MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly (original) (raw)
Abstract
When T cells are activated, the expression of the CD95 ligand is elevated, with the purpose of inducing apoptosis in target cells and to later eliminate the activated T cells. We have shown previously that mitogen-activated protein kinase (MAPK or ERK) signaling suppresses CD95-mediated apoptosis in different cellular systems. In this study we examined whether MAPK signaling controls the persistence and CD95-mediated termination of an immune response in activated T cells. Our results show that activation of Jurkat T cells through the T cell receptor immediately suppresses CD95-mediated apoptosis, and that this suppression is mediated by MAPK activation. During the phase of elevated MAPK activity, the activation of caspase-8 and Bid is inhibited, whereas the assembly of a functional death-inducing signaling complex (DISC) is not affected. These results explain the resistance to CD95 responses observed during the early phase of T cell activation and suggest that MAPK-activation deflects DISC signaling from activating caspase-8 and Bid. The physiological relevance of the results was confirmed in activated primary peripheral T cells, in which inhibition of MAPK signaling markedly sensitized the cells to CD95-mediated apoptosis.
Keywords: apoptosis/death receptor/MAP kinase/T cell activation/T cell receptor
Introduction
Apoptosis is essential in the control of immune responses, embryonic development and tissue homeostasis in the adult. In particular, CD95 (Fas/APO-1)-mediated apoptosis plays an important role in the immune system, both in the negative selection of T cells and in eliminating target cells as well as unwanted lymphocytes during the immune response (Nagata and Golstein, 1995). CD95 is a 48 kDa large glycoprotein (Itoh et al., 1991; Oehm et al., 1992) belonging to the tumor necrosis factor receptor (TNF-R) superfamily. Oligomerization of CD95 by the CD95 ligand (CD95L/FasL; Suda et al., 1993) activates the apoptotic pathway (Itoh et al., 1991; Oehm et al., 1992) by recruiting a set of proteins that form the death-inducing signaling complex (DISC; Kischkel et al., 1995). The DISC contains several proteins with distinct functions, among them a cytosolic adaptor protein, Fas-associated protein with death domain (FADD or MORT1; Chinnaiyan et al., 1995; Boldin et al., 1996). The death domain (DD) of CD95 binds to the C-terminal DD of FADD, whose N-terminal death effector domain (DED) in turn recruits caspase-8 (Chinnaiyan et al., 1995; Muzio et al., 1996), which is thereby autoproteolyzed and activated (Medema et al., 1997). Recently, a molecular link between DISC-activated caspase-8 and the mitochondria was described (Li et al., 1998; Luo et al., 1998). In these studies, the Bcl-2 family protein Bid was shown to be a direct target for caspase-8. Cleavage of the p22 Bid by caspase-8 results in the formation of a truncated p15 Bid protein, which directly affects mitochondria and releases cytochrome c. Several effector caspases are then cleaved and activated, thereby amplifying the caspase cascade (Cryns and Yuan, 1998). This mitochondrial activation pathway has been shown to be essential for CD95-mediated apoptosis in certain cell types, termed type II cells (Scaffidi et al., 1999b).
Resting peripheral T cells are activated during an immune response by repeated stimulation of both the T cell receptor (TCR) and the co-receptors, with specific antigens, mitogens or antibodies. This activation results in increased expression of several genes, including CD95 and CD95L (Klas et al., 1993; Brunner et al., 1995). A major role for the latter is activation of apoptosis in unwanted target cells, such as tumor cells (Nagata and Golstein, 1995). When the activated T cells have accomplished their task of killing self-altered or virus-infected cells, they themselves undergo CD95L-mediated activation- induced cell death (AICD). AICD is induced within 1 day of OKT3 stimulation in Jurkat T cells and in several murine T cell hybridoma cell lines, and within 5–7 days in peripheral T cells (Brunner et al., 1995; Dhein et al., 1995; Ju et al., 1995). AICD is a major mechanism for removing activated T cells after they have accomplished the task of clearing virus-infected or tumorigenic cells from the host. Therefore, the precise regulation of CD95-mediated apoptosis is of crucial importance to ensure elimination of target cells as well as termination of the immune response. Since AICD occurs long after the CD95L levels start to increase, there has to be specific mechanisms responsible for protecting the cells against CD95 stimulation. Upregulation of the apoptosis-inhibitory protein c-FLIP (Irmler et al., 1997; Tschopp et al., 1999) and anti-apoptotic Bcl-family members (Peter et al., 1997) has been indicated to protect activated T cells from CD95-mediated apoptosis, but their expression does not always correlate with CD95 sensitivity (Scaffidi et al., 1999a). Therefore, it is difficult to develop a paradigm based solely on the expression of these proteins, which would explain the protection observed during the early phase of T cell activation. While there is evidence for a role of phosphorylation-mediated signaling in regulation of CD95 responses (Holmström et al., 1998, 1999), little is known about the role of phosphorylation in the suppression of CD95-mediated apoptosis during T cell activation.
A possible signaling candidate that could protect activated T cells from AICD is the mitogen-activated protein kinase (MAPK or ERK) pathway, a major regulator of cell activation, proliferation and differentiation (Lewis et al., 1998). Ligation of the TCR activates two major signaling cascades, namely the MAPK and calcineurin pathways, both of which are required for T-cell-specific transcriptional activation. The paradigm of MAPK as an inhibitory pathway in regulation of CD95-mediated apoptosis is supported by studies showing suppression of apoptosis by MAPK activation (Xia et al., 1995) and by our studies showing that MAPK specifically protects cells from CD95-induced apoptosis (Holmström et al., 1998, 1999). Furthermore, signaling through the TCR has previously been shown to antagonize CD95-mediated apoptosis (Klas et al., 1993; Peter et al., 1997). It is also important to know where in the CD95-signaling pathway TCR signaling is exerting its effects. This is needed in order to determine whether the cells still maintain their normal functions, or whether they shift to some other functional state when CD95 is stimulated but no apoptotic response is triggered. We wanted, therefore, to investigate more closely the role of the MAPK pathway in protecting recently activated T cells from CD95-mediated apoptosis. In the present study we show that TCR-mediated MAPK activation suppresses CD95-mediated apoptosis during the early phase of T cell activation. This inhibition is protein synthesis independent and occurs after assembly of a functional DISC but before overall caspase-8 activation and cleavage of Bid. These results imply that direct MAPK-mediated signaling, in addition to the known inhibitory proteins, also participates in regulating the functions and responses of CD95.
Results
Activation of Jurkat T cells with the agonistic CD3 antibody, OKT3, suppresses CD95-mediated apoptosis
To determine how activation of the CD3–TCR complex affects CD95-mediated apoptosis, we pre-treated Jurkat T cells with an immobilized agonistic CD3 antibody, OKT3, and analyzed whether OKT3 suppressed two characteristic signs of apoptosis: the formation of oligonucleosomal DNA fragments (Figure 1A) and phosphatidylserine exposure on the cell membrane (Figure 1B). Pre-treatment of Jurkat T cells with OKT3 clearly suppressed the amount of apoptotic cells by ∼50%. Furthermore, the duration of OKT3-induced suppression of CD95-mediated apoptosis was analyzed and shown to last for 10 h (Figure 1C).
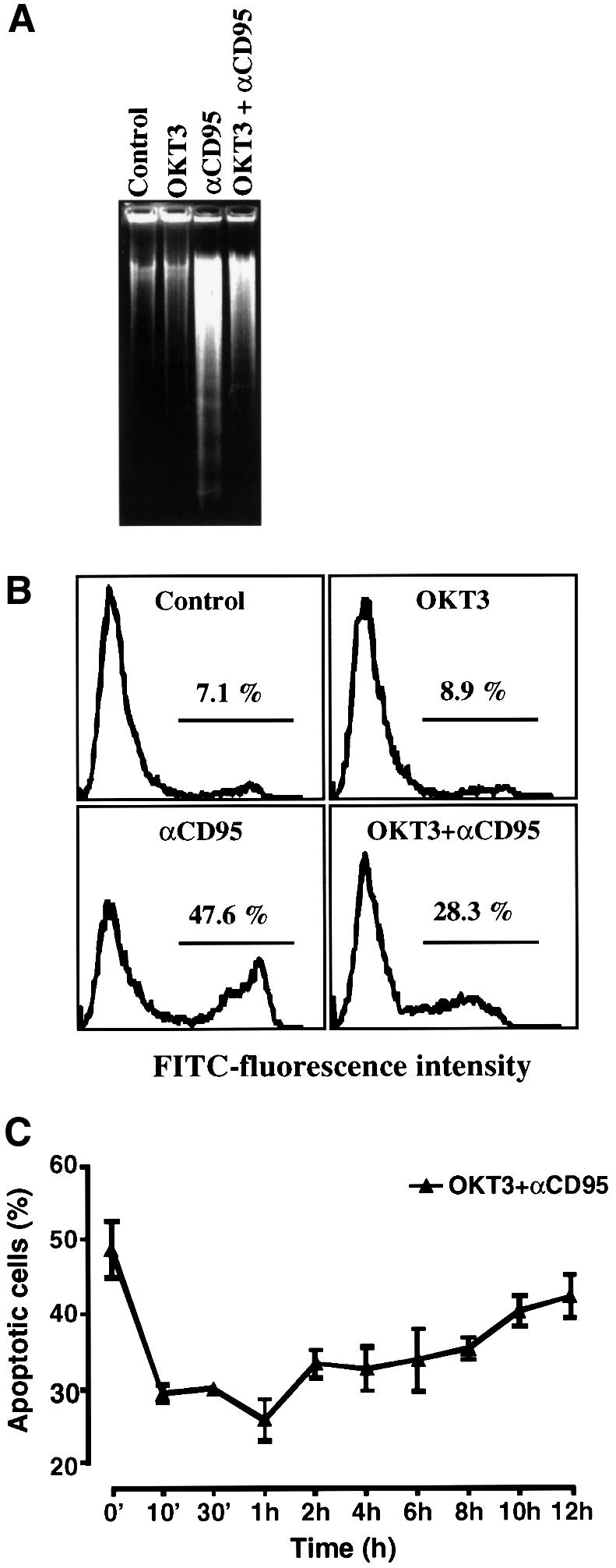
Fig. 1. Activation of Jurkat T cells with an agonistic CD3 antibody, OKT3, suppresses CD95-mediated apoptosis. Jurkat T cells were incubated with medium alone, immobilized anti-CD3 (OKT3) antibody (100 µg/ml), anti-CD95 (anti-Fas, 100 ng/ml) or pre-incubated for 30 min with immobilized OKT3 before addition of anti-CD95. After 2 h aliquots of cells were analyzed by either (A) one-stage DNA gel electrophoresis or (B) annexin V–FITC staining, and the proportion of apoptotic nuclei was determined using a FACScan flow cytometer. Bars indicate the percentage of cells with exposed phoshatidylserine (FITC positive), a feature that is characteristic of apoptotic cells. (C) The OKT3-mediated suppression of CD95-induced apoptosis lasts for 10–12 h after stimulation. Jurkat T cells were pre-incubated with immobilized OKT3 for the indicated time periods prior to incubation with anti-CD95 for 2 h and analyzed as in (A). The data represent mean values (mean ± SEM) from a minimum of three separate experiments.
OKT3-mediated MAPK activation corresponds to the observed suppression of CD95-mediated apoptosis
Since the TCR is a potential target for MAPK, we compared the kinetics of MAPK activation with the observed suppression of CD95-mediated apoptosis by OKT3. A MAPK assay showed that stimulation of Jurkat T cells with OKT3 induces a rapid increase in MAPK activity starting 10 min after stimulation of CD3 and lasting up to 10–12 h (Figure 2A). The MAPK activity peaked at 1–4 h, being ∼2.5-fold higher than the control values (Figure 2B). This activation was not due to increased synthesis of MAPK as the MAPK protein levels remained constant following CD95 stimulation (Figure 2B). The OKT3-induced MAPK activation was completely inhibited by pre-treatment of Jurkat T cells with the specific MKK1 inhibitor, PD 98059 (Alessi et al., 1995), before triggering CD95 (Figure 2A). Furthermore, there was a close correlation between OKT3-mediated MAPK activation and suppression of CD95-mediated apoptosis (Figure 2C).

Fig. 2. The kinetics of MAPK activation in Jurkat T cells after OKT3 stimulation corresponds to the observed suppression of CD95-mediated apoptosis. MAPK activation was followed in cell extracts incubated with medium alone, in the presence of immobilized OKT3 or in the presence of the MKK1 inhibitor PD 98059 (30 µM) + OKT3, and the activation was determined by an immunocomplex kinase assay. (A and B) A representative autoradiograph of the immunocomplex kinase assay together with an immunoblot of the immunoprecipitated ERK2, to verify presence of the MAPK, is shown. Multiple samples at the different time points were quantified by phosphoimager analysis. (C) A close correlation between MAPK activity and the amount of apoptotic cells was obtained when the data from the MAPK assays and the percentage of apoptotic cells, treated as described above, were plotted against the incubation time with OKT3. (B and C) The data represent mean values (mean ± SEM) from a minimum of three separate experiments.
The OKT3-mediated suppression of CD95-mediated apoptosis is MAPK dependent and independent of protein synthesis
As we have shown previously that MAPK signaling suppresses CD95-mediated apoptosis (Holmström et al., 1998, 1999), we wanted to determine whether MAPK activation is responsible for the observed resistance of recently activated Jurkat T cells to CD95-mediated apoptosis, and if this suppression requires protein synthesis. The MAPK dependence was demonstrated by pre-treatment of the cells with PD 98059, which abolished the protective effect of OKT3 on CD95-mediated apoptosis (Figure 3). However, cycloheximide (CHX) treatment had no effect, as cells pre-treated with immobilized OKT3 were equally insensitive to CD95-mediated apoptosis both in the presence and absence of CHX (Figure 3), indicating that the process is independent of protein synthesis. The MAPK-dependent protection was further corroborated by transient transfection of Jurkat T cells with a dominant-negative MKK1 containing a hemagglutinin (HA) tag (Mansour et al., 1996). The transfected cells were then visualized by immunofluorescence staining of HA and the nuclear morphology by DNA staining (Figure 4A). The protective effect of OKT3 on CD95-mediated apoptosis was abolished in the cells expressing dominant-negative MKK1 (Figure 4B). Taken together, these results show that the observed suppression of CD95 is MAPK dependent and protein synthesis independent.

Fig. 3. OKT3-mediated suppression of CD95-induced apoptosis in Jurkat T cells is independent of protein synthesis and mediated through MAPK activation. Annexin V–FITC staining was used as an indicator for the amount of apoptotic cells. Cells were cultured for 2 h with medium alone, in the presence of anti-CD95 (anti-Fas), immobilized OKT3, PD 98059, CHX (100 µg/ml), OKT3 + anti-CD95, CHX + anti-CD95, CHX + OKT3 + anti-CD95 or PD 98059 + OKT3 + anti-CD95. The cells were pre-incubated with immobilized OKT3 as previously described and/or for 10 min with CHX prior to incubation with anti-CD95. The data represent mean values (mean ± SEM) from a minimum of three separate experiments.


Fig. 4. Dominant-negative MKK1 abolishes the anti-CD3-mediated protective effect on CD95-mediated apoptosis. (A) Representative immunofluorescence micrographs of cells transfected with a dominant-negative (MKK1-8E) construct and incubated for 2 h with anti-CD95 in the absence or presence of OKT3. Hoechst staining was used to detect alterations in the nuclei and a monoclonal HA antibody linked to an FITC-conjugated secondary antibody was used to detect the presence of the HA-tagged MKK1 in transfected cells. The arrows indicate the transfected cells. Mock transfected cells were treated as indicated above with or without stimulation of CD95. (B) Percentage of apoptosis in transfected cells after treatment with anti-CD95. The data represent mean values (mean ± SEM) from a minimum of three separate experiments.
Inhibition of MAPK signaling sensitizes activated primary T cells to CD95-mediated apoptosis
Resting T cells express low levels of CD95 on the cell surface. One day after activation of peripheral T cells the levels of CD95 are upregulated, and on day 5–7, when grown continuously in the presence of interleukin-2, the cells show a CD95-sensitive phenotype (Klas et al., 1993; Peter et al., 1997). It is therefore interesting to examine whether MAPK signaling is able to modulate the sensitivity of T cells to CD95-mediated apoptosis. For this purpose we activated peripheral blood T cells with 1 µg/ml phytohemagglutinin (PHA) for 5–6 days prior to triggering CD95 in the absence or presence of PD 98059 (Figure 5). Quite interestingly, the specific MKK1 inhibitor, PD 98059, clearly sensitized T cells to CD95-mediated apoptosis. Our results imply that primary T cells are relatively insensitive to CD95-mediated apoptosis for 5 days after activation. Suppression of MAPK activation, however, sensitizes them to CD95-mediated apoptosis (Figure 5).

Fig. 5. Suppression of MAPK activation sensitizes activated human peripheral T cells to CD95-mediated apoptosis. Annexin V–FITC staining was used as an indicator for the amount of apoptotic cells. Activated T cells (day 5–6; 1 × 106 cells/ml) were cultured for 6 or 24 h with medium alone, in the presence of anti-CD95 (anti-Fas, 500 ng/ml), immobilized OKT3, PD 98059, OKT3 + anti-CD95 or PD 98059 + OKT3 + anti-CD95. The cells were pre-incubated with immobilized OKT3 as previously described prior to incubation with anti-CD95. The data represent mean values (mean ± SEM) from a minimum of three separate experiments.
Activation of Jurkat T cells by OKT3 does not affect the levels of apoptotic inhibitory proteins, Bcl-2, Bcl-xL and FLIP
Although our results indicate that the OKT3-mediated suppression of apoptosis is protein synthesis-independent, we wanted to examine more closely the effect of OKT3 treatment on some of the known anti-apoptotic proteins. In order to examine whether Bcl-2, Bcl-xL and FLIP protein levels were increasing, we performed immunoblotting of the respective proteins after treatment of Jurkat T cells with OKT3 (Figure 6A). Immunoblotting of Bcl-2, Bcl-xL and FLIP showed that all these proteins are constitutively expressed in Jurkat T cells, and that there was no significant increase in the amount of these proteins. This indicates that these proteins are not affected by OKT3 treatment and it is unlikely that they are involved in the observed suppression of CD95-mediated apoptosis in Jurkat T cells.

Fig. 6. OKT3 stimulation does not affect Bcl-xL, Bcl-2, c-FLIPL and CD95 levels. (A) Jurkat T cells were treated with immobilized OKT3 for the indicated time points and the amount of proteins known to affect apoptosis was assessed by immunoblotting with specific antibodies to Bcl-xL, Bcl-2 and c-FLIPL. Equal loading was confirmed by immunoblotting of the same gel with an actin-specific antibody. A representative immnunoblot from three experiments is shown. (B) To measure the amount of CD95 on the cell surface after MAPK activation, Jurkat T cells were treated as indicated in the figure. The amount of CD95 was then assessed by staining with a specific CD95 antibody (anti-APO-1) followed by an FITC-conjugated secondary antibody and analyzed by flow cytometry. As a negative control we used cells stained with the secondary antibody only.
Activation of MAPK by TPA does not alter the distribution of the CD95
One possible mechanism by which MAPK activation could affect the CD95 response is by altering the levels of the CD95 protein (Peli et al., 1999) or by changing the cellular compartmentalization of the receptor. It has indeed been indicated that MAPK signaling leads to phosphorylation and internalization of the TNF-R1 (Cottin et al., 1999). Our previous studies, however, did not indicate any alterations in the level or distribution of CD95 after activation of the MAPK signaling pathway (Holmström et al., 1999). To exclude the possibility that MAPK activation would affect the levels of receptor protein, we analyzed the presence of CD95 on the cell surface by FACS analysis in tetradecanoylphorbol 13-acetate (TPA)-treated samples, as TPA is a very efficient MAPK activator and also an efficient inhibitor of CD95-mediated apoptosis (Holmström et al., 1998). According to our results, CD95 levels on the cell surface were not altered by TPA treatment (Figure 6B). These results exclude alterations of CD95 levels or cellular distribution of CD95 as a mechanism behind MAPK-mediated suppression of CD95-induced apoptosis.
MAPK activation does not affect formation of a functional DISC but suppresses overall activation of caspase-8
Previous studies have indicated that only a small amount of caspase-8 cleavage and activation is required to trigger the CD95-mediated apoptotic pathway in Jurkat T cells (Scaffidi et al., 1998, 1999b). In order to examine at which level in the CD95-signaling pathway MAPK activity is suppressing apoptosis, we immunoprecipitated CD95 followed by immunoblotting for caspase-8 from the immunoprecipitates. Jurkat T cells are type II cells that require the mitochondrial amplification loop for execution of CD95-mediated apoptosis, presumably because they form only low amounts of the DISC (Scaffidi et al., 1998, 1999b). The caspase-8 immunoblot from CD95 immunoprecipitates, using both unstimulated control cells and CD95-stimulated cells, shows that caspase-8 is recruited to the DISC (Figure 7A). However, there were no significant differences in recruitment of caspase-8 to the DISC even when MAPK was activated by TPA (Figure 7A). Hence, the DISC seemed to assemble regardless of whether the cells are undergoing apoptosis or not. To test whether the DISC in TPA-treated Jurkat T cells had the capacity to convert procaspase-8 into active caspase-8 subunits, we performed an in vitro caspase-8 cleavage assay (Medema et al., 1997) with the DISC isolated from Jurkat T cells (Figure 7B). In this assay the DISC of unlabeled cells is immunoprecipitated, and 35S-labeled caspase-8/a is added. After incubation for 24 h at 4°C, caspase-8/a will be processed into active fragments if the DISC is active. By western blotting, procaspase-8, as part of the DISC, was barely detectable in the Jurkat T cells (Figure 7A). However, the caspase-8 enzymatic activity present in the immunoprecipitated DISC was sufficient to process and activate caspase-8 at the receptor level (Figure 7B), demonstrating that also the DISC of Jurkat T cells pre-treated with TPA prior to triggering of CD95 was functionally active. However, while the DISC is functional even following MAPK activation, the elevated MAPK suppressed overall caspase-8 activation, as measured by analysis of caspase-8 cleavage in the whole-cell lysates (Figure 7C). The immunoblot of caspase-8 showed a clear decrease in the amount of cleaved caspase-8 in cells pre-treated with OKT3 prior to triggering of CD95 when compared with CD95-stimulation alone, indicating that MAPK activation suppresses a general cleavage and activation of caspase-8 (Figure 7C). This observation is in accordance with our previous studies, indicating that elevated MAPK activity suppresses caspase activation (Holmström et al., 1998, 1999). In conclusion, we could not detect any difference in DISC assembly nor activity after MAPK activation, whereas activation of the cytoplasmic pool of caspase-8 was clearly inhibited.
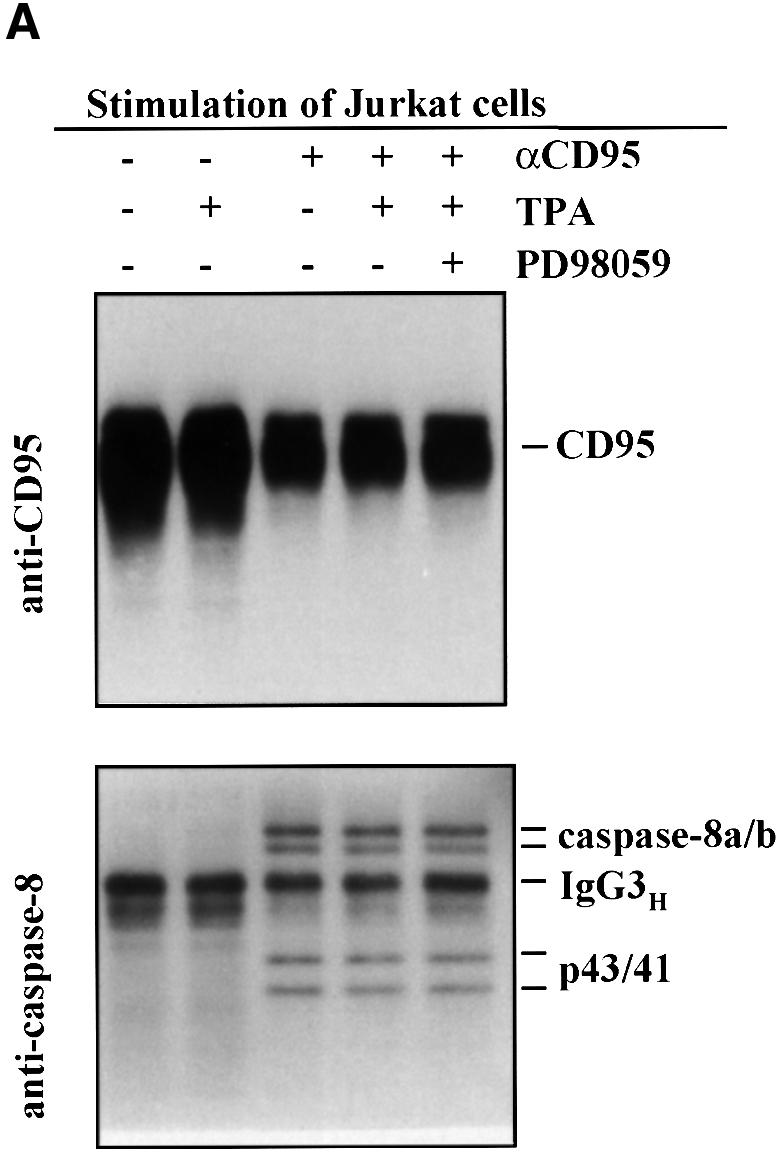
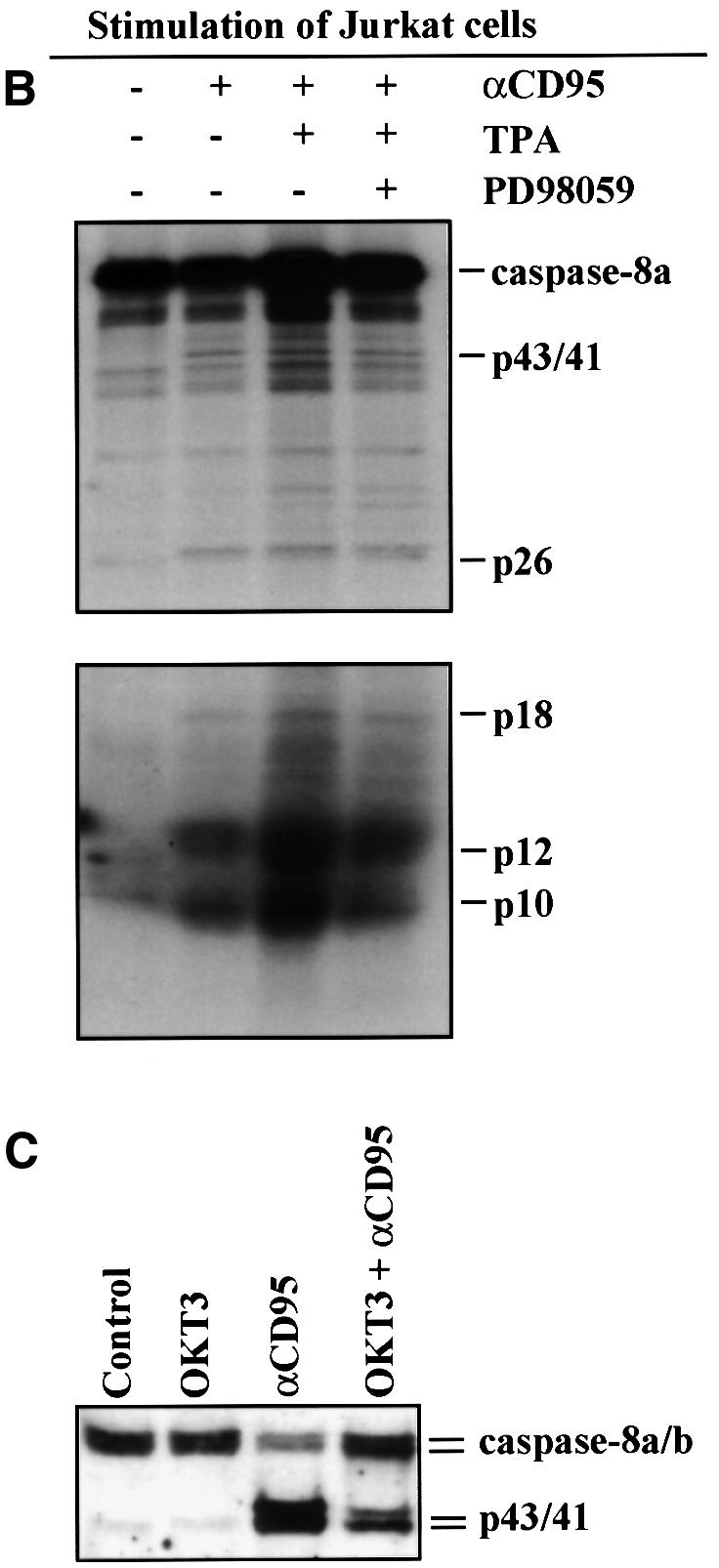
Fig. 7. MAPK activation suppresses overall cleavage of caspase-8 but does not affect formation of a functional DISC. (A) Jurkat T cells were treated as indicated in the figure prior to immunoprecipitation of CD95 from the cell extracts. Subsequently the levels of immunoprecipitated CD95 and associated caspase-8 were detected by western blotting with appropriate antibodies after the respective treatments. A representative immunoblot from three experiments is shown. (B) Jurkat T cells were treated as indicated in the figure prior to immunoprecipitation of CD95 from the cell extracts. Immunoprecipitates were washed four times and incubated with in vitro translated 35S-labeled caspase-8/a. After 24 h the samples were analyzed on 15% SDS–PAGE. The upper part of the gel was exposed for 24 h and the lower part was exposed for 5 days. (C) Jurkat cells were treated as indicated in the figure and the degree of caspase-8 processing in the cells was monitored by western blotting with a caspase-8-specific antibody. Caspase activation can be observed as the appearance of the p43/41 active intermediate fragments of the caspase proforms.
MAPK activation suppresses cleavage of Bid
To narrow down the entry point of the MAPK-mediated inhibitory signal into the CD95 apoptotic signaling pathway, we tested whether the cleavage of Bid would be affected by MAPK activation. Cleavage of the p22 Bid by caspase-8 results in the formation of a p15 Bid protein, which directly affects mitochondria and releases cytochrome c (Li et al., 1998; Luo et al., 1998). To this end, we treated the cells with TPA prior to CD95 triggering and tested whether the effect of TPA could be abolished by pre-treatment with the MKK1 inhibitor PD 98059 (Figure 8A). Activation of MAPK clearly reduced the formation of the p15 Bid formed at 2 h and this protective effect was abolished when the samples were pre-treated with PD 98059, suggesting that MAPK suppresses activation of Bid itself or a target that affects the processing of Bid. A further indication that the Bid-mediated mitochondrial amplification loop is inhibited came from experiments showing that the mitochondrial membrane potential stays unaffected in CD95-stimulated cells in the presence of TPA (data not shown).

Fig. 8. MAPK activation suppresses cleavage of Bid and phosphorylates Bad on Ser112. (A) MAPK-mediated suppression of Bid cleavage. Jurkat T cells were treated as indicated in the figure for 2 h prior to immunoblotting for Bid. The amount of full-length Bid compared with caspase-cleaved p15 was determined by western-blotting. (B) The phosphorylation of Bad on Ser112 was measured by immunoblotting using a phospho-Bad Ser112 antibody after 30 min stimulation with TPA or PD 98059 + TPA. The relative induction (control = 1) was measured by densitometric measurement of the amount of Bad as visualized by an immunoblot using control Bad antibody to the amount of Bad phosphorylated on Ser112.
MAPK activation phosphorylates Bad on Ser112
In addition to suppressing activation and cleavage of Bid, MAPK could also have an effect on other proteins in the mitochondrial apoptosis signaling pathway. An interesting cytosolic substrate for the MAPK pathway is the pro-apoptotic protein, Bad. Bad is known to heterodimerize with Bcl-xL and Bcl-2, both of which suppress CD95-mediated apoptosis downstream of caspase-8 activation in Jurkat T cells (Li et al., 1998; Luo et al., 1998; Scaffidi et al., 1998, 1999b). Bad has recently been shown to be phosphorylated by the MAPK pathway on Ser112, thereby inactivating Bad. This inactivated Bad is held by the 14-3-3 protein, freeing Bcl-xL and Bcl-2 to promote survival (Bonni et al., 1999; Scheid et al., 1999). We therefore wanted to study whether MAPK activation would result in phosphorylation of Bad on Ser112 in Jurkat T cells. The level of Bad phosphorylation on the Ser112 residue was assessed by immunoblotting using a Ser112 phospho-specific Bad antibody. The lowest band in the Ser112 phospho-specific Bad immunoblot (Figure 8B, upper panel) corresponds to the specific band on the Bad immunoblot (Figure 8B, lower panel). Our results clearly show that TPA induces an increase in the phosphorylation of Bad on Ser112, which is abolished when the cells are pre-treated with the specific MKK1 inhibitor PD 98059 (Figure 8B). This indicates that the TPA-generated phosphorylation of Bad on Ser112 is MAPK dependent.
Discussion
The sensitivity of T cells towards TCR-induced AICD varies during the course of an immune response. In the early phase, peripheral T cells are resistant to CD95-mediated apoptosis. However, after successful completion of this process, most of the activated T cells are removed by AICD. This implies that the resistance of T cells to CD95-mediated apoptosis is tightly regulated to ensure homeostasis in the immune system. In our model system, the activated Jurkat T cells showed MAPK-dependent resistance to CD95-mediated apoptosis immediately after T cell activation, independently of the expression levels of the previously implicated CD95 inhibitors FLIP and Bcl-xL. In this regard, post-translational regulation of receptor functions is likely to be significantly faster and more dynamic than regulation through protein synthesis dependent mechanisms. This resistance to CD95-mediated apoptosis is important, as the recently activated T cell will encounter other T cells already expressing the CD95L. If the recently activated T cells would not be protected, they would undergo apoptosis before they had accomplished their tasks. In fact, CD95 stimulation during the early phases of T cell activation may be beneficial, as indicated by studies showing that stimulation of CD95 together with suboptimal stimulation of the TCR induces mitogenic stimulation (Alderson et al., 1993) and that maximal proliferation of T cells through the TCR requires co-stimulatory signals through CD95 (Suzuki and Fink, 1998). In further support for a role of CD95 in T cell growth are studies showing that T cells lacking FADD or expressing a dominant-negative mutant FADD show impaired TCR-stimulated proliferation and AICD (Newton et al., 1998; Walsh et al., 1998; Zhang et al., 1998).
MAPK activation suppresses CD95-mediated apoptosis during T cell activation
When the time-frame of MAPK activation was related to the suppression of CD95-mediated apoptosis in Jurkat T cells, it was obvious that OKT3 induced a rapid MAPK activation and suppression of CD95-mediated apoptosis, both of which lasted as long as the MAPK activity stayed elevated. Interestingly, OKT3 stimulation for 24 h induced CD95L-mediated autocrine suicide in Jurkat T cells (Dhein et al., 1995). These observations are consistent with the hypothesis that T cell activation has a dual role in both inhibiting and promoting apoptosis. Our results indicate that MAPK activation mediates a protective signal during the initial phases of activation, which is turned off towards the end of the immune response to allow the apoptotic CD95 signal to be executed. The physiological significance of MAPK-dependent protection against CD95-mediated apoptosis was further corroborated by our results demonstrating that the specific MKK1 inhibitor, PD 98059, sensitizes peripheral blood T cells to CD95-mediated apoptosis. These data imply that MAPK activation plays an important role in suppressing CD95-mediated apoptosis during T cell activation, and in regulating the duration and strength of the immune response. This may also explain the above-mentioned implications of synergistic effects between CD95 and the TCR.
MAPK-mediated protection of CD95-induced apoptosis is not dependent on protein synthesis
It is well established that many cell types resistant to CD95- or TNF-R1-mediated apoptosis can be sensitized by pre-incubation with protein synthesis inhibitors such as CHX. Accordingly, it has been suggested that stimulation of CD95 and TNF-R1 can elicit a distinct protein synthesis-dependent protective signal (Wallach, 1997) and that viral as well as cellular proteins suppress CD95-mediated apoptosis (Tschopp et al., 1999). In particular, upregulation of c-FLIP (Irmler et al., 1997; Yeh et al., 1998) and Bcl-xL (Peter et al., 1997) during T cell activation has been indicated to suppress CD95-mediated apoptosis. In this respect, it has been suggested that MAPK suppresses CD95-mediated apoptosis through elevated expression of c-FLIP (Yeh et al., 1998). However, it should be noted that this study is based solely on RNA data and therefore does not reveal the possible effects on actual protein levels. Furthermore, a recent study indicates that the role of FLIP in protecting activated T cells is still unclear (Scaffidi et al., 1999a), which is in accordance with the present study, as we could not find a major role for FLIP. Apart from the protein inhibitor-based modulation of CD95 responses, there are reports demonstrating signals protecting from CD95-mediated apoptosis that occur post-translationally through phosphorylation-based signaling pathways, such as MAPK (Holmström et al., 1998, 1999; Ruiz-Ruiz et al., 1999; Wilson et al., 1999) and PI-3K (Häusler et al., 1998; Varadhachary et al., 1999). It is quite plausible that activation of MAPK would generate both a protein synthesis-dependent and -independent mechanism. In this scenario, both the protein synthesis-dependent and -independent effect would be important during different stages of T cell activation. This hypothesis is in accordance with a recent study showing both a protein synthesis-dependent and -independent MAPK-mediated protective effect of serum withdrawal-induced apoptosis in neurons (Bonni et al., 1999). The results presented in this study, showing that OKT3 suppresses CD95-mediated apoptosis in the presence of CHX, preclude the requirement for protein synthesis in the MAPK-mediated protection against CD95-induced apoptosis, which is in agreement with our previous studies (Holmström et al., 1998, 1999). Thus, a high constitutive MAPK activity seems to be sufficient for inhibition of the CD95-mediated apoptotic signal, which does not rule out additional protein synthesis-dependent protective mechanisms that could regulate CD95 responses during the later phase of T cell activation. Recently, FLIP has been shown to promote MAPK signaling following CD95 activation (Kataoka et al., 2000) in resistant T cells. It is tempting to speculate that the initial CD3-induced MAPK activation would tune FLIP into a MAPK-directed signaling mode.
MAPK activation does not affect formation of a functional DISC but suppresses overall cleavage of caspase-8 and Bid
It is important to determine where in the CD95-mediated apoptotic signaling pathway MAPK is exerting its protective effect, in order to assess whether the cells still maintain their normal functions when CD95 is stimulated but the apoptotic response is not triggered. Our results show that MAPK activation does not suppress formation of a functional DISC. However, MAPK inhibits DISC-mediated activation of overall caspase-8 activity. This could take place by inhibition of caspase-8 processing, although the DISC is assembled, resulting in inhibition of both overall caspase activation and activation of the mitochondrial amplification loop through Bid. Interest ingly, the PI-3K pathway has similarily been shown to restrict caspase-8 processing in CD95-resistant Th-2 cells, although the DISC is assembled (Varadhachary et al., 1999). Jurkat T cells are assumed to be type II cells in terms of the CD95 response, and in these cells caspase-8 is mainly activated after the mitochondrial amplification loop (Scaffidi et al., 1998). Therefore, the lack of caspase-8 activation hints at a defect in the mitochondrial amplification loop. In this respect, we found further evidence that MAPK acts upstream of mitochondria, as we observed that the cleavage of Bid by caspase-8 is inhibited in a MAPK-dependent manner, as well as the CD95-induced loss of the mitochondrial membrane potential (data not shown). Recently, it has been shown that Bid is a direct target of caspase-8 (Li et al., 1998; Luo et al., 1998) and that this cleavage can be inhibited by TPA treatment (Scaffidi et al., 1999b). Processed p15 Bid translocates to mitochondria causing release of cytochrome c. The assumption that MAPK activation inhibits the mitochondrial amplification loop in CD95-mediated apoptosis is further strengthened by data showing that TPA only inhibits CD95-mediated apoptosis in cells that require release of cytochrome c for the apoptotic response (type II cells), whereas TPA does not affect CD95-mediated apoptosis in cells where the apoptotic response depends solely on caspases (type I cells; Scaffidi et al., 1999b). The inhibition of apoptosis at the early steps in the apoptotic pathway is likely to be desirable, as only this way could cells escape from any harmful effects of a partially activated apoptotic effector machinery. This type of regulation would also be useful in situations where initiator caspase activity is required for signaling other than activation of apoptotic effector caspases. In fact, it has recently been shown that caspase activation would be required for normal T cell development (Kennedy et al., 1999), an observation that would warrant a direct regulatory mechanism of caspases that can be dynamically modified to enable direction of the signal according to a given situation.
Possible downstream targets of MAPK affecting CD95-mediated apoptosis
An interesting cytosolic substrate for MAPK is Bad, a pro-apoptotic protein that functions in the regulation of the transmembrane potential in mitochondria. Bad is known to be phosphorylated by the PI-3K pathway on Ser112 and Ser136 (Datta et al., 1997; del Peso et al., 1997). Phosphorylation of Bad at these sites inhibits binding of Bad to Bcl-xL and induces binding of Bad to the 14-3-3 proteins, thereby inactivating Bad (Datta et al., 1997; del Peso et al., 1997) and freeing Bcl-xL and Bcl-2 to promote survival. Recent reports indicate that cell survival by the MAPK signaling pathway would be due to a similar mechanism. In these reports the MAPK pathway is demonstrated to phosphorylate Bad on Ser112, thereby dissociating it from Bcl-xL (Bonni et al., 1999; Scheid et al., 1999). Our results indicate a similar MAPK-dependent phosphorylation of Bad on Ser112, thus implying MAPK-mediated phosphorylation of Bad as a possible parallel mechanism for promoting cell survival. While there is no previous evidence of Bad being involved in direct regulation of CD95 responses, its phosphorylation could add another level of cell survival promoting activity in T cells, i.e. one inhibitory signal is targeted directly at CD95 and another at the general mitochondrial activation mechanism. In comparison, it has been shown that the PI-3K pathway is able to phosphorylate and affect the activity of several proteins regulating the apoptotic process (Datta et al., 1999).
While our study conclusively shows that CD95 responses are regulated by direct MAPK-mediated signaling, further characterization and understanding of the biological roles of phosphorylation-based signaling mechanisms in CD95 functions will be important in order to utilize these mechanisms for modulation of CD95 responses.
Materials and methods
Cell culture
The human leukemic T cell line, Jurkat (clone EG-1; ATCC, Manassas, VA), was cultured in RPMI 1640 medium supplemented with 10% inactivated fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin in a humidified incubator with 5% CO2 in air at 37°C. The cells were kept at a density of 0.5–1.0 × 106/ml. To test the effects of TCR activation, the cells were incubated at a density of 2 × 106 with 100 ng/ml mouse anti-human CD95 IgM antibody (Kamiya Biomedical Company, Thousand Oaks, CA), or 1 µg/ml anti-mouse CD95 IgG3 antibody (Jo2, PharMingen, San Diego, CA) for the indicated time periods, in the absence or presence of 100 µg/ml immobilized OKT3 (a kind gift from the R.W.Johnson Pharmaceutical Research Institute, Bassersdorf, Switzerland). OKT3 was immobilized by pre-incubating the wells with 100 µg/ml protein A (Sigma, St Louis, MO) in a 50 mM Na2HCO3/NaH2CO3 buffer pH 9.8 for 4 h in 37°C followed by incubation with 100 µg/ml OKT3 for 4 h at 37°C. The effect of MAPK activation was determined by pre-treating the cells with 20 nM TPA (Sigma), in the presence and absence of 100 µM CHX (Sigma), or by pre-treating the cells for 30 min with 30 µM PD 98059 (Calbiochem, La Jolla, CA) before CD95 stimulation.
Human peripheral T cells were prepared as described elsewhere (Klas et al., 1993). Resting peripheral T cells were separated from whole blood using Hypaque medium, cultured in RPMI medium (10% FCS), at a density of 0.5–1.0 × 106/ml, activated by addition of 1 µg/ml PHA (Sigma) for 24 h, and further stimulated 2 days later by addition of 50 U/ml interleukin-2 (Sigma).
Analysis of DNA fragmentation and phosphatidylserine exposure
Detection of DNA fragmentation into oligonucleosomal DNA fragments by agarose gel electrophoresis was performed as described previously (Holmström et al., 1998). To detect phosphatidylserine exposure by flow cytometry, Jurkat T cells and the primary peripheral T cells were washed once with phosphate-buffered saline (PBS) and incubated for 10 min on ice in 400 µl of binding buffer (2.5 mM HEPES–NaOH pH 7.4, 35 mM NaCl, 0.625 mM CaCl2) with 1 µl annexin V–FITC (Alexis, Laufelfingen, Switzerland) and 10 µg/ml propidium iodide (Molecular Probes, Eugene, OR) and analyzed on a FACScan flow cytometer (Becton Dickinson, NJ).
Analysis of CD95 expression
The effect of MAPK activation on CD95 expression on the cell surface was followed by fixing TPA-treated Jurkat T cells (15 and 30 min) with 3% paraformaldehyde, and incubating the cells in the presence of an anti-APO-1 antibody, followed by an FITC-conjugated secondary antibody (Zymed, San Francisco, CA) and detection by using a FACScan flow cytometer.
Immunoblotting techniques
The levels of Bad, Bcl-2, Bcl-xL and FLIP were followed by immunoblotting the respective proteins after treatment with 100 µg/ml immobilized OKT3 for different times. Phosphorylation of Bad on Ser112 and cleavage of Bid were followed after the respective treatments that affected MAPK activity.
Immunoblotting was performed by lysing cells in Laemmli sample buffer and then resolving the proteins on 12.5% SDS–PAGE. The separated proteins were transferred to a nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany) and probed with the specific antibody to Bad (Transduction Laboratories, Lexington, KY), phospho-Bad Ser112 (New England Biolabs, Boston, MA), Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), Bcl-xL (Transduction laboratories), Bid (Santa Cruz Biotechnology) and c-FLIPL (Alexis), respectively. The proteins were visualized by the ECL system (Amersham, Buckinghamshire, UK) after addition of appropriate HRP-conjugated secondary antibodies.
MAPK activity assays
The MAPK assay was carried out as described previously (Holmström et al., 1998). After immunoprecipitation of the kinase, it was incubated in the presence of 1 mg/ml myelin basic protein (MBP; Sigma) as substrate, using a kinase assay buffer with 25 µM ATP, 2.5 µCi of [γ-32P]ATP (Amersham). The reaction was carried out for 15 min at 37°C and stopped by addition of 3× Laemmli sample buffer. The samples were resolved on 12.5% SDS–PAGE, and MBP phosphorylation was quantified with a phosphoimager (Bio-Rad Laboratories, Hercules, CA). In parallel with the immunocomplex kinase assays, we determined the amount of MAPK2 in the immunoprecipitates by using a rabbit anti-human MAPK1 and 2 antibody (New England Biolabs). After coupling to a secondary antibody (Zymed), the proteins were visualized with the ECL system.
Transfection studies
Cells were transiently transfected by using DEAE dextran (Pharmacia LKB, Stockholm, Sweden) as described previously (Holmström et al., 1998). Cells were allowed to rest for 48 h before the respective treatments. The DNA construct used was pMCL-HA-MKK1-K97M, encoding an HA-tagged dominant-negative form of the MKK1 (Mansour et al., 1996), a kind gift from Natalie Ahn (University of Colorado, CO). For detection of transfected cells, the cells were collected by centrifugation, resuspended in PBS, and fixed on ice for 1 h with 3% formaldehyde in PBS. The cells were then washed with PBS and permeabilized with 0.2% NP-40 (Sigma) for 10 min at room temperature. Detection of the transfected cells was performed with 10 µg/ml monoclonal HA-specific antibody (12CA5, Boehringer Mannheim, Germany) followed by an FITC-conjugated anti-mouse secondary antibody and 10 mg/ml Hoechst (Molecular Probes). Cells were mounted in 50% glycerol and viewed under a Leica RMB epifluorescence microscope. Mock transfections were carried out using a pIRES-EGFP plasmid (Clontech Inc., Palo Alto, CA).
Analysis of caspase-8 recruited to the DISC by western blotting
The amount of DISC-associated caspase-8 was determined as follows: 107 cells were either treated with 2 µg/ml anti-APO-1 antibody for 5 min at 37°C and then lysed in lysis buffer [30 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), small peptide inhibitors 1% Triton X-100 (Sigma) and 10% glycerol] (stimulated condition) or lysed and then supplemented with anti-APO-1 (unstimulated condition). The CD95 DISC was then precipitated for 2 h at 4°C with protein A–Sepharose. After immunoprecipitation the beads were washed five times with 1 ml of lysis buffer. Caspase-8 was detected from the immunoprecipitates by western blotting with a caspase-8-specific antibody.
In vitro translation and in vitro cleavage assay of caspase-8
Caspase-8/a was in vitro translated using a T7 polymerase-directed reticulate lysate system (TNT, Promega, Madison, WI). In vitro cleavage assays were performed as follows. CD95 DISC was immunoprecipitated from 5 × 107 cells as described above. Subsequently, the beads (containing the DISC) were incubated in 50 µl of reaction buffer (50 mM HEPES pH 7.4, 100 mM NaCl, 0.1% CAPS, 10 mM dithiothreitol and 20% sucrose) for 24 h at 4°C with 0.5 µl of in vitro translated caspase-8/a. After boiling for 3 min in a standard reducing sample buffer the resulting products were analyzed on 15% SDS–PAGE with subsequent amplification (Amplify, Amersham Pharmacia Biotech), drying of the gels and autoradiography.
Acknowledgments
Acknowledgements
We thank Natalie Ahn (University of Boulder, CO) for the MKK1 plasmid and the R.W.Johnson Pharmaceutical Research Institute (Bassersdorf, Switzerland) for providing the OKT3 antibody. Financial support from the Academy of Finland (grant 35718), the Sigrid Jusélius Foundation, the Erna and Victor Hasselblad Foundation, the Finnish Cancer Foundation and the Cell Signaling Program of Åbo Akademi University is gratefully acknowledged.
References
- Alderson M.R., Armitage,R.J., Maraskovsky,E., Tough,T.W., Roux,E., Schooley,K., Ramsdell,F. and Lynch,D.H. (1993) Fas transduces activation signals in normal human T lymphocytes. J. Exp. Med., 178, 2231–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R., Cuenda,A., Cohen,P., Dudley,D.T. and Saltiel,A.R. (1995) PD 098059 is a specific inhibitor of the mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem., 270, 27489–27494. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Goncharov,T.M., Goltsev,Y.V. and Wallach,D. (1996) Involvement of MACH a novel MORT/Fadd-interacting protease in Fas/APO-1 and TNF receptor induced cell death. Cell, 85, 803–815. [DOI] [PubMed] [Google Scholar]
- Bonni A., Brunet,A., West,A.E., Datta,S.R., Takasu,M.A. and Greenberg,M.E. (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science, 286, 1358–1362. [DOI] [PubMed] [Google Scholar]
- Brunner T. et al. (1995) Cell autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature, 373, 441–444. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O’Rourke,K., Tewari,M. and Dixit,V.M. (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell, 81, 505–512. [DOI] [PubMed] [Google Scholar]
- Cottin V., Van Linden,A. and Riches,D.W.H. (1999) Phosphorylation of tumor necrosis factor receptor CD120a (p55) by p42mapk/erk2 induces changes in its subcellular localization. J. Biol. Chem., 274, 32975–32987. [DOI] [PubMed] [Google Scholar]
- Cryns V. and Yuan,J. (1998) Proteases to die for. Genes Dev., 12, 1551–1570. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek,H., Tao,X., Masters,S., Fu,H., Gotoh,Y. and Greenberg,M.E. (1997) Akt phosphorylation of BAD couples survival signals to cell-intrinsic death machinery. Cell, 91, 231–241. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet,A. and Greenberg,M.E. (1999) Cellular survival: a play in three Akts. Genes Dev., 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- del Peso L., Gonzalez-Garcia,M., Page,C., Herrera,R. and Nuñez,G. (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science, 278, 687–689. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak,H., Bäumler,C., Debatin,K.-M. and Krammer,P.H. (1995) Autocrine T-cell suicide mediated by APO-1(/Fas/CD95). Nature, 373, 438–440. [DOI] [PubMed] [Google Scholar]
- Häusler P., Papoff,G., Eramo,A., Reif,K., Cantrell,D.A. and Ruberti,G. (1998) Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur. J. Immunol., 28, 57–69. [DOI] [PubMed] [Google Scholar]
- Holmström T.H., Chow,S.C., Elo,I., Coffey,E., Orrenius,S., Sistonen,L. and Eriksson,J.E. (1998) Suppression of Fas/APO-1-mediated apoptosis by the p42/p44 mitogen-activated kinases. J. Immunol., 160, 2626–2636. [PubMed] [Google Scholar]
- Holmström T.H., Tran,S.E.F., Johnson,V.L., Ahn,N., Chow,S.C. and Eriksson,J.E. (1999) Inhibition of mitogen-activated kinase signaling sensitizes to Fas-receptor-mediated apoptosis. Mol. Cell. Biol., 19, 5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M. et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature, 388, 190–194. [DOI] [PubMed] [Google Scholar]
- Itoh N., Yonehara,S., Ishii,A., Yonehara,M., Mizushima,S.-I., Sameshima,M., Hase,A., Seto,Y. and Nagata,S. (1991) The polypeptide encoded by the cDNA for human cell surface antigen can mediate apoptosis. Cell, 66, 233–243. [DOI] [PubMed] [Google Scholar]
- Ju S.-T., Panka,D.J., Cui,H., Ettinger,R., El-Khatib,M., Sherr,D.H., Stanger,B.Z. and Marshak-Rothstein,A. (1995) Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature, 373, 444–448. [DOI] [PubMed] [Google Scholar]
- Kataoka T. et al. (2000) The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol., 10, 640–648. [DOI] [PubMed] [Google Scholar]
- Kennedy N.J., Kataoka,T., Tschopp,J. and Budd,R.C. (1999) Caspase activation is required for T cell proliferation. J. Exp. Med., 190, 1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt,S., Behrmann,I., Germer,M., Pawlita,M., Krammer,P.H. and Peter,M.E. (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J., 14, 5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klas C., Debatin,K.M., Jonker,R.R. and Krammer,P.H. (1993) Activation interferes with the APO-1 pathway in mature human T cells. Int. Immunol., 5, 625–630. [DOI] [PubMed] [Google Scholar]
- Lewis T.S., Shapiro,P.S. and Ahn,N.G. (1998) Signal transduction through MAP kinase cascades. Adv. Cancer Res., 74, 49–139. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu,H., Xu,C.J. and Yuan,J. (1998) Cleavage of BID by caspase-8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell, 94, 491–501. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo,I., Zou,H., Slaughter,C. and Wang,X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell, 94, 481–490. [DOI] [PubMed] [Google Scholar]
- Mansour S.J., Candia,J.M., Matsuura,J.E., Manning,M.C. and Ahn,N.G. (1996) Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry, 35, 15529–15536. [DOI] [PubMed] [Google Scholar]
- Medema J.P., Scaffidi,C., Kischkel,F.C., Shevchenko,A., Mann,M., Krammer,P.H. and Peter,M.E. (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J., 16, 2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M. et al. (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell, 85, 817–827. [DOI] [PubMed] [Google Scholar]
- Nagata S. and Golstein,P. (1995) The Fas death factor. Science, 267, 1449–1456. [DOI] [PubMed] [Google Scholar]
- Newton K., Harris,A.W., Bath,M.L., Smith,K.G. and Strasser,A. (1998) A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J., 17, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehm A. et al. (1992) Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor superfamily. Sequence identity with the Fas antigen. J. Biol. Chem., 267, 10709–10726. [PubMed] [Google Scholar]
- Peli J., Schroter,M., Rudaz,C., Hahne,M., Meyer,C., Reichmann,E. and Tschopp,J. (1999) Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J., 18, 1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M.E., Kischkel,F.C., Scheuerpflug,C.G., Medema,J.P., Debatin,K.-M. and Krammer,P.H. (1997) Resistance of cultured peripheral T cells towards activation-induced cell death involves a lack of recruitment of FLICE (MACH/caspase 8) to the CD95 death-inducing signaling complex. Eur. J. Immunol., 27, 1207–1212. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ruiz M.C., Robledo,G., Font,J., Izquierdo,M. and Lopez-Rivas,A. (1999) Protein kinase C inhibits CD95 (Fas/APO-1)-mediated apoptosis by at least two different mechanisms in Jurkat T-cells. J. Immunol., 163, 4737–4746. [PubMed] [Google Scholar]
- Scaffidi C., Fulda,S., Srinivasan,A., Friesen,C., Li,F., Tomaselli,K.J., Debatin,K.-M., Krammer,P.H. and Peter,M.E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J., 17, 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz,I., Krammer,P.H. and Peter,M.E. (1999a) The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem., 274, 1541–1548. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz,I., Zha,J., Korsmeyer,S.J., Krammer,P.H. and Peter,M.E. (1999b) Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem., 274, 22532–22538. [DOI] [PubMed] [Google Scholar]
- Scheid M.P., Schubert,K.M. and Duronio,V. (1999) Regulation of Bad phosphorylation and association with Bcl-xL by the MAPK/Erk kinase. J. Biol. Chem., 274, 31108–31113. [DOI] [PubMed] [Google Scholar]
- Suda T., Takahashi,T., Golstein,P. and Nagata,S. (1993) Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell, 75, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Suzuki I. and Fink,P.J. (1998) Maximal proliferation of cytotoxic T lymphocytes reguires reverse signaling through Fas ligand. J. Exp. Med., 187, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J., Martinon,F. and Hofmann,K. (1999) Apoptosis: silencing the death receptors. Curr. Biol., 9, 381–384. [DOI] [PubMed] [Google Scholar]
- Varadhachary A.S., Peter,M.E., Perdow,S.N., Krammer,P.H. and Salgame,P. (1999) Selective up-regulation of phosphatidylinositol 3-kinase activity in Th2 cells inhibits caspase-8 cleavage at the death-inducing signaling complex: a mechanism for Th2 resistance from Fas-mediated apoptosis. J. Immunol., 163, 4772–4779. [PubMed] [Google Scholar]
- Wallach D. (1997) Cell death induction by TNF: a matter of self control. Trends Biochem. Sci., 22, 107–109. [DOI] [PubMed] [Google Scholar]
- Walsh C.M., Wen,B.G., Chinnaiyan,A.M., O’Rourke,K., Dixit,V.M. and Hedrick,S.M. (1998) A role for FADD in T-cell activation and development. Immunity, 8, 439–449. [DOI] [PubMed] [Google Scholar]
- Wilson D.J., Alessandrini,A. and Budd,R.A. (1999) MEK1 activation rescues Jurkat T-cells from Fas-induced apoptosis. Cell. Immunol., 194, 67–77. [DOI] [PubMed] [Google Scholar]
- Xia Z., Dickens,M., Raingeaud,J., Davis,R.J. and Greenberg,M.E. (1995) Opposing effects of ERK and p38 MAP kinases on apoptosis. Science, 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yeh J.-H., Hsu,S.-C., Han,S.-H. and Lai,M.-Z. (1998) Mitogen-activated protein kinase kinase antagonized Fas-associated death domain protein-mediated apoptosis by induced FLICE-inhibitory protein expression. J. Exp. Med., 188, 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Cado,D., Chen,A., Kabra,N.H. and Winoto,A. (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature, 392, 296–300. [DOI] [PubMed] [Google Scholar]