mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 16.
Published in final edited form as: Science. 2010 Aug 20;329(5994):959–964. doi: 10.1126/science.1190287
Abstract
The rapid antidepressant response after ketamine administration in treatment resistant depressed patients suggests a possible new approach for treating mood disorders compared to the weeks or months required for standard medications. However, the mechanisms underlying this action of ketamine have not been identified. We observed that ketamine rapidly activated the mammalian target of rapamycin (mTOR) pathway, leading to increased synaptic signaling proteins and increased number and function of new spine synapses in the prefrontal cortex of rat. Moreover, blockade of mTOR signaling completely blocked ketamine-induction of synaptogenesis and behavioral responses in models of depression. Our results demonstrate that these effects of ketamine are opposite to the synaptic deficits that result from exposure to stress and could contribute to the fast antidepressant actions of ketamine.
Major depressive disorder (MDD) afflicts approximately 17 percent of the population and is one of the leading causes of total disability and economic burden (1). Although there are medications that alleviate depressive symptoms, these have serious limitations. Most notably, available treatments require weeks or months to produce a therapeutic response, and only about one third of patients respond to the first medication prescribed (2). In contrast, recent studies demonstrate that a single, low dose of a glutamate N-methyl-D-aspartic acid (NMDA) receptor antagonist produces a rapid (within hours) antidepressant response that lasts for up to 7 days (3, 4), and is effective in MDD patients who are resistant to traditional antidepressants (5). The mechanisms underlying rapid antidepressant actions are likely more complicated than simple NMDA receptor blockade, and so far have not been identified. We carried out a series of studies to examine the cellular signaling pathways that mediate the behavioral actions of NMDA receptor blockade, focusing on signaling cascades known to rapidly influence synaptic plasticity
The drug used for clinical trials is ketamine, a nonselective NMDA receptor antagonist (6). A low dose of ketamine (10 mg/kg), which is reported to have antidepressant actions in behavioral models of depression (6), rapidly activated the mammalian target of rapamycin (mTOR) signaling pathway in the prefrontal cortex (PFC) of rats (Fig. 1A). Activation of mTOR signaling was observed in a preparation enriched in synaptoneurosomes (see fig. S1), and included increased levels of the phosphorylated and activated forms of eukaryotic initiation factor 4E binding protein 1 (4E-BP1), p70S6 kinase (p70S6K), and mTOR (Fig. 1A). Increased phosphorylation of 4E-BP1, p70S6K, and mTOR is transient, returning to basal levels by two hours after ketamine administration (Fig. 1A). In contrast, other antidepressants tested, including electroconvulsive seizure, imipramine, or fluoxetine, did not significantly influence mTOR signaling (fig. S2). Ketamine produced a similar rapid and transient increase in the phosphorylated and activated forms of extracellular signal-regulated kinase (ERK, including ERK1 and ERK2) and protein kinase B (PKB/Akt) (fig. S3A), growth factor signaling pathways that have been linked to activation of mTOR signaling (7). The activation of 4E-BP1, p70S6K, mTOR (Fig. 1B), ERK, and Akt (fig. S3B) was dose dependent, occurring at relatively low doses (5 to 10 mg/kg) that produce antidepressant behavioral actions, but not at a higher anesthetic dose (6).
Fig. 1.

Ketamine transiently and dose-dependently activates mTOR signaling in rat prefrontal cortex (PFC). (A) Time course of ketamine (10 mg/kg, i.p.) induced mTOR signaling determined by Western blot analysis of phospho-mTOR (pmTOR), phospho-4E-BP1 (p4E-BP1), and phospho-p70S6K (pp70S6K) in synaptoneurosomes of PFC. Levels of total mTOR, GAPDH and p70S6K were also determined. (B) Dose-dependent activation, determined 1 hr after ketamine administration, of pmTOR, p4E-BP1 and pp70S6K. (C) Pre-treatment (10 min) with NBQX (10 mg/kg, i.p.) blocked ketamine (10 mg/kg, i.p.) activation of pmTOR, p4E-BP1, and pp70S6K, as well as upstream signaling kinases phospho-ERK (pERK) and phospho-Akt (pAkt) (analyzed 1 hr after ketamine). Levels of pERK1 and pERK2 were similarly regulated and were combined for quantitative analysis. (D) Pre-treatment (30 min) with inhibitors of ERK (U0126, 20 nmol, ICV) or PI-3k/Akt (LY294002, 20 nmol, ICV) abolished ketamine (10 mg/kg, i.p.) activation of mTOR signaling proteins (analyzed 1 hr after ketamine administration). Values represent mean ± SEM [n = 4 animals; * P < 0.05; ** P < 0.01, Analysis of Variance (ANOVA)].
The antidepressant actions of ketamine have been reported to require glutamate a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA) receptors (6). In line with this, we found that pre-treatment (10 minutes) with a selective AMPA receptor inhibitor, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) completely blocked the induction of phosphorylated, 4E-PB1, p70S6K and mTOR, as well as ERK and Akt in the PFC (Fig. 1C, fig. S4). We assessed the possible involvement of ERK and Akt pathways in the activation of mTOR by intracerebroventricular (ICV) infusion of specific pharmacological antagonists. Inhibition of either ERK (U0126) or Akt (via inhibition of the upstream Akt activator PI3 kinase by LY294002) blocked ketamine-induction of phosphorylated 4E-BP1, mTOR, and p70S6K (Fig. 1D, fig. S5).
Activation of mTOR has been functionally linked with local protein synthesis in synapses, resulting in the production of proteins required for the formation, maturation and function of new spine synapses (7). To determine if ketamine-induction of mTOR produces similar effects, levels of several synaptic proteins were analyzed in synaptoneurosomes from PFC. Ketamine administration increased levels of the postsynaptic proteins PSD95 and GluR1, as well as the presynaptic protein synapsin I (Fig. 2, A and B). Ketamine-induced increases in these synaptic proteins was delayed relative to ERK, Akt and mTOR, peaking at 2 to 6 hrs, but remained elevated up to 72 hrs. Ketamine also induced expression of activity regulated cytoskeletal-associated protein (Arc), with an intermediate (peak at 1–2 hrs), but transient time course. The functional connection between mTOR activation and synaptic protein synthesis was examined by ICV infusion of selective mTOR inhibitor rapamycin, which completely blocked the induction of PSD95, GluR1 and synapsin I (Fig. 2B, fig. S6).
Fig. 2.

Ketamine rapidly increases synaptic proteins and spine number. (A) Time course for ketamine (10 mg/kg, ip) induction of synaptic proteins ARC, synapsin I, PSD95, and GluR1 in synaptoneurosomes of prefrontal cortex (PFC), and (B) blockade by pre-treatment (30 min) with a selective mTOR inhibitor rapamycin (0.2 nmol, ICV) (values represent mean ± SEM, n = 4 – 6 animals; *P < 0.05; **P < 0.01, ANOVA.) (C) Ketamine increased spine density in medial PFC, analyzed by two-photon microscopy 24 hr after treatment. Representative images are shown of high magnification Z-stack projections of apical tuft segments of Neurobiotin-labeled layer V pyramidal cells (Scale: 5 μm). (D) Results are the mean ± SEM (8 cells from 4 rats in each group; *P < 0.05; **P < 0.01, _t_-test). (E) Ketamine enhanced mPFC layer V pyramidal cell EPSC responses. Sample whole cell voltage-clamp recordings of 5-HT and hypocretin-induced EPSCs in slices (24 hr post-ketamine). (F) Cumulative probability distributions showing significant increases in amplitude (p < 0.0001, KS-z vale = 6.5 for 5-HT and 6.7 for Hcrt) and (H) frequency of 5-HT- and hypocretin-induced EPSCs (n = 12 neurons/group; *P < 0.05, _t_-test). Ketamine-induction of spine density and function were blocked by rapamycin infusions (C–G)
An increase in synapse-associated proteins indicates that ketamine administration rapidly increases synapse/spine formation. We therefore examined the influence of ketamine on spine number and morphology by two-photon imaging of the apical tuft of prelabeled layer V mPFC pyramidal neurons. Analysis of the images shows that ketamine administration increased spine density in both distal and proximal segments of the apical tuft 24 hrs after ketamine injection (Fig. 2, C and D). Ketamine also increased the population of mushroom (large-diameter) spines (fig. S7), an indication of increased spine maturation and synaptic strengthening (8). Synaptic strengthening is supported by analysis of the electrophysiological properties of the same cells, demonstrating that EPSC frequency responses to 5-HT, which are mediated predominantly by cortical-cortical synapses in the apical tuft (9), are significantly increased by ketamine administration (Fig. 2, E–G). Responses to hypocretin/orexin, which occur selectively at apical thalamocortical synapses (10), are also increased (Fig. 2, E–G). In both cases, there is a significant increase in EPSC amplitude (Fig. 2F), consistent with the elevation in mushroom spines (fig. S7) and GluR1. Ketamine-induction of spines and 5-HT and hypocretin-stimulated EPSPs are blocked by infusion of rapamycin (Fig. 2, C–G; fig. S7).
We next determined if activation of mTOR signaling is required for the antidepressant behavioral actions of ketamine. As previously reported, a low dose of ketamine produced a rapid antidepressant effect in two well-established behavioral models of despair, the forced swim test (FST) and learned helplessness (LH) in response to inescapable stress (IES), as well as a model of anxiety, the novelty suppressed feeding test (NSFT) (Fig. 3, A–C). Traditional antidepressants are effective in these tests after acute (1 d, FST), subchronic (6 d, LH), or chronic (21 d, NSFT) treatment, while a single dose of ketamine was effective in all three tests. Moreover, infusion (ICV) of rapamycin completely abolished the antidepressant actions of ketamine in all three tests (Fig. 3, A–C). The role of mTOR signaling in the actions of ketamine was further examined by local infusion of rapamycin into the medial PFC, which resulted in a complete blockade of the behavioral effects of ketamine in FST and NSFT tests (Fig. 3, D and E) (location of rapamycin infusions into the medial PFC is shown in Fig. 3F). The effects of inhibitors of ERK and PI3k/Akt, which mediate ketamine-induction of mTOR, were also examined. Infusion (ICV) of selective ERK (U0126) or PI3k/Akt (LY294002) inhibitors blocked the antidepressant actions of ketamine in the FST and NSFT (Fig. 3, G and H). Finally the behavioral actions of ketamine in the FST occurred with a dose-response profile similar to that for induction of ERK, Akt and mTOR (Fig. 3I). This addresses a major point of discussion in the field as to whether an anesthetic dose of ketamine could produce an antidepressant effect.
Fig. 3.

Rapid behavioral actions of ketamine require mTOR signaling. Rapamycin was infused (0.2 nmol, ICV) 30 min prior to ketamine (10 mg/kg, ip), and responses in the FST (A), NSFT (B), or LH (C) paradigms was determined. Infusion of rapamycin (0.01 nmol) into the medial prefrontal cortex (PFC) blocked the antidepressant actions of ketamine (10 mg/kg, ip) in the FST (D) and NSFT (E). (F) Location of rapamycin infusions in the PFC (infusion sites are the same on the right and left sides due to the use of bilateral cannulae). Pre-treatment with inhibitors of ERK (U0126, 20 nmol, ICV) or PI3 kinase/Akt (LY294002, 20 nmol, ICV) blocked the behavioral effects of ketamine in FST (G) and NSFT (H). (I) Low (10 mg/kg) but not a high anesthetic dose (80 mg/kg) of ketamine produced an antidepressant action in the FST. Values represent mean ± SEM (n = 6–8 animals; *P < 0.05; **P < 0.01, ANOVA).
Basic research and clinical studies demonstrate that stress and depression can cause atrophy of PFC pyramidal neurons in rodent models and in postmortem tissue of depressed subjects (9, 11), and brain imaging studies report a decrease in PFC volume in depressed patients (12). Exposure to stress in the LH paradigm decreased levels of synapatic proteins GluR1, PSD95, and synapsin I, and a single dose of ketamine reversed this deficit in a rapamycin-sensitive manner (fig. S8). These results indicate that ketamine-induction of synapse-associated protein synthesis provides a mechanism for rapid reversal of stress- and/or depression-mediated deficits, and could enhance PFC mediated connectivity and function.
Ketamine is a psychotomimetic drug with abuse potential and a more selective agent would be desirable for clinical antidepressant use. There have been reports that selective NR2B antagonists produce antidepressant actions similar to ketamine in both preclinical models and clinical trials (6, 13). Therefore we examined the influence of Ro 25-6981, a NR2B selective compound, on mTOR signaling and rapamycin-sensitive behaviors. Ro25-6981, at a dose (10 mg/kg) identified in the FST to be optimal (fig. S9), produced a transient activation of mTOR signaling (increased phosphorylation of 4E-BP1, p70S6K, and mTOR), as well as ERK and Akt signaling in synaptoneurosomes from PFC (Fig. 4, A and B). Ro 25-6981 administration also increased levels of the synaptic proteins, Arc, PSD95, GluR1, and synapsin I in synaptoneurosomes (Fig. 4, A and B). Induction of Arc was more rapid and transient, while PSD95, GluR1, and synapsin 1 were delayed, similar to the delay observed following ketamine administration. Finally, Ro 25-6981 administration produced rapid antidepressant effects (24 hr) in the FST and NSFT, which were blocked by pre-infusion (ICV) of rapamycin (Fig. 4, C and D).
Fig. 4.
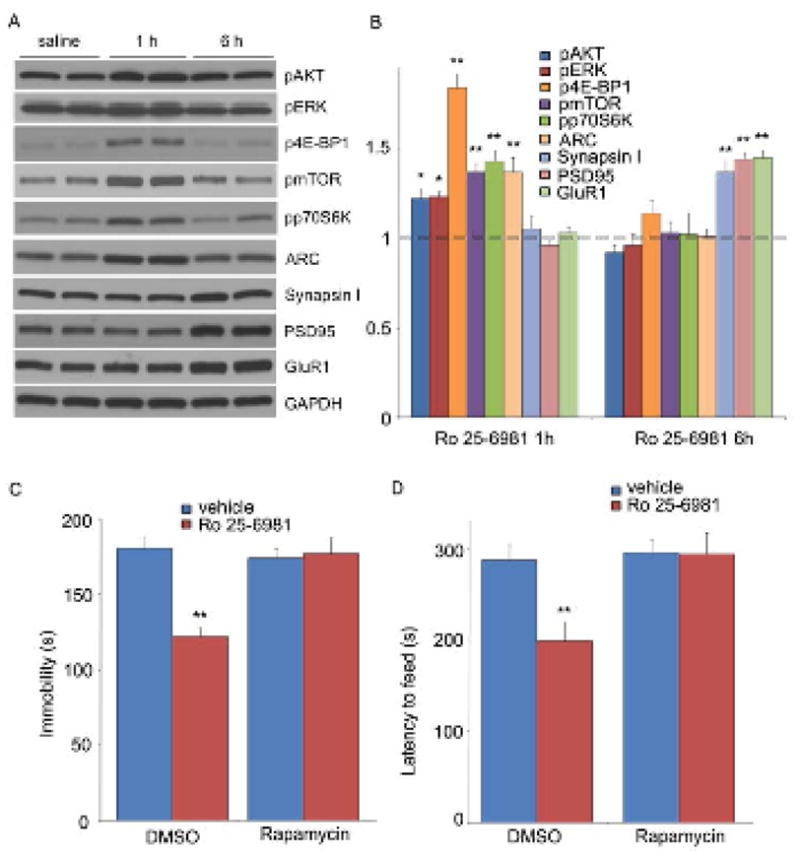
Selective NR2B antagonist Ro 25-6981 activates mTOR signaling and produces rapamycin-sensitive behavioral effects. (A, B) Effects of Ro 25-6981 (10 mg/kg, ip) on pmTOR, p4E-BP1, pp70S6K, pERK, pAkt, Arc, synapsin I, PSD95, and GluR1 in prefrontal cortex. (C–D) Pre-treatment with rapamycin (0.2 nmol, ICV) abolished the actions of Ro 25-6981 in FST (C) and NSFT (D). Values represent mean ± SEM (n = 6–8 animals; *P < 0.05; **P < 0.01, ANOVA).
Fast activation of mTOR signaling resulting in rapid and sustained elevation of synapse associated proteins and spine number in the PFC represents a mechanism for the rapid antidepressant actions of ketamine. These effects result in elevated 5-HT neurotransmission, a primary target of traditional antidepressants, although the latter can take weeks or months to induce a therapeutic response (2). The mechanisms underlying the induction of mTOR signaling are unclear, but the requirement for glutamate-AMPA receptor activation is consistent with the hypothesis that there is a subset of NMDA receptors, possibly on GABAergic interneurons, that when activated lead to disinhibition of glutamate signaling (14). Further characterization of these actions of NMDA receptor blockade and the signaling pathways that stimulate mTOR signaling and mediate the rapid induction of synapses in the PFC will provide novel therapeutic targets for antidepressant drug development.
Supplementary Material
supplementary data
References and Notes
- 1.Kessler RC, et al. JAMA. 2003;289:3095. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Trivedi MH, et al. Am J Psychiatry. 2006;163:28. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 3.Berman RM, et al. Biol Psychiatry. 2000;47:351. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 4.Zarate CA, Jr, et al. Arch Gen Psychiatry. 2006;63:856. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 5.Krystal JH. Swiss Med Wkly. 2007;137:215. doi: 10.4414/smw.2007.11932. [DOI] [PubMed] [Google Scholar]
- 6.Maeng S, et al. Biol Psychiatry. 2008;63:349. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 7.Hoeffer CA, Klann E. Trends Neurosci. 2010;33:67. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshihara Y, De Roo M, Muller D. Curr Opin Neurobiol. 2009;19:146. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Liu RJ, Aghajanian GK. Proc Natl Acad Sci U S A. 2008;105:359. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambe EK, Aghajanian GK. Neuroscience. 2007;145:900. doi: 10.1016/j.neuroscience.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajkowska G, et al. Biol Psychiatry. 1999;45:1085. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 12.Drevets WC. Annu Rev Med. 1998;49:341. doi: 10.1146/annurev.med.49.1.341. [DOI] [PubMed] [Google Scholar]
- 13.Preskorn SH, et al. J Clin Psychopharmacol. 2008;28:631. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- 14.Farber NB, Newcomer JW, Olney JW. Prog Brain Res. 1998;116:421. doi: 10.1016/s0079-6123(08)60453-7. [DOI] [PubMed] [Google Scholar]
- 15.This work is supported by USPHS grants MH45481 and 2P01 MH25642 and by the Connecticut Mental Health Center.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary data