Inflammatory Concepts of Obesity (original) (raw)
Abstract
Obesity, long considered a condition characterized by the deposition of inert fat, is now recognized as a chronic and systemic inflammatory disease, where adipose tissue plays a crucial endocrine role through the production of numerous bioactive molecules, collectively known as adipokines. These molecules regulate carbohydrate and lipid metabolism, immune function and blood coagulability, and may serve as blood markers of cardiometabolic risk. Local inflammatory loops operate in adipose tissue as a consequence of nutrient overload, and crosstalk among its cellular constituents-adipocytes, endothelial and immune cells-results in the elaboration of inflammatory mediators. These mediators promote important systemic effects that can result in insulin resistance, dysmetabolism and cardiovascular disease. The understanding that inflammation plays a critical role in the pathogenesis of obesity-derived disorders has led to therapeutic approaches that target different points of the inflammatory network induced by obesity.
1. Introduction
Atherothrombosis is the basis of most coronary, peripheral, and cerebral arterial disease and is a critical health burden and major cause of death worldwide [1]. Despite the undeniable importance of cardiovascular disease in morbidity and mortality in most regions of the world, control of risk factors and advances in the treatment of atherothrombosis have significantly reduced age-adjusted cardiovascular events in USA and Western Europe. However, all this progress in the war against cardiovascular disease has been threatened by the dramatic increase in the prevalence of obesity, an important risk factor for both atherogenesis and increased coagulability [1].
The significant advance of the obesity epidemic worldwide and the association between atherothrombosis and obesity have attracted great interest from the scientific community, contributing importantly to increase the understanding of the pathophysiology of excess adiposity. Indeed, several concepts related to obesity pathophysiology have changed in the last 2 decades [2]. The hypothesis of obesity as a low-grade chronic and systemic inflammatory disease gradually replaced the idea of a mere lipid deposit disease characterized by inert adipose tissue and passive accumulation of fat in the context of weight gain [2, 3]. Several research groups demonstrated that adipose tissue of obese animals and humans produces increased amounts of inflammatory mediators and presents higher number of inflammatory cells compared to adipose tissue of lean controls [2, 3]. This recently recognized endocrine role of adipose tissue likely provides a crucial mechanistic link between obesity and atherothrombosis.
2. Inflammatory Mechanisms of Obesity
2.1. The Local Inflammatory Network in Adipose Tissue
The first clues supporting the involvement of inflammation in obesity came to light almost half a century ago, including a report that described increased plasma levels of fibrinogen in obese patients [4]. However, the inflammatory view of obesity started attracting interest in the 1990s, particularly after the demonstration of enhanced expression of tumor necrosis factor-alpha (TNF-α) in adipose tissue of obese rodents and the amelioration of insulin resistance after neutralization of this potent cytokine [5, 6]. Since the publication of these reports, several other groups demonstrated the production and secretion of multiple cytokines, chemokines, hormones, and other inflammatory mediators by adipose tissue, collectively referred to as adipokines, culminating with the recognition of adipose tissue as one of the greatest endocrine organs in the body [7]. Besides the aforementioned cytokine TNF-α, macrophage chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1), interleukin-6 (IL-6), leptin, and adiponectin are remarkable examples of adipokines differentially expressed by obese adipose tissue, with potentially important roles in the pathophysiology of obesity, either locally or systemically [7–11].
Adipocytes constitute the major cell type of adipose tissue, being a major source of several bioactive products secreted by this tissue. Indeed, they are responsible for the production of several adipokines, among which adiponectin and leptin are likely exclusively elaborated in adipocytes. Pioneer work published in 2003 demonstrated that adipocytes are not alone and that macrophages also accumulate in adipose tissue of obese animals (Table 1 and Figure 1), coinciding with increased expression of inflammatory markers and preceding a significant increase in circulating insulin levels [12, 13]. Since then, extensive work has supported the involvement of macrophages in the inflammatory network of adipose tissue. Studies on the contribution of MCP-1 and its receptor, chemokine (C-C motif) receptor 2 (CCR2), both important mediators of macrophage recruitment to sites of inflammation (Figure 1), demonstrated reduced macrophage accumulation in adipose tissue of diet-induced obese mice deficient in either of those genes [22, 23]. These knockout animals exhibit decreased insulin resistance and hepatic steatosis, suggesting that adipose tissue macrophage influx contributes to the local and systemic metabolic effects of obesity [22, 23]. However, the role of the MCP-1_/_CCR2 duo in obesity-induced inflammation remains incompletely understood, as not all studies found the influence of MCP-1 deficiency relevant to macrophage accumulation in adipose tissue or insulin sensitivity [24, 25].
Table 1.
Inflammatory cell types in adipose tissue.
| Inflammatory cells in adipose tissue network of obesity | Comments |
|---|---|
| Macrophages | Accumulate in obese versus lean AT. Presence correlated with ↑ expression of inflammatory mediators in AT and metabolic disturbances [12, 13]. |
| Infiltrative macrophages and M1 markers predominate over resident macrophages and M2 markers in obese AT [14]. | |
| T cells | Fewer than macrophages, they also accumulate in obese versus lean AT [15–17]. |
| Depletion of CD8 lymphocytes from DIO-mice ↓ macrophage accumulation in AT and ↓ systemic IR [18]. | |
| CD4+ Foxp3+ T regulatory cells (Treg) decrease in AT of obese versus lean mice. Treg cells may keep homeostasis and limit inflammation in lean AT [19]. | |
| Mast cells | Genetic deficiency of mast cells or their pharmacological stabilization in DIO-mice ↓ weight gain, AT, and systemic inflammation, and improve glucose metabolism and energy expenditure [20]. |
| NKT cells | DIO-mice lacking NKT cells present less AT inflammation and glucose intolerance than wild-type control animals [21]. |
Figure 1.
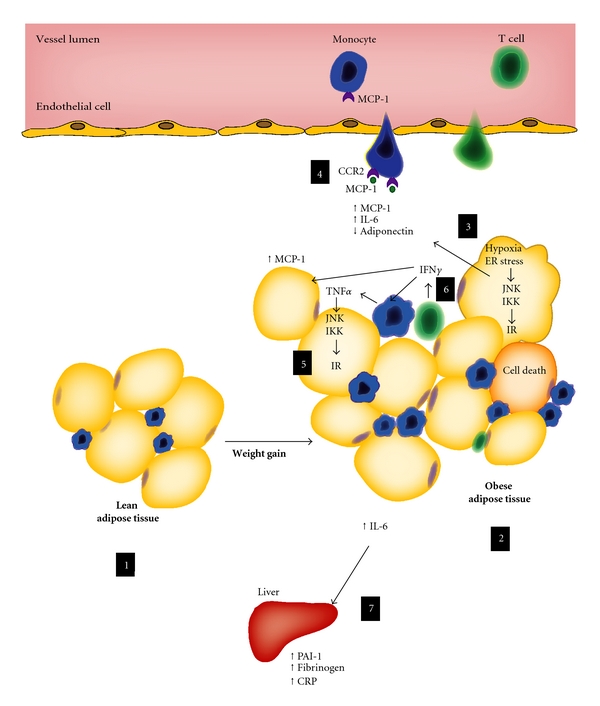
Adipose tissue inflammation in obesity. Whereas lean adipose tissue contains a population of resident inflammatory cells (1) and secretes various active substances, the obese adipose tissue (2) accumulates higher numbers of macrophages and T cells, producing copious amounts of inflammatory mediators, such as monocyte chemoattractant protein-1 (MCP-1) and interleukin-6 (IL-6), and less adiponectin (3). In the context of nutrient surplus and hypoxia, expanding adipocytes present endoplasmic reticulum (ER) stress (3), important trigger of inflammatory kinases, such as JNK and IKK, which can ultimately inhibit insulin signaling (further detail in the text) and activate inflammatory cascades and the production of inflammatory mediators. Existing evidence suggests that higher production of chemokines, such as MCP-1, within the obese adipose tissue could enhance local macrophage accumulation (4). Once in the tissue, monocyte-derived macrophages can be a fundamental source of tumor necrosis factor-alpha (TNF_α_), among other mediators. Cytokines like TNF_α_ and other stimuli can cause further activation of inflammatory kinases (5). Several studies have demonstrated that T cells also accumulate in adipose tissue in the obese state (6). Interferon-gamma (IFN_γ_), a typical T-helper 1 cytokine, likely regulates local expression of TNF_α_, MCP-1, and other inflammatory mediators, suggesting a role for adaptive immunity in obesity pathophysiology. The spillover of adipokines, such as IL-6, into the circulation can also promote important systemic effects (7), such as increased production of liver-derived acute-phase inflammatory mediators and coagulation-related factors, most of them likely correlated with atherothrombosis.
Macrophages are not all equal in the inflamed adipose tissue, according to differential phenotypic patterns and chemokine receptor usage [26]. Whereas the so-called resident macrophages, the dominant subtype in lean fat tissue, predominantly express markers of alternative activation or M2 (such as mannose receptor C type I), the infiltrative macrophages, widely present within the obese adipose tissue, are characterized by their enhanced expression of classic activation or M1 markers, such as TNF-α and inducible NO synthase (iNOS) [14]. Interestingly, adipose tissue-derived macrophages from obese CCR2-deficient mice present significantly less expression of M1 markers than their wild-type counterparts, with M2 markers at levels comparable to those from lean mice [14]. Thus, although CCR2-deficient animals have a less prominent subset of infiltrative macrophages within their adipose tissue, their population of adipose tissue-resident macrophages remains intact, suggesting the usage of distinct chemokine receptors by different subsets of macrophages and local operation of various chemotactic systems [14].
In the last five years, other inflammatory cell types have also been gathering attention in the pathophysiology of obesity. T cells, although less numerous than macrophages, also accumulate in adipose tissue of obese mice (Table 1 and Figure 1). We and others showed the presence of both T cell subpopulations, CD4+ and CD8+, in fat tissue [15–18]. Nishimura and colleagues reported that mice fed a high-fat diet have an increased number of CD8 cells in adipose tissue and that depletion of these cells reduced macrophage infiltration and adipose tissue inflammation and improved systemic insulin resistance [18].
The classic Th1 cytokine interferon-gamma (IFN-γ) also figures importantly in the inflammatory circuit that operates in obese adipose tissue (Figure 1). In one study, high-fat diet promoted a progressive IFN-γ bias among adipose tissue-derived T cells in mice [27]. Furthermore, IFN-γ or IFN-_γ_-receptor deficiencies lower adipose tissue expression of inflammatory genes and ameliorate metabolic parameters in obese animals [16]. In humans, a positive association of CD3+cells and IFN-γ mRNA expression in adipose tissue with waist circumference in a cohort of patients with type 2 diabetes mellitus suggests the involvement of the Th1 arm of adaptive immunity in obesity-related metabolic disorders [17].
Whereas proinflammatory T cells appear enriched in obese adipose tissue, the pool of anti-inflammatory T cells, CD4+ Foxp3+ T regulatory (Treg) cells, decreases in fat tissue of obese animals compared to their lean counterparts [19] (Table 1). Using loss-of-function and gain-of-function approaches, Feuerer and colleagues revealed that Treg cells influence the inflammatory state of adipose tissue and insulin resistance. The higher number of Treg cells in lean fat tissue may be one important factor to restrain inflammation and keep local homeostasis [19, 27]. In humans, Th1 cells expressing the transcription factor Tbet outnumber Foxp3+ T cells with a ratio of approximately 12 : 1 in visceral adipose tissue of obese individuals, compared to 6 : 1 in lean ones [27].
Recent studies highlighted potential roles for other immune cells, such as mast cells and natural killer T (NKT) cells, in adipose tissue inflammation [20, 21] (Table 1). Genetic deficiency of mast cells or their pharmacological stabilization in diet-induced obese mice reduce weight gain, adipose tissue, and systemic inflammation and improve glucose metabolism and energy expenditure [20]. Mice lacking NKT also present less adipose tissue inflammation and glucose intolerance than wild-type control animals when fed a high-fat diet [21]. A recent study has reported an important role of eosinophils in the maintenance of metabolic homeostasis [28]. These cells are major producers of IL-4 that contribute to sustain alternatively activated macrophages in adipose tissue [28].
2.2. Systemic Inflammatory Loops
The global endocrine profile of adipose tissue appears to reflect the interactions among its paracrine loops. In other words, local crosstalks involving adipocytes, endothelial cells, and immune cells result in the production of a wide repertoire of bioactive substances that can act in a paracrine fashion, further amplifying inflammation within the adipose tissue [29]. At the same time, the spillover of adipokines into the circulation can also promote important systemic effects, and specific mediators such as adiponectin, leptin, plasminogen activator inhibitor-1 (PAI-1), and IL-6 may even serve as blood markers of cardiometabolic risk.
2.2.1. Adiponectin
The most abundant and one of the most extensively studied adipokines is adiponectin. Unlike most other adipokines, adiponectin plasma levels are lower in obese than in nonobese individuals [30]. It circulates in the plasma at levels of 3–30 mg/mL and forms three major oligomeric complexes with distinct biological functions: trimer, hexamer, and high-molecular-mass form, the latter likely being the most bioactive form in vascular cells (reviewed in [31]). There is also a bioactive proteolytic product of adiponectin that includes its C1q-like globular domain, which circulates at low concentration in plasma [32]. The adiponectin receptors, AdipoR1 and AdipoR2, activate signaling molecules such as AMP-activated protein kinase (AMPK), peroxisome-proliferator-activated receptor (PPAR)-α, and p38 mitogen-activated protein kinase (MAPK) [33]. Targeted disruption of AdipoR1 and AdipoR2 simultaneously abrogates adiponectin binding, causing insulin resistance and glucose intolerance [34].
Numerous experimental studies support the idea that adiponectin has antidiabetic properties [31]. Adiponectin-deficient (APN−/−) mice exhibited late clearance of free fatty acids from plasma and diet-induced insulin resistance [35], whereas adiponectin delivery via adenovirus in those knockout animals improved insulin sensitivity [35]. In another study, ob/ob mice overexpressing adiponectin showed improved glucose tolerance and reduced triglyceride levels compared to their nontransgenic ob/ob littermates in spite of being morbidly obese [36]. Nevertheless, the interpretation of these results is often difficult from a mechanistic standpoint, because different studies used distinct forms of the recombinant protein [32, 37].
Vast literature also suggests anti-inflammatory and antiatherogenic properties of adiponectin. Studies have shown anti-inflammatory effects of adiponectin on most cells involved in atherogenesis, including endothelial cells and macrophages [38, 39]. Physiological levels of adiponectin attenuate the attachment of monocytes to the endothelium in culture by reducing TNF-_α_-induced expression of adhesion molecules [38]. Pretreatment of human macrophages with adiponectin attenuates lipopolysaccharide- (LPS-) induced expression of TNF-α, and of a trio of T-lymphocyte chemoattractants associated with atherogenesis: interferon- (IFN-) inducible protein 10 (IP-10/CXCL10), IFN-inducible T-cell alpha chemoattractant (I-TAC/CXCL11), and monokine induced by IFN-gamma (MIG/CXCL9) [39]. Whereas the anti-inflammatory mechanisms elicited by adiponectin are not completely understood, recent work from our laboratory shed some light on this arena. We demonstrated that pretreatment of human macrophages with adiponectin inhibits phosphorylation of nuclear factor _κ_B inhibitor (I_κ_B), c-Jun N-terminal kinase (JNK), and p38 MAPK induced by either LPS or TNF-α as well as signal transducer and activation of transcription 3 (STAT3) phosphorylation induced by IL-6 [40]. Interestingly, treatment of human macrophages with adiponectin alone induced sustained phosphorylation of I_κ_B, JNK, p38, and STAT3 but prevented further activation of these signaling molecules upon addition of pro-inflammatory agonists [40]. These findings and others from additional studies suggest that adiponectin may induce some degree of inflammatory activation that likely mediates tolerance to further treatment with pro-inflammatory stimuli [40–42].
Several animal studies have confirmed the anti-inflammatory and antiatherogenic properties of adiponectin. Adenovirus-mediated delivery of adiponectin to apolipoprotein-E-deficient (ApoE−/−) mice, an atherosclerotic murine model, reduced plaque formation in the aortic sinus [43]. Transgenic mice expressing globular adiponectin crossed with ApoE−/− mice also had less atherosclerotic burden than ApoE−/− control animals, despite similar plasma glucose and lipid levels44. Additionally, APN−/− mice exhibited a 5-fold increase in leukocyte adhesion in the microcirculation, in association with decreased NO levels and augmented expression of adhesion molecules in the endothelium [44]. Yet, a recent study that used APN−/− mice and transgenic mice with chronically elevated adiponectin levels crossed to ApoE−/− or LDLR−/− mice found no correlation between adiponectin levels and atheroma development [45].
Multiple clinical studies have correlated hypoadiponectinemia with insulin resistance and type II diabetes mellitus in various populations [46, 47]. Recently, a meta-analysis of prospective studies that involved 14,598 subjects demonstrated that higher adiponectin concentrations associate with lower risk of type II diabetes [48]. A recent study that compared metabolic parameters in insulin-resistant versus insulin-sensitive obese individuals demonstrated that the strongest predictors of insulin sensitivity were macrophage infiltration in adipose tissue and circulating levels of adiponectin [49]. Genetic studies have also provided a link between adiponectin and metabolic disorders. Genetic mutations that likely reduce adiponectin plasma concentrations [50, 51] and adiponectin multimerization [52] associate closely with type II diabetes. These findings, although correlational in nature and thus not proving direct causality, support a primary role for adiponectin in preventing metabolic disease in humans.
Abundant data from several epidemiological studies have also reported an inverse correlation between adiponectin plasma levels and incidence of hypertension [53], dyslipidemia [54, 55], and cardiovascular disease [38, 56, 57]. Patients with coronary artery disease (CAD) have lower adiponectinemia than controls [38]. Similarly, men with both type II diabetes and CAD have lower circulating levels of adiponectin than patients with diabetes without CAD [56]. Another study showed a 2-fold increase in the prevalence of CAD among male patients with low plasma concentrations of adiponectin, independent of classic risk factors [57]. Prospective data have also demonstrated that high plasma adiponectin concentrations associate with lower risk of myocardial infarction in healthy men [58], and decreased CAD risk in diabetic men [59]. However, there are prospective studies that have not observed a correlation between adiponectinemia and risk of future CAD [60, 61]. Furthermore, a large prospective study and meta-analysis found a weak association between CAD and plasma adiponectin levels [62]. Intriguingly, two secondary prevention studies associated adiponectin with an increased risk of recurrent cardiovascular events [63, 64]. These conflicting studies indicate that further prospective analyses of various populations are still necessary to examine whether adiponectinemia can independently predict cardiovascular disease.
2.2.2. Leptin
Leptin, a 16 kDa hormone product of the ob gene [65], is predominantly released by adipocytes [66] to control body weight centrally through its cognate receptor in the hypothalamus [67]. Leptin circulates in the plasma at levels proportional to total body adiposity [68] and immediate nutritional state. In fed states, leptin levels increase and, via a central action in the brain, inhibit appetite and stimulate thyroid-mediated thermogenesis and fatty acid oxidation. In a fasting situation, leptin levels fall, and thus appetite increases, and thermogenesis becomes limited.
Leptin deficiency associates with increased appetite and marked obesity in mice and humans, a scenario completely abrogated by treatment with recombinant leptin [69]. Interestingly, human obesity only rarely associates with leptin deficiency or leptin receptor mutations. In the common form of obesity, leptin concentrations are actually increased in proportion to body adiposity [70], and the response of body weight to recombinant leptin is modest [71], defining a state of leptin resistance [72].
In addition to its role in energy homeostasis, leptin participates in other energy-demanding physiological processes such as reproduction [73], hematopoiesis [74], and angiogenesis [75]. Several studies have also shown that leptin has an important immunomodulatory role [76]. In monocytes or macrophages, it increases phagocytic function and proinflammatory cytokine production [77]. In polymorphonuclear cells of healthy subjects, it stimulates the production of reactive oxygen species [78] and chemotaxis [79]. Leptin is also involved in processes of cell development, proliferation, activation, and cytotoxicity of NK cells [80]. In adaptive immunity, leptin polarizes Th cytokine production toward a proinflammatory phenotype (Th1) [76].
Leptin signaling occurs typically via Janus tyrosine kinases (JAKs) and STATs. After binding to its functional receptor (ObRb), which is expressed not only in hypothalamus, but also in all cell types of innate and adaptive immunity [76, 81, 82], leptin recruits JAKs and activates the receptor, which serves as a docking site for adaptors such as STATs [83]. STATs translocate to the nucleus and induce gene expression. Several studies in human peripheral blood mononuclear cells have demonstrated that although the JAK-2-STAT-3 pathway constitutes an important pathway mediating leptin's function on immune cells, there are other pathways involved in this activity, such as the MAPK, the insulin receptor substrate 1, and the phosphatidyl-inositol 3′-kinase pathways [84].
2.2.3. Plasminogen Activator Inhibitor (PAI-1)
Obesity and metabolic syndrome promote a hyperthrombotic state through distinct pathways that involve hypofibrinolysis, hypercoagulability, and platelet activation [85]. One of the most remarkable abnormalities of the haemostatic system in the context of obesity and metabolic syndrome is the increased circulating concentrations of PAI-1 [86, 87], which seems particularly associated with higher production of this factor by the fatty liver, ectopic adipose tissues, and dysfunctional endothelium. PAI-1 correlates with all components of the insulin resistance syndrome, and weight loss associates with a significant decrease in PAI-1 levels [88].
PAI-1 is a serpin that regulates both tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA), likely representing the principal physiological inhibitor of plasminogen activation [89]. Thus, the increase in PAI-1 levels observed in obesity and metabolic syndrome impairs fibrinolysis and may influence the risk of atherothrombosis.
In addition to hypofibrinolysis, obesity and metabolic syndrome appear associated with enhanced platelet activity [87]. Moreover, higher concentrations of vitamin K-dependent coagulation factors and fibrinogen may also accompany excess adiposity, at least partially due to its inflammatory component [86]. Endothelial dysfunction, likely an important element in obesity, may also contribute to the thrombotic diathesis observed in this condition [87], through increased expression of proinflammatory and haemostatic products such as microparticles and von Willebrand factor.
2.2.4. Interleukin-6 (IL-6)
IL-6 is an inflammatory cytokine with distinct pathophysiologic roles in humans. It stimulates the hypothalamic-pituitary-adrenal axis, being under negative control by glucocorticoids and positive regulation by catecholamines [90]. IL-6 is a potent inducer of the acute-phase reaction [90] that circulates at high levels during stress, inflammatory and infectious diseases. In mice, the metabolic role of IL-6 is not fully understood. Mice genetically deficient in IL-6 develop mature-onset obesity, partly reversed with IL-6 administration, and abnormalities in carbohydrate and lipid metabolism [91]. On the other hand, mice chronically exposed to IL-6 develop hepatic insulin resistance [92]. In either case though, IL-6 is likely a critical metabolic modulator.
Several immune cell types, including but not limited to monocytes, constitute typical sources of IL-6 [93]. In the last decades, the study of adipose tissue as an endocrine organ unraveled its abundant production of IL-6. Indeed, subcutaneous adipose tissue appears to release approximately 25% of circulating IL-6 in humans [94]. The obese state associates with increased secretion of IL-6 and, therefore, higher hepatic release of acute-phase reactants, C-reactive protein (CRP) likely being the major one [95]. A large bulk of evidence, including experimental and cross-sectional data, suggests that both IL-6 and CRP correlate with hyperglycemia, insulin resistance, and type 2 diabetes mellitus [96–100]. A prospective study found strong correlation between baseline IL-6 and CRP levels and risk of developing type 2 diabetes mellitus [101]. After multivariate analysis and adjustment for adiposity measurements, there was significant attenuation of the association between IL-6 and risk of diabetes, but CRP persisted as an independent predictor of incident diabetes [101].
Excess adiposity, particularly central adiposity, also associates with elevated levels of CRP. Indeed, besides the higher CRP levels in obese than in nonobese individuals, there is a positive correlation between CRP and waist-to-hip ratio (a measurement of visceral obesity) even after adjustment for BMI [102]. CRP, a liver-derived pentraxin, also correlates well with other risk factors and increased cardiovascular risk in the absence of acute inflammation. It has emerged as one of the most promising biomarkers for future cardiovascular events since 1997, when the Physicians Health Study demonstrated a strong and independent correlation between systemic levels of CRP and future occurrence of myocardial infarction and stroke in apparently healthy men [103]. After this work, several other prospective studies corroborated the CRP capacity of predicting cardiovascular events, even beyond traditional risk factors [104–110]. Besides the evidence in primary prevention, several studies also suggested that CRP levels predict efficiently new cardiovascular events in individuals who already had a myocardial infarction [111, 112]. CRP levels also seem to correlate with cerebrovascular events [113] and development of peripheral vascular disease [114].
Despite the large bulk of evidence on the CRP prediction capacity, the utility of this biomarker in risk stratification beyond traditional risk factors is not consensual, with opponents arguing that it only has a modest ability of stratifying the risk beyond conventional scores according to the C statistic [115, 116].
A recent meta-analysis including individual records of 160,309 people without a history of vascular disease from 54 long-term prospective studies showed that log(e) CRP concentration was linearly associated with several conventional risk factors and inflammatory markers, and nearly log-linearly with the risk of ischemic vascular disease and nonvascular mortality [117]. However, the risk ratios for coronary heart disease, ischemic stroke, vascular mortality, and nonvascular mortality per 1-SD higher log(e) CRP concentration decreased when adjusted for age and sex and even further for conventional risk factors [117]. These results suggest that a great proportion of the vascular risk associated with CRP depends on its correlation with traditional risk factors. On the other hand, CRP still kept its ability of predicting vascular events (and even non-vascular mortality) despite the adjustment for several conventional risk factors [117]. Interestingly, after further adjustment for other markers of inflammation, such as fibrinogen, the relative risk declined significantly, suggesting that CRP might be indeed an efficient marker of systemic inflammation but probably not a causal factor of atherothrombosis per se. A genetic study on the polymorphisms in the CRP gene also suggests a noncausal link between CRP levels and the risk of ischemic vascular disease [118]. In this work, genetic variants associated with lifelong elevations in circulating CRP, and therefore, with a theoretically predicted increase in the risk of ischemic vascular disease, failed to show the expected risk [118].
Research has also tested the utility of CRP beyond risk stratification. JUPITER (justification for the use of statins in primary prevention: an intervention trial evaluating rosuvastatin) for example, tested the value of CRP as a therapeutic guide for the initiation of statins [119]. This study found a significant reduction in cardiovascular events among individuals with low LDL-cholesterol levels (LDL <130 mg_/dL), but high CRP levels (CRP >2 mg/_L) receiving Rosuvastatin 20 mg per day compared to placebo [119]. The noninclusion of a group of people with low LDL-cholesterol and low CRP levels and the early interruption of the study figure among its limitations and most frequent critics.
Multiple studies have also showed that several other adipokines, such as resistin, retinoid binding protein 4 (RBP4), visfatin, and omentin, present varied potential effects on glucose homeostasis. Extensive reviews on these bioactive mediators exist elsewhere [120, 121].
3. Potential Links between Inflammation and Insulin Resistance
Ample literature supports that insulin resistance precedes the development of overt hyperglycemia and type 2 diabetes [122]. Genetic burden and acquired conditions, including obesity as one of the most relevant, may play a role in the origin of insulin resistance [122]. While pancreatic _β_-cells properly respond to insulin resistance with substantial secretion of insulin, glucose metabolism stays equilibrated. Once the pancreas fails in overcoming insulin resistance with more insulin release, hyperglycemia, and eventually diabetes mellitus ensue.
In adipose tissue and skeletal muscle, insulin promotes cellular glucose uptake through a system of intracellular substrates [123]. Upon insulin binding to its receptor at the cell surface, members of the insulin receptor substrate (IRS) family become tyrosine phosphorylated. This process leads to downstream signaling which triggers translocation of GLUT-4 from intracellular stores to the cellular membrane and, therefore, glucose transport into the cell [124]. The phosphorylation of particular serine residues of IRS-1 may instead abrogate the association between IRS-1 and the insulin receptor, impairing insulin signaling [125]. Inflammatory mediators such as TNF-α or elevated levels of free fatty acids may activate serine kinases, such as c-Jun NH2-terminal kinase (JNK) and I_κ_B kinase (IKK) (Figure 1), which phosphorylate serine residues of IRS-1 and disrupt insulin signaling [126–129]. Both JNK and IKK, which are members of two major pro-inflammatory cascades, are likely activated in insulin-resistant states, and thus provide potential connections between inflammation and insulin resistance [127, 130, 131].
Experimental research with JNK-1-deficient mice showed substantial reduction of IRS-1 serine phosphorylation and amelioration of insulin sensitivity [127]. Besides serine phosphorylation of IRS-1, IKK-β can also influence insulin function through phosphorylation of the NF_κ_B inhibitor (I_κ_B), leading to activation of NF_κ_B. The stimulation of this potent inflammatory pathway culminates with further production of several inflammatory substances including TNF-α, which can maintain and potentiate inflammatory activation [132]. Experiments involving rodents with targeted disruption of IKK-β demonstrated significant attenuation of insulin resistance [130]. There are also other kinases, such as mammalian target of rapamycin (mTOR), protein kinase R (PKR), and protein kinase θ (PK_θ_) that can induce inhibitory phosphorylation of IRS-1, and thus contribute to nutrient and inflammation-related disruption of insulin signaling [133].
Although obesity-associated inflammation probably initiates in adipocytes, where nutrient overload is first sensed, adipose tissue is not the only organ characterized by local inflammation, nor is it solely responsible for systemic inflammation and glucose metabolism abnormalities in obesity [3]. The liver, as another major metabolic site in the body, also participates importantly in the systemic inflammatory networks of obesity by experiencing activation of inflammatory pathways within its local cells [3]. The liver contains a resident population of macrophage-like cells, the Kupffer cells, which can secrete inflammatory mediators upon activation. However, unlike adipose tissue, the liver does not accumulate macrophages or other immune cells in the context of obesity. Animal studies involving liver-specific gain-of-function or loss-of-function of IKK-β increased our understanding on the role of obese liver in inflammation-related insulin resistance [132, 134]. Hepatocyte-specific deletion of IKK-β protects the liver but not muscle or adipose tissue of obese mice from insulin resistance [134]. On the other hand, transgenic mice with constitutively active IKK-β in hepatocytes display increased hepatic production of inflammatory cytokines, severe hepatic insulin resistance, and moderate systemic insulin resistance [132]. Intriguingly, specific deletion of JNK-1 in hepatocytes deteriorated glucose homeostasis in mice [135], whereas administration of a cell-permeable JNK-inhibitory peptide had the opposite effect, improving insulin sensitivity and glucose tolerance in diabetic mice [136]. In summary, although adipose tissue may be the organ where energy surplus exerts its first effects, presenting a pivotal role in the inflammatory and metabolic derangements of obesity, other metabolic organs, such as the liver, also seem to participate in this loop although in a somehow more localized manner.
4. The Origin of the Inflammatory Response in Obesity
Despite the large bulk of data available on the local and systemic inflammatory networks operating in obesity, the precise triggers of inflammation in this condition remain unidentified. However, several recent studies have brought potential answers to fundamental questions in this fascinating area of research. A reasonable hypothesis posits that nutrient overload in metabolic cells such as adipocytes induces intracellular stress which results in activation of inflammatory cascades [3, 137] (Figure 1). The endoplasmic reticulum (ER), an organelle specialized in protein folding, maturation, storage, and transport, senses nutrient levels in the cell. Under conditions of cellular stress induced by nutrient surplus, misfolded or unfolded proteins accumulate in the ER and activate the so-called unfolded protein response (UPR) pathway [138]. The UPR functioning depends essentially on three main ER sensors: PKR-like eukaryotic initiation factor 2_α_ kinase (PERK), inositol requiring enzyme 1 (IRE-1), and activating transcription factor 6 (ATF-6) [139]. Once activated, the UPR leads to increased activity of the kinases JNK and IKK-β, serine-phosphorylation of IRS-1, and activation of the NF_κ_B pathway, leading to enhanced proinflammatory cytokine expression and impaired insulin signaling [3, 140–142].
The ER also responds to other conditions of cellular stress, such as hypoxia, another element of obese adipose tissue [124]. As obesity evolves and enlargement of adipose tissue ensues, the expanding adipocytes get relatively hypoxic. Regions of microhypoxia within the adipose tissue also exhibit increased activation of inflammatory pathways [143, 144].
It is also possible that, in excess, nutrients can themselves directly activate immune pathogen-sensors in the cell such as toll-like receptors (TLRs), which likely respond to excess fatty acids and may contribute to insulin resistance [145]. TLR4 deficiency improves insulin sensitivity in high-fat diet-induced obese mice [145, 146]. PKR, another pathogen-sensor in the cell, is likely activated in mice during lipid infusion and in obesity [147] and, as discussed above, is also able to activate JNK and IKK and disrupt insulin signaling.
5. Obesity, Inflammation, and CardiovascularDisease
There are numerous potential mechanisms by which increased adiposity may induce atherothrombosis and cardiovascular disease [85]. Obesity, predominantly visceral, often associates with other morbid conditions, such as insulin resistance, glucose and lipid abnormalities, and hypertension, each one an independent cardiovascular risk factor per se. Moreover, the inflammatory state that characterizes obesity is likely an important connection between this condition and the other cardiovascular risk factors, and at the same time, a possible direct link to atherothrombosis [85]. Indeed, as aforementioned, excess adiposity often correlates with abnormal production of several mediators which often associate with cardiovascular events. The adipokine imbalance characterizing obesity, including low levels of adiponectin, high levels of leptin, inflammatory mediators (IL-6 and TNF-α) and antifibrinolytic factors (PAI-1) may induce oxidative stress and endothelial dysfunction, initial steps of atherogenesis. Moreover, the insulin-resistant state of obesity frequently involves high circulating levels of non-esterified fatty acids, which cause lipotoxicity, and therefore further oxidative stress and endothelial dysfunction. As described above, numerous studies also support an independent association between circulating levels of CRP, an inflammatory marker potently induced by IL-6 and TNF-α (in excess in the obesity state), and cardiovascular events.
6. Insights into Anti-Inflammatory Therapies
All the evidence linking obesity-related metabolic disorders to inflammation raises the question whether modulation of inflammatory pathways would have a beneficial impact on metabolism. Several studies have targeted inflammation at different points of the inflammatory network of obesity, ranging from inhibition of circulating cytokines to suppression of intracellular inflammatory cascades and ER stress.
The approval of anti-TNF-α compounds for clinical use in patients with specific inflammatory conditions motivated studies with these reagents in obese and insulin-resistant subjects. These trials have yielded conflicting results on the improvement of insulin sensitivity by TNF-α blockers, such as etanercept [148, 149], and final conclusions warrant further studies.
Another promising anti-inflammatory approach in obesity and insulin-resistant states targets kinases and intracellular inflammatory pathways. Salsalates, nonacetylated members of the group of salicylates, appear to modulate inflammation through suppression of IKK action [29]. Clinical studies have shown decreased inflammation and improved metabolism, including glucose and lipid parameters, in diabetic subjects under therapy with salsalates [150]. Studies involving JNK antagonists, available for mice but still lacking for human treatment, also yielded positive results in glucose metabolism [136].
Since ER stress may represent one major source of inflammatory signals within the cell, it provides a potential therapeutic target for modulation of inflammation and metabolic derangements. Indeed, attenuation of ER stress through use of chemical chaperones restores glucose homeostasis in a mouse model of type 2 diabetes [151]. In humans, studies are still limited, but already reveal positive metabolic results [152].
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor-γ (PPAR_γ_) agonists, which possess significant anti-inflammatory effects and insulin-sensitizing properties [153]. PPAR_γ_ is a nuclear transcription factor, member of the nuclear-receptor superfamily, with important regulatory effects on inflammatory processes. As a synthetic ligand of PPAR_γ_, TZDs may act via repression of inflammatory gene promoters [154]. They can also exert PPAR_γ_-independent stimulation of glucocorticoid receptors [155]. By activating PPAR_γ_, these compounds can also regulate genes related to adipocyte differentiation, lipid metabolism, and glucose uptake, each of which can contribute to their beneficial metabolic effects. However, changes in fluid balance and in myocardial function and predisposition to fractures render the therapeutic use of TZDs, more specifically rosiglitazone, challenging in specific groups of patients, particularly in patients with heart failure or increased cardiovascular risk [156–158].
The use of nutrients with anti-inflammatory properties may constitute another promising weapon against obesity. Mice receiving high fat diet supplemented with omega-3 polyunsaturated fatty acids (n-3 PUFAs) had reduced adipose tissue inflammation and improved insulin sensitivity [159]. In type 2 diabetic individuals, supplementation with omega-3 fatty acids has beneficial effects on serum triglycerides, HDL-cholesterol, lipid peroxidation, and antioxidant enzymes, which may lead to reduced rate of occurrence of vascular complications in those patients [160]. In diabetic women, high consumption of fish and long-chain omega-3 fatty acids supplementation associated with decreased cardiovascular risk [161].
Finally, cell-based immunomodulation of obesity inflammation has recently attracted attention although it is still incipient. Several animal studies, including some discussed above, demonstrated significant metabolic benefits as a consequence of depletion or stimulation of specific cell pools. Whereas deficiency or inhibition of CD11c-positive cells [162], CD8 T cells [18], mast cells [20] and NKT cells [21] improves metabolic parameters, an increase in the pool of CD4+ Foxp3+ T cells following CD3-specific antibody treatment likely produce similar beneficial metabolic effects [27].
7. Conclusions
Far beyond a mere inert lipid storage, adipose tissue represents a site of plural activities involving secretion of a wide gamma of active substances, a great proportion of them portending inflammatory actions and/or highly regulated by inflammation. By imposing a state of nutrient overload, obesity significantly boosts inflammation in adipose tissue and other metabolic organs, with consequent impairment of insulin signaling and abnormalities in glucose metabolism. Progressive understanding of all inflammatory mechanisms operating in obesity is mandatory for further therapeutic advances.
Abbreviations
CCR2:
C-C receptor 2
CRP:
C-reactive protein
ER:
Endoplasmic reticulum
IKK:
I_κ_B kinase
IR:
Insulin resistance
IFN-γ:
Interferon-gamma
IL-6:
Interleukin-6
JNK:
c-Jun NH2-terminal kinase
MCP-1:
Monocyte chemoattractant protein-1
PAI-1:
Plasminogen activator inhibitor-1.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2010;123(4):E18–E29. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nature reviews. 2009;6(6):399–409. doi: 10.1038/nrcardio.2009.55. [DOI] [PubMed] [Google Scholar]
- 3.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 4.Ogston D, Mcandrew GM. FIbrinolysis in Obesity. The Lancet. 1964;2(7371):1205–1207. doi: 10.1016/s0140-6736(64)91042-6. [DOI] [PubMed] [Google Scholar]
- 5.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 6.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor α inhibits signaling from the insulin receptor. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4854–4858. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rocha VZ, Libby P. The multiple facets of the fat tissue. Thyroid. 2008;18(2):175–183. doi: 10.1089/thy.2007.0296. [DOI] [PubMed] [Google Scholar]
- 8.Di Gregorio GB, Yao-Borengasser A, Rasouli N, et al. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes. 2005;54(8):2305–2313. doi: 10.2337/diabetes.54.8.2305. [DOI] [PubMed] [Google Scholar]
- 9.Mertens I, Van Gaal LF. New international diabetes federation (idf) and national cholesterol education program adult treatment panel III (ncep-atpIII) criteria and the involvement of hemostasis and fibrinolysis in the metabolic syndrome. Journal of Thrombosis and haemostasis. 2006;4(5):1164–1166. doi: 10.1111/j.1538-7836.2006.01919.x. [DOI] [PubMed] [Google Scholar]
- 10.Spranger J, Kroke A, Möhlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European prospective investigation into cancer and nutrition (EPIC)-Potsdam study. Diabetes. 2003;52(3):812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 11.Müller S, Martin S, Koenig W, et al. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia. 2002;45(6):805–812. doi: 10.1007/s00125-002-0829-2. [DOI] [PubMed] [Google Scholar]
- 12.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. Journal of Clinical Investigation. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115(8):1029–1038. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 16.Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-γ, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circulation Research. 2008;103(5):467–476. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(7):1304–1310. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 19.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nature Medicine. 2009;15(8):940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohmura K, Ishimori N, Ohmura Y, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(2):193–199. doi: 10.1161/ATVBAHA.109.198614. [DOI] [PubMed] [Google Scholar]
- 22.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. Journal of Clinical Investigation. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirk EA, Sagawa ZK, McDonald TO, O’Brien KD, Heinecke JW. Monocyte chemoattractant protein deficiency fails to restrain macrophage infiltration into adipose tissue [corrected] Diabetes. 2008;57(5):1254–1261. doi: 10.2337/db07-1061. [DOI] [PubMed] [Google Scholar]
- 25.Inouye KE, Shi H, Howard JK, et al. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes. 2007;56(9):2242–2250. doi: 10.2337/db07-0425. [DOI] [PubMed] [Google Scholar]
- 26.Gordon S. Macrophage heterogeneity and tissue lipids. Journal of Clinical Investigation. 2007;117(1):89–93. doi: 10.1172/JCI30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nature Medicine. 2009;15(8):921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu D, Molofsky AB, Liang HE, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and Biophysical Research Communications. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto Y, Kihara S, Funahashi T, Matsuzawa Y, Libby P. Adiponectin: a key adipocytokine in metabolic syndrome. Clinical Science. 2006;110(3):267–278. doi: 10.1042/CS20050182. [DOI] [PubMed] [Google Scholar]
- 32.Fruebis J, Tsao TS, Javorschi S, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(4):2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature Medicine. 2007;13(3):332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 35.Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nature Medicine. 2002;8(7):731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 36.Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. Journal of Clinical Investigation. 2007;117(9):2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nature Medicine. 2001;7(8):947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 38.Ouchi N, Kihara S, Arita Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100(25):2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto Y, Folco EJ, Minami M, et al. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circulation Research. 2008;102(2):218–225. doi: 10.1161/CIRCRESAHA.107.164988. [DOI] [PubMed] [Google Scholar]
- 40.Folco EJ, Rocha VZ, López-Ilasaca M, Libby P. Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. Journal of Biological Chemistry. 2009;284(38):25569–25575. doi: 10.1074/jbc.M109.019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsatsanis C, Zacharioudaki V, Androulidaki A, et al. Adiponectin induces TNF-α and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochemical and Biophysical Research Communications. 2005;335(4):1254–1263. doi: 10.1016/j.bbrc.2005.07.197. [DOI] [PubMed] [Google Scholar]
- 42.Park PH, McMullen MR, Huang H, Thakur V, Nagy LE. Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-α (TNF-α) expression via ERK1/2 activation and Egr-1 expression: role of TNF-α in adiponectin-stimulated interleukin-10 production. Journal of Biological Chemistry. 2007;282(30):21695–21703. doi: 10.1074/jbc.M701419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamoto Y, Kihara S, Ouchi N, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106(22):2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 44.Ouedraogo R, Gong Y, Berzins B, et al. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. Journal of Clinical Investigation. 2007;117(6):1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nawrocki AR, Hofmann SM, Teupser D, et al. Lack of association between adiponectin levels and atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(6):1159–1165. doi: 10.1161/ATVBAHA.109.195826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindsay RS, Funahashi T, Hanson RL, et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet. 2002;360(9326):57–58. doi: 10.1016/S0140-6736(02)09335-2. [DOI] [PubMed] [Google Scholar]
- 47.Snehalatha C, Mukesh B, Simon M, Viswanathan V, Haffner SM, Ramachandran A. Plasma adiponectin is an independent predictor of type 2 diabetes in Asian Indians. Diabetes Care. 2003;26(12):3226–3229. doi: 10.2337/diacare.26.12.3226. [DOI] [PubMed] [Google Scholar]
- 48.Li S, Shin HJ, Ding EL, van Dam RM. Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis. Journal of the American Medical Association. 2009;302(2):179–188. doi: 10.1001/jama.2009.976. [DOI] [PubMed] [Google Scholar]
- 49.Kloting N, Fasshauer M, Dietrich A, et al. Insulin-sensitive obesity. American Journal of Physiology, Endocrinology and Metabolism. 2010;299(3):E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 50.Kondo H, Shimomura L, Matsukawa Y, et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes. 2002;51(7):2325–2328. doi: 10.2337/diabetes.51.7.2325. [DOI] [PubMed] [Google Scholar]
- 51.Kara K, Boutin P, Mori Y, et al. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes. 2002;51(2):536–540. doi: 10.2337/diabetes.51.2.536. [DOI] [PubMed] [Google Scholar]
- 52.Waki H, Yamauchi T, Kamon J, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. Journal of Biological Chemistry. 2003;278(41):40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- 53.Adamczak M, Wiecek A, Funahashi T, Chudek J, Kokot F, Matsuzawa Y. Decreased plasma adiponectin concentration in patients with essential hypertension. American Journal of Hypertension. 2003;16(1):72–75. doi: 10.1016/s0895-7061(02)03197-7. [DOI] [PubMed] [Google Scholar]
- 54.Matsubara M, Maruoka S, Katayose S. Decreased plasma adiponectin concentrations in women with dyslipidemia. Journal of Clinical Endocrinology and Metabolism. 2002;87(6):2764–2769. doi: 10.1210/jcem.87.6.8550. [DOI] [PubMed] [Google Scholar]
- 55.Kazumi T, Kawaguchi A, Hirano T, Yoshino G. Serum adiponectin is associated with high-density lipoprotein cholesterol, triglycerides, and low-density lipoprotein particle size in young healthy men. Metabolism. 2004;53(5):589–593. doi: 10.1016/j.metabol.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 56.Hotta K, Funahashi T, Arita Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(6):1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 57.Kumada M, Kihara S, Sumitsuji S, et al. Association of hypoadiponectinemia with coronary artery disease in men. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(1):85–89. doi: 10.1161/01.atv.0000048856.22331.50. [DOI] [PubMed] [Google Scholar]
- 58.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. Journal of the American Medical Association. 2004;291(14):1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 59.Schulze MB, Shai I, Rimm EB, Li T, Rifai N, Hu FB. Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes. 2005;54(2):534–539. doi: 10.2337/diabetes.54.2.534. [DOI] [PubMed] [Google Scholar]
- 60.Lindsay RS, Resnick HE, Zhu J, et al. Adiponectin and coronary heart disease: the strong heart study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(3):E15–E16. doi: 10.1161/01.ATV.0000153090.21990.8c. [DOI] [PubMed] [Google Scholar]
- 61.Lawlor DA, Smith GD, Ebrahim S, Thompson C, Sattar N. Plasma adiponectin levels are associated with insulin resistance, but do not predict future risk of coronary heart disease in women. Journal of Clinical Endocrinology and Metabolism. 2005;90(10):5677–5683. doi: 10.1210/jc.2005-0825. [DOI] [PubMed] [Google Scholar]
- 62.Sattar N, Wannamethee G, Sarwar N, et al. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation. 2006;114(7):623–629. doi: 10.1161/CIRCULATIONAHA.106.618918. [DOI] [PubMed] [Google Scholar]
- 63.Cavusoglu E, Ruwende C, Chopra V, et al. Adiponectin is an independent predictor of all-cause mortality, cardiac mortality, and myocardial infarction in patients presenting with chest pain. European Heart Journal. 2006;27(19):2300–2309. doi: 10.1093/eurheartj/ehl153. [DOI] [PubMed] [Google Scholar]
- 64.Schnabel R, Messow CM, Lubos E, et al. Association of adiponectin with adverse outcome in coronary artery disease patients: results from the AtheroGene study. European Heart Journal. 2008;29(5):649–657. doi: 10.1093/eurheartj/ehn009. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 66.Maffei M, Fei H, Lee GH, et al. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(15):6957–6960. doi: 10.1073/pnas.92.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flier JS. The adipocyte: storage depot or node on the energy information superhighway? Cell. 1995;80(1):15–18. doi: 10.1016/0092-8674(95)90445-x. [DOI] [PubMed] [Google Scholar]
- 68.Frederich RC, Lollmann B, Hamann A, et al. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. Journal of Clinical Investigation. 1995;96(3):1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. Journal of Clinical Investigation. 2002;110(8):1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New England Journal of Medicine. 1996;334(5):292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 71.Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Journal of the American Medical Association. 1999;282(16):1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 72.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116(2):337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 73.Ahlma RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382(6588):250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 74.Bennett BD, Solar GP, Yuan JQ, Mathias J, Thomas GR, Matthews W. A role for leptin and its cognate receptor in hematopoiesis. Current Biology. 1996;6(9):1170–1180. doi: 10.1016/s0960-9822(02)70684-2. [DOI] [PubMed] [Google Scholar]
- 75.Sierra-Honigmann MR, Nath AK, Murakami C, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281(5383):1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 76.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation- induced immunosuppression. Nature. 1998;394(6696):897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 77.Mancuso P, Gottschalk A, Phare SM, Peters-Golden M, Lukacs NW, Huffnagle GB. Leptin-deficient mice exhibit impaired host defense in Gram-negative pneumonia. Journal of Immunology. 2002;168(8):4018–4024. doi: 10.4049/jimmunol.168.8.4018. [DOI] [PubMed] [Google Scholar]
- 78.Caldefie-Chezet F, Poulin A, Tridon A, Sion B, Vasson MP. Leptin: a potential regulator of polymorphonuclear neutrophil bactericidal action? Journal of Leukocyte Biology. 2001;69(3):414–418. [PubMed] [Google Scholar]
- 79.Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radical Research. 2003;37(8):809–814. doi: 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 80.Tian Z, Sun R, Wei H, Gao B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: leptin as a critical regulator in NK cell development and activation. Biochemical and Biophysical Research Communications. 2002;298(3):297–302. doi: 10.1016/s0006-291x(02)02462-2. [DOI] [PubMed] [Google Scholar]
- 81.Zhao Y, Sun R, You L, Gao C, Tian Z. Expression of leptin receptors and response to leptin stimulation of human natural killer cell lines. Biochemical and Biophysical Research Communications. 2003;300(2):247–252. doi: 10.1016/s0006-291x(02)02838-3. [DOI] [PubMed] [Google Scholar]
- 82.Zarkesh-Esfahani H, Pockley G, Metcalfe RA, et al. High-dose leptin activates human leukocytes via receptor expression on monocytes. Journal of Immunology. 2001;167(8):4593–4599. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 83.Baumann H, Morella KK, White DW, et al. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(16):8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin-Romero C, Sanchez-Margalet V. Human leptin activates PI3K and MAPK pathways in human peripheral blood mononuclear cells: possible role of Sam68. Cellular Immunology. 2001;212(2):83–91. doi: 10.1006/cimm.2001.1851. [DOI] [PubMed] [Google Scholar]
- 85.van Gaal LF, Mertens IL, de Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444(7121):875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 86.Sakkinen PA, Wahl P, Cushman M, Lewis MR, Tracy RP. Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. American Journal of Epidemiology. 2000;152(10):897–907. doi: 10.1093/aje/152.10.897. [DOI] [PubMed] [Google Scholar]
- 87.Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thrombosis and Haemostasis. 2008;99(6):995–1000. doi: 10.1160/TH07-11-0682. [DOI] [PubMed] [Google Scholar]
- 88.Mertens I, van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obesity Reviews. 2002;3(2):85–101. doi: 10.1046/j.1467-789x.2002.00056.x. [DOI] [PubMed] [Google Scholar]
- 89.Rau JC, Beaulieu LM, Huntington JA, Church FC. Serpins in thrombosis, hemostasis and fibrinolysis. Journal of Thrombosis and Haemostasis. 2007;5(supplement 1):102–115. doi: 10.1111/j.1538-7836.2007.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papanicolaou DA, Wilder RL, Manolagas SC, Chrousos GP. The pathophysiologic roles of interleukin-6 in human disease. Annals of Internal Medicine. 1998;128(2):127–137. doi: 10.7326/0003-4819-128-2-199801150-00009. [DOI] [PubMed] [Google Scholar]
- 91.Wallenius V, Wallenius K, Ahrén B, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nature Medicine. 2002;8(1):75–79. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 92.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52(11):2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 93.Papanicolaou DA, Vgontzas AN. Interleukin-6: the endocrine cytokine. Journal of Clinical Endocrinology and Metabolism. 2000;85(3):1331–1333. doi: 10.1210/jcem.85.3.6582. [DOI] [PubMed] [Google Scholar]
- 94.Mohamed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. International Journal of Obesity and Related Metabolic Disorders. 1998;22(12):1145–1158. doi: 10.1038/sj.ijo.0800770. [DOI] [PubMed] [Google Scholar]
- 95.Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(4):972–978. doi: 10.1161/01.atv.19.4.972. [DOI] [PubMed] [Google Scholar]
- 96.Sandler S, Bendtzen K, Eizirik DL, Welsh M. Interleukin-6 affects insulin secretion and glucose metabolism of rat pancreatic islets in vitro. Endocrinology. 1990;126(2):1288–1294. doi: 10.1210/endo-126-2-1288. [DOI] [PubMed] [Google Scholar]
- 97.Kanemaki T, Kitade H, Kaibori M, et al. Interleukin 1β and interleukin 6, but not tumor necrosis factor α, inhibit insulin-stimulated glycogen synthesis in rat hepatocytes. Hepatology. 1998;27(5):1296–1303. doi: 10.1002/hep.510270515. [DOI] [PubMed] [Google Scholar]
- 98.Tsigos C, Papanicolaou DA, Kyrou I, Defensor R, Mitsiadis CS, Chrousos GP. Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. Journal of Clinical Endocrinology and Metabolism. 1997;82(12):4167–4170. doi: 10.1210/jcem.82.12.4422. [DOI] [PubMed] [Google Scholar]
- 99.Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the insulin resistance atherosclerosis study (IRAS) Circulation. 2000;102(1):42–47. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- 100.Fröhlich M, Imhof A, Berg G, et al. Association between C-reactive protein and features of the metabolic syndrome. Diabetes Care. 2000;23(12):1835–1839. doi: 10.2337/diacare.23.12.1835. [DOI] [PubMed] [Google Scholar]
- 101.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. Journal of the American Medical Association. 2001;286(3):327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 102.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. Journal of the American Medical Association. 1999;282(22):2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 103.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. New England Journal of Medicine. 1997;336(14):973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 104.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. New England Journal of Medicine. 2000;342(12):836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 105.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. New England Journal of Medicine. 2002;347(20):1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 106.Pai JK, Pischon T, Ma J, et al. Inflammatory markers and the risk of coronary heart disease in men and women. New England Journal of Medicine. 2004;351(25):2599–2610. doi: 10.1056/NEJMoa040967. [DOI] [PubMed] [Google Scholar]
- 107.Koenig W, Sund M, Fröhlich M, et al. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (monitoring trends and determinants in cardiovascular disease) Augsburg cohort study, 1984 to 1992. Circulation. 1999;99(2):237–242. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 108.Koenig W, Löwel H, Baumert J, Meisinger C. C-reactive protein modulates risk prediction based on the framingham score: implications for future risk assessment: results from a large cohort study in Southern Germany. Circulation. 2004;109(11):1349–1353. doi: 10.1161/01.CIR.0000120707.98922.E3. [DOI] [PubMed] [Google Scholar]
- 109.Ballantyne CM, Hoogeveen RC, Bang H, et al. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the atherosclerosis risk in communities (ARIC) study. Circulation. 2004;109(7):837–842. doi: 10.1161/01.CIR.0000116763.91992.F1. [DOI] [PubMed] [Google Scholar]
- 110.Boekholdt SM, Hack CE, Sandhu MS, et al. C-reactive protein levels and coronary artery disease incidence and mortality in apparently healthy men and women: the EPIC-Norfolk prospective population study 1993–2003. Atherosclerosis. 2006;187(2):415–422. doi: 10.1016/j.atherosclerosis.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 111.Ridker PM, Rifai N, Pfeffer MA, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CARE) investigators. Circulation. 1998;98(9):839–844. doi: 10.1161/01.cir.98.9.839. [DOI] [PubMed] [Google Scholar]
- 112.Retterstol L, Eikvar L, Bohn M, Bakken A, Erikssen J, Berg K. C-reactive protein predicts death in patients with previous premature myocardial infarction—a 10 year follow-up study. Atherosclerosis. 2002;160(2):433–440. doi: 10.1016/s0021-9150(01)00595-0. [DOI] [PubMed] [Google Scholar]
- 113.Rost NS, Wolf PA, Kase CS, et al. Plasma concentration of C-reactive protein and risk of ischemic stroke and transient ischemic attack: the Framingham Study. Stroke. 2001;32(11):2575–2579. doi: 10.1161/hs1101.098151. [DOI] [PubMed] [Google Scholar]
- 114.Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. Journal of the American Medical Association. 2001;285(19):2481–2485. doi: 10.1001/jama.285.19.2481. [DOI] [PubMed] [Google Scholar]
- 115.Wang TJ, Gona P, Larson MG, et al. Multiple biomarkers for the prediction of first major cardiovascular events and death. New England Journal of Medicine. 2006;355(25):2631–2639. doi: 10.1056/NEJMoa055373. [DOI] [PubMed] [Google Scholar]
- 116.Melander O, Newton-Cheh C, Almgren P, et al. Novel and conventional biomarkers for prediction of incident cardiovascular events in the community. Journal of the American Medical Association. 2009;302(1):49–57. doi: 10.1001/jama.2009.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaptoge S, di Angelantonio E, Lowe G, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375(9709):132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zacho J, Tybjærg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. New England Journal of Medicine. 2008;359(18):1897–1908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 119.Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New England Journal of Medicine. 2008;359(21):2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 120.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444(7121):847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology. 2007;132(6):2103–2115. doi: 10.1053/j.gastro.2007.03.058. [DOI] [PubMed] [Google Scholar]
- 122.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 123.White MF. The insulin signalling system and the IRS proteins. Diabetologia. 1997;40(supplement 2):S2–S17. doi: 10.1007/s001250051387. [DOI] [PubMed] [Google Scholar]
- 124.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. Journal of Clinical Investigation. 2008;118(9):2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87(1):99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 126.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 127.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 128.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) Journal of Biological Chemistry. 2000;275(12):9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 129.Gao Z, Hwang D, Bataille F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor κB kinase complex. Journal of Biological Chemistry. 2002;277(50):48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 130.Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. International Journal of Obesity and Related Metabolic Disorders. 2003;24(supplement 3):S49–S52. doi: 10.1038/sj.ijo.0802501. [DOI] [PubMed] [Google Scholar]
- 131.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science. 2001;293(5535):1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 132.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nature Medicine. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. American Journal of Physiology - Endocrinology and Metabolism. 2009;296(4):E581–E591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- 134.Arkan MC, Hevener AL, Greten FR, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 135.Sabio G, Cavanagh-Kyros J, Ko HJ, et al. Prevention of steatosis by hepatic JNK1. Cell Metabolism. 2009;10(6):491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kaneto H, Nakatani Y, Miyatsuka T, et al. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nature Medicine. 2004;10(10):1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 137.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 138.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 139.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 140.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 141.Deng J, Lu PD, Zhang Y, et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and Cellular Biology. 2004;24(23):10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gregor MF, Hotamisligil GS. Adipocyte stress: the endoplasmic reticulum and metabolic disease. Journal of Lipid Research. 2007;48(9):1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- 143.Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 144.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. American Journal of Physiology, Endocrinology and Metabolism. 2007;293(4):E1118–E1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- 145.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. Journal of Clinical Investigation. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saberi M, Woods NB, de Luca C, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metabolism. 2009;10(5):419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nakamura T, Furuhashi M, Li P, et al. Double-stranded rna-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140(3):338–348. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and vascular effects of tumor necrosis factor-α blockade with etanercept in obese patients with type 2 diabetes. Journal of Vascular Research. 2005;42(6):517–525. doi: 10.1159/000088261. [DOI] [PubMed] [Google Scholar]
- 149.Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, et al. Anti-tumor necrosis factor-α blockade improves insulin resistance in patients with rheumatoid arthritis. Clinical and Experimental Rheumatology. 2006;24(1):83–86. [PubMed] [Google Scholar]
- 150.Goldfine AB, Silver R, Aldhahi W, et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clinical and Translational Science. 2008;1(1):36–43. doi: 10.1111/j.1752-8062.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115(4):518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- 154.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature. 2005;437(7059):759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ialenti A, Grassia G, di Meglio P, Maffia P, Di Rosa M, Ianaro A. Mechanism of the anti-inflammatory effect of thiazolidinediones: relationship with the glucocorticoid pathway. Molecular Pharmacology. 2005;67(5):1620–1628. doi: 10.1124/mol.104.004895. [DOI] [PubMed] [Google Scholar]
- 156.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007;356(24):2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 157.Singh S, Loke YK, Furberg CD. Thiazolidinediones and heart failure: a teleo-analysis. Diabetes Care. 2007;30(8):2148–2153. doi: 10.2337/dc07-0141. [DOI] [PubMed] [Google Scholar]
- 158.Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. Canadian Medical Association Journal. 2009;180(1):32–39. doi: 10.1503/cmaj.080486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kesavulu MM, Kameswararao B, Apparao Ch, Kumar EGTV, Harinarayan CV. Effect of ω-3 fatty acids on lipid peroxidation and antioxidant enzyme status in type 2 diabetic patients. Diabetes and Metabolism. 2002;28(1):20–26. [PubMed] [Google Scholar]
- 161.Hu FB, Cho E, Rexrode KM, Albert CM, Manson JE. Fish and long-chain ω-3 fatty acid intake and risk of coronary heart disease and total mortality in diabetic women. Circulation. 2003;107(14):1852–1857. doi: 10.1161/01.CIR.0000062644.42133.5F. [DOI] [PubMed] [Google Scholar]
- 162.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metabolism. 2008;8(4):301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]