosomal cargo proteins such as the lysosome-associated membrane protein 2 and mediates their direct transport to lysosomes without passing via the plasma membrane. In Madin-Darby canine kidney cells (MDCK), the μ4 subunit interacts with different cargo proteins destined for the basolateral membrane. In the brain, the AP4 complex has been involved in the transport of amyloid precursor protein (APP) from the trans-Golgi network to early endosomes.27 Ap4b1-deficient mice were fertile, exhibited no anatomical brain abnormalities, and had normal life spans, body weight, and grip power. They exhibited no ataxia but a significantly poorer rotorod performance than wild-type mice did. There was no information about learning ability of the Ap4b1-deficient mice. Analysis of those mice demonstrated that Ap4 mediates the trans-Golgi network to the postsynaptic somatodendritic domain transport of d2 and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors in both cerebellar Purkinje cells and hippocampal neurons.28 We thus propose that motor disturbances observed in patients with mutations in the AP4 complex might be because of cerebellar dysfunction caused by mislocalization of glutamate receptors. Similarly, defective AMPA receptor sorting might impact synaptic plasticity in hippocampal neurons and cause ID.
Mutations in other AP complexes have already been linked to human disorders. Mutations of AP1S2 (encoding the σ small subunit of the adaptor complex 1 [MIM 300629]) cause an X-linked intellectual disability syndrome associating muscular hypotonia, delayed walking, speech delay, aggressive behavior, brain calcifications, and elevated cerebrospinal fluid protein levels (MIM 300630).29,30 Mutations in AP3B1 (encoding the β subunit of the adaptor complex 3 [MIM 603401]) cause Hermansky-Pudlak syndrome type 2 (HPS [MIM 608233]), a disease characterized by hypopigmentation of the eyes and skin, prolonged bleeding, and lysosomal ceroid storage.27
Interestingly, homozygosity for a splice-donor site mutation in AP4M1 (encoding the μ subunit of the AP4 complex [MIM 602296]) was found to be associated with an autosomal-recessive spastic tetraplegia with intellectual disability and white matter anomalies (MIM 612936).31 Moreover, a 192-kb-long deletion encompassing SPPL2A (MIM 608238) and AP4E1 (MIM 607244) was also found to be associated with autosomal-recessive cerebral palsy, microcephaly, and intellectual disability (no MIM number has been assigned).32 Patients carrying mutations in AP4M1 and AP4E1 share many clinical features with those patients described in our study (Table 1). Primarily, all patients had an infantile muscular hypotonia that progressed to spasticity, hypertonia, and paralysis and became unable to walk. They presented with a severe ID, absent or markedly delayed speech, stereotypic laughter, and growth retardation. In addition to those common symptoms, several other features were observed in some of the patients: microcephaly, epilepsy, waddling gait, joint hyperlaxity (in patients with AP4B1 mutation), and feet deformity (Table 1). Yet, despite this phenotypic variability, our data suggest that disruption of any of the four AP4 subunits causes a clinically recognizable AP4-deficiency syndrome. This was also supported by the fact that three other individuals in families MR061 and MR071 have a clinical phenotype of ID different from the AP4 syndrome and consistently do not carry the familial mutations homozygously. Heterozygous carriers of the mutations have no ID.
In conclusion, our findings illustrate the power of combining systematic autozygosity mapping with large-scale sequencing for unraveling the molecular bases of autosomal-recessive ID. More importantly, they suggest the existence of a clinically recognizable AP4-deficiency syndrome characterized by the association of severe ID, growth retardation, stereotypic laughter, progressive spasticity, and inability to walk. Finally, this study provides further support to the hypothesis of a crucial role of AP4-mediated trafficking in brain development and functioning.
Acknowledgments
We are grateful to the families for their participation in the study. We also thank Angelika Diem, Petra Rothe, Bianca Schmick, and Anna Benet-Pagès for the excellent technical support. This study was supported by the Centre National de la Recherche Scientifique (CNRS), the Agence Nationale de la Recherche (ANR-08-MNP-010), the Ministère de la Recherche et de l'Enseignement Supérieur, the German Intellectual disability Network (MRNET) through a grant from the German Ministry of Research and Education to A. Reis and T. Strom (01GS08160 and 01GR0804-4), the European Commission 7th Framework Program, Project N. 261123, GEUVADIS, and by the Deutsche Forschungsgemeinschaft (DFG) (AB393/1-2).
Contributor Information
Rami Abou Jamra, Email: rami.aboujamra@uk-erlangen.de.
Laurence Colleaux, Email: laurence.colleaux@inserm.fr.
Supplemental Data
Document S1. One Figure and One Table
Web Resources
The URLs for data presented herein are as follows:
- Ensembl Genome Browser, http://www.ensembl.org/
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
- National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
- Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 2.Rauch A., Hoyer J., Guth S., Zweier C., Kraus C., Becker C., Zenker M., Hüffmeier U., Thiel C., Rüschendorf F. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 3.Ropers H.H., Hamel B.C. X-linked mental retardation. Nat. Rev. Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 4.Çalışkan M., Chong J.X., Uricchio L., Anderson R., Chen P., Sougnez C., Garimella K., Gabriel S.B., dePristo M.A., Shakir K. Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum. Mol. Genet. 2011;20:1285–1289. doi: 10.1093/hmg/ddq569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garshasbi M., Hadavi V., Habibi H., Kahrizi K., Kariminejad R., Behjati F., Tzschach A., Najmabadi H., Ropers H.H., Kuss A.W. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2008;82:1158–1164. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgins J.J., Pucilowska J., Lombardi R.Q., Rooney J.P. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63:1927–1931. doi: 10.1212/01.wnl.0000146196.01316.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mir A., Kaufman L., Noor A., Motazacker M.M., Jamil T., Azam M., Kahrizi K., Rafiq M.A., Weksberg R., Nasr T. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:909–915. doi: 10.1016/j.ajhg.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mochida G.H., Mahajnah M., Hill A.D., Basel-Vanagaite L., Gleason D., Hill R.S., Bodell A., Crosier M., Straussberg R., Walsh C.A. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molinari F., Foulquier F., Tarpey P.S., Morelle W., Boissel S., Teague J., Edkins S., Futreal P.A., Stratton M.R., Turner G. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am. J. Hum. Genet. 2008;82:1150–1157. doi: 10.1016/j.ajhg.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molinari F., Rio M., Meskenaite V., Encha-Razavi F., Augé J., Bacq D., Briault S., Vekemans M., Munnich A., Attié-Bitach T. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 11.Motazacker M.M., Rost B.R., Hucho T., Garshasbi M., Kahrizi K., Ullmann R., Abedini S.S., Nieh S.E., Amini S.H., Goswami C. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2007;81:792–798. doi: 10.1086/521275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philippe O., Rio M., Carioux A., Plaza J.M., Guigue P., Molinari F., Boddaert N., Bole-Feysot C., Nitschke P., Smahi A. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:903–908. doi: 10.1016/j.ajhg.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 15.Lindner T.H., Hoffmann K. easyLINKAGE: A PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 16.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 17.Abou Jamra R., Wohlfart S., Zweier M., Uebe S., Priebe L., Ekici A., Giesebrecht S., Abboud A., Al-Khateeb M., Fakher M. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 2011 doi: 10.1038/ejhg.2011.98. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue):W593–599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehm M., Bonifacino J.S. Genetic analyses of adaptin function from yeast to mammals. Gene. 2002;286:175–186. doi: 10.1016/s0378-1119(02)00422-5. [DOI] [PubMed] [Google Scholar]
- 23.Boehm M., Aguilar R.C., Bonifacino J.S. Functional and physical interactions of the adaptor protein complex AP-4 with ADP-ribosylation factors (ARFs) EMBO J. 2001;20:6265–6276. doi: 10.1093/emboj/20.22.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dell'Angelica E.C., Mullins C., Bonifacino J.S. AP-4, a novel protein complex related to clathrin adaptors. J. Biol. Chem. 1999;274:7278–7285. doi: 10.1074/jbc.274.11.7278. [DOI] [PubMed] [Google Scholar]
- 25.Hirst J., Bright N.A., Rous B., Robinson M.S. Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Kilimann M.W. Identification of two new mu-adaptin-related proteins, mu-ARP1 and mu-ARP2. FEBS Lett. 1997;402:57–61. doi: 10.1016/s0014-5793(96)01500-1. [DOI] [PubMed] [Google Scholar]
- 27.Burgos P.V., Mardones G.A., Rojas A.L., daSilva L.L., Prabhu Y., Hurley J.H., Bonifacino J.S. Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell. 2010;18:425–436. doi: 10.1016/j.devcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuda S., Miura E., Matsuda K., Kakegawa W., Kohda K., Watanabe M., Yuzaki M. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron. 2008;57:730–745. doi: 10.1016/j.neuron.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Borck G., Mollà-Herman A., Boddaert N., Encha-Razavi F., Philippe A., Robel L., Desguerre I., Brunelle F., Benmerah A., Munnich A., Colleaux L. Clinical, cellular, and neuropathological consequences of AP1S2 mutations: Further delineation of a recognizable X-linked mental retardation syndrome. Hum. Mutat. 2008;29:966–974. doi: 10.1002/humu.20531. [DOI] [PubMed] [Google Scholar]
- 30.Tarpey P.S., Stevens C., Teague J., Edkins S., O'Meara S., Avis T., Barthorpe S., Buck G., Butler A., Cole J. Mutations in the gene encoding the Sigma 2 subunit of the adaptor protein 1 complex, AP1S2, cause X-linked mental retardation. Am. J. Hum. Genet. 2006;79:1119–1124. doi: 10.1086/510137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verkerk A.J., Schot R., Dumee B., Schellekens K., Swagemakers S., Bertoli-Avella A.M., Lequin M.H., Dudink J., Govaert P., van Zwol A.L. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet. 2009;85:40–52. doi: 10.1016/j.ajhg.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreno-De-Luca A., Helmers S.L., Mao H., Burns T.G., Melton A.M., Schmidt K.R., Fernhoff P.M., Ledbetter D.H., Martin C.L. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet. 2011;48:141–144. doi: 10.1136/jmg.2010.082263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. One Figure and One Table