Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP (original) (raw)
. 2000 Feb 1;14(3):272–277.
Abstract
Mice with monoallelic inactivation of the CBP gene develop highly penetrant, multilineage defects in hematopoietic differentiation and, with advancing age, an increased incidence of hematologic malignancies. The latter are characterized, at least in some cases, by loss of heterozygosity (LOH) at the CBP locus. No such pathology was observed in wild-type or p300 heterozygous null mice of the same age and genetic background. Thus, a full complement of CBP, but not p300, is required for normal hematopoietic differentiation. These results also provide the first experimental evidence for the hypothesis that CBP has tumor-suppressing activity.
Keywords: CBP, p300, hematopoiesis, LoH, tumor suppressor
The CREB-binding protein (CBP) and the highly related protein, p300, are transcriptional coactivators that facilitate gene expression through three different mechanisms. First, CBP and p300 function as molecular scaffolds coupling a multitude of different transcription factors (Eckner 1996) to the core transcriptional machinery (Nakajima et al. 1997). Second, CBP and p300 further facilitate transcription through chromatin remodeling by histone acetyltransferase (HAT) activity inherent to CBP/p300 (Bannister and Kouzarides 1996; Ogryzko et al. 1996) and/or to two associated proteins—the SRC-1 family (Yao et al. 1996) and pCAF (Yang et al. 1996). Third, acetylation of associated proteins by CBP/p300 contributes to the activation of at least two CBP/p300-interacting transcription factors, p53 (Gu and Roeder 1997) and the hematopoietic differentiation factor, GATA-1 (Blobel et al. 1998).
CBP/p300 are integrators of key events in disparate signal transduction pathways (Shikama et al. 1997), and both are required for normal development, as mouse embryos nullizygous for either gene die by embryonic day 10.5 (E10.5) (Yao et al. 1998; Oike et al. 1999b). The importance of CBP during development is further underscored by the fact that, in humans, monoallelic mutation of the CBP locus is the genetic basis for Rubinstein-Taybi syndrome (RTS) (Petrij et al. 1995), a disease characterized by craniofacial, skeletal, and cardiac defects, as well as growth and mental retardation (Giles et al. 1998). Mice carrying a single inactivated CBP allele recapitulate many of the phenotypic characteristics of RTS patients (Tanaka et al. 1997; Oike et al. 1999a).
CBP and p300 are targeted by the viral oncoproteins adenovirus E1A and SV40 large T antigen (Whyte et al. 1989; Eckner et al. 1994). Interaction of these proteins with p300/CBP is essential for the full expression of their transforming function, although a molecular understanding of how these interactions contribute to neoplastic transformation has been unclear. By analogy with the nuclear pocket proteins (pRB, p103, and p130) which are inactivated by binding of E1A and large T antigen, there has been speculation that CBP/p300 function as tumor suppressors (Giles et al. 1998). The fact that RTS patients have an increased risk of cancer (Miller and Rubinstein 1995) and the finding of biallelic mutations of p300 in certain gastrointestinal tumors (Muraoka et al. 1996) are consistent with the speculation. To date, however, there has been no reported experimental evidence for this hypothesis.
Results
CBP is required during murine embryogenesis
The murine CBP gene was targeted by homologous recombination with a vector in which exons encoding the CH1 domain (amino acids 340–443) were deleted and replaced by a neomycin resistance cassette (Fig. 1A). Correct targeting was verified by Southern blot analysis with probes flanking the targeted sequences (Fig. 1B). To confirm that the CBP allele had been inactivated, we analyzed protein levels in E9.0 embryos resulting from mating between CBP heterozygous parents. Consistent with the results of the DNA analysis, the CBP protein level was reduced in heterozygous mutant embryos and absent from homozygous mutant embryos when compared with wild-type (WT) embryos from the same litter (Fig. 1C). Western blot analysis with monospecific antibodies to the amino terminus of CBP (Fig. 1C) or to an internal epitope (data not shown) demonstrated no truncated protein products in embryos carrying one or two mutant alleles. Thus, we concluded that targeting of the CBP gene had generated a null allele.
Figure 1.



Generation of CBP knockout mice. (A) Schematic representation of the CBP WT allele, targeting vector, and the predicted mutant CBP allele resulting from homologous recombination. (A) _Asp_718; (Bg) _Bgl_II; (M) _Mun_I; (Nc) _Nco_I; (S) _Sal_I. (B) Southern blot analysis of genomic DNA derived from WT (+/+),CBP+/− and CBP−/− embryos. The sizes (kb) of the expected fragments corresponding to the WT and mutant (MUT) alleles are indicated. (C) CBP amino-terminal-specific immunoblot analysis of standardized quantities of cell extracts derived from embryos of the indicated genotypes. Immunoblots for GAPDH and p300 are shown for loading normalization.
Mating CBP heterozygotes failed to produce viable CBP−/− offspring. At E9.5, embryos of all three genotypes were fully represented, but, by E10.5, all CBP−/− embryos were either dead or moribund (data not shown). As reported previously, CBP−/− embryos displayed severe open neural tube defects similar to their p300−/− counterparts (Yao et al. 1998). Thus, nullizygosity at CBP led to embryonic lethality. In keeping with this conclusion, lethality has also been noted among embryos homozygous for a mutant CBP allele encoding a truncated protein product (Oike et al. 1999b).
Phenotypic characteristics of CBP+/− mice
Our CBP+/− mice demonstrated many of the features reported previously for such mice (Tanaka et al. 1997; Oike et al. 1999a) including growth retardation and craniofacial abnormalities (data not shown) reminiscent of patients with RTS (Giles et al. 1998). To further define the spectrum of phenotypic abnormalities resulting from hemizygosity at the CBP locus, CBP+/− animals were followed with advancing age, along with their WT littermates and p300+/− mice (Yao et al. 1998) of the same background strain (C57BL/6 × 129).
Necropsy of cohorts of randomly selected CBP+/−, p300+/−, and WT mice at 12–18 months of age revealed dramatic splenomegaly in most CBP+/− animals. On average, the spleens of CBP+/− animals weighed 1130 mg (range 150–5200 mg, n = 15), compared with 145 mg (range 60–270 mg, n = 8) for spleens from age-matched, WT littermates, and 150 mg (range 102–244, n = 6) in age- and strain-matched p300+/− animals. Splenomegaly, defined as a grossly enlarged spleen, or spleen weighing >300 mg, was present in 73% of CBP heterozygotes in the above-noted cohort. In contrast, no WT or p300+/− animals had splenomegaly. FACS analysis of spleen cells from mice of each genotype revealed increased cells of myeloid (MAC-1+/Gr-1+) and erythroid (TER119+) lineages in CBP+/− mice with splenomegaly, which was histologically correlated with the finding of extramedullary hematopoiesis (data not shown). Because extramedullary hematopoiesis is a reflection of abnormal hematopoiesis, we further analyzed the hematopoietic system in these animals.
Hematopoietic differentiation defects in CBP+/− mice
CBP+/− mice with splenomegaly, as defined above, were compared with age-matched CBP+/− mice without splenomegaly, to WT littermates, and to age and strain-matched p300+/− mice. The average age of each cohort was 17.1 ± 3.3 for CBP+/−, 18.9 ± 2.9 for WT, and 22.2 ± 6.8 months for p300+/− animals.
Overall bone marrow cellularity (Fig. 2A) and the abundance of all hematopoietic cell types (Fig. 2C) were significantly diminished in CBP+/− animals with splenomegaly (statistically significant differences denoted by asterisk). In keeping with these findings, these animals had deficiencies in pre-B cell and myeloid colony-forming progenitor cells (Fig. 2D). Peripheral blood (PB) analysis demonstrated a deficit in B cells, as well as a statistically significant increase in myeloid cells (Fig. 2B). CBP+/− mice without splenomegaly did not manifest the full spectrum of abnormalities seen in CBP+/− mice with splenomegaly, except for defects in the bone marrow B cell compartment (Fig. 2C,D). In contrast, p300+/− animals demonstrated no measurable hematopoietic differentiation abnormalities (Fig. 2A–D).
Figure 2.

Defective hematopoiesis in CBP+/− mice. Analysis of hematopoietic subpopulations in PB and bone marrow from WT (open bars), p300+/− (hatched bars), CBP+/− mice without splenomegaly (shaded bars), and CBP+/− mice with splenomegaly (solid bars). (A) Total mononuclear cell counts; (C) phenotypic analysis; (D) analysis of the number of CFCs from bone marrow cell suspensions. The actual numbers of myeloid (M) colonies are 10 times the values presented in D. (B) Quantification of B and T lymphocytes, myeloid, and erythroid (E) cells in PB. Values shown represent the mean ± s.d. of three or more separate experiments. Significant differences (P<0.05) are indicated with an asterisk.
To determine whether the hematopoietic abnormalities in CBP+/− mice were age dependent, analysis of young (<3 months of age) CBP+/− mice and their WT littermates was undertaken. The average age of the WT animals was 10.3 ± 1.4 weeks, compared with 8.7 ± 0.3 for the CBP+/− animals. There was no splenomegaly in any of the mice. Overall bone marrow cellularity was similar between young CBP+/− animals and their WT littermates (Table 1). However, there was a subtle, but significant deficit in B and T cells in the bone marrow of the former, although this did not extend to a deficit in colony-forming pre-B cells (Table 1).
Table 1.
Hematopoietic subpopulations in the bone marrow of young CBP+/− compared to WT mice
| | WT (n = 4) | CBP+/− (n = 7) | | | ----------------------- | ----------------- | --------------------- | | Number of cells (×106) | | | | Total | 18.7 (3.5) | 15.4 (3.6) | | B (B220+) | 4.6 (0.3) | 3.3 (1.3)a | | T (Ly-1+) | 0.4 (0.04) | 0.2 (0.05)a | | M (Gr-1/Mac-1+) | 8.0 (2.8) | 7.5 (1.8) | | E (Ter119+) | 4.4 (3.2) | 2.6 (1.3) | | Number of CFU per femur | | | | B | 17342 (10075) | 13792 (9873) | | M | 80208 (28900) | 69960 (19976) | | E | 7571 (2793) | 6143 (3047) |
Taken together, these results demonstrate that a full complement of CBP is required for normal hematopoietic differentiation. Abnormalities in B and T cell differentiation were apparent by 3 months of age in the heterozygotes, with more pervasive multilineage defects and bone marrow hypocellularity developing later in life. Splenomegaly in these animals likely resulted from compensatory extramedullary hematopoiesis secondary to bone marrow failure. No such abnormalities were observed in matched p300+/− mice.
Hematologic malignancies in CBP+/− mice
The pervasive defects in hematopoietic differentiation noted in the CBP heterozygotes and the increased risk of cancers in RTS patients (Miller and Rubinstein 1995) prompted us to search for tumor formation in these animals. Although no tumors were found in CBP heterozygotes <1 year of age (_n_ > 10), hematologic neoplasia were noted in older animals. Among a random cohort of 18 CBP+/− mice between 10–21 months of age, 4 animals presented with gross tumors at necropsy (Table 2). Histiocytic sarcomas (HSs), a tumor of hematopoietic origin (Frith et al. 1993), were noted in two animals (Fig. 3D), and leukemias in two others (Table 2), myelogenous in one case and lymphocytic in the other (Fig. 3F). No hematologic tumors were found in a random cohort of age-matched, WT littermates (n = 20) or in p300 heterozygous mutants of the same age and strain (n = 15).
Table 2.
Hematologic malignancies in CBP+/− mice
| Mouse no. | Age (months) | Splenomegaly | Primary tumora | Transplantedb | Tumor in recipientsa |
|---|---|---|---|---|---|
| 1 | 14 | N | HS | N | |
| 2 | 18 | Y | none | N | |
| 3 | 15 | N | none | N | |
| 4 | 15 | N | none | Y | HS |
| 5 | 15 | Y | none | Y | MM |
| 6 | 20 | Y | HS | N | |
| 7 | 20 | Y | none | N | |
| 8 | 17 | N | none | Y | |
| 9 | 11 | Y | none | Y | |
| 10 | 17 | Y | none | Y | HS |
| 11 | 15 | Y | none | Y | |
| 12 | 18 | Y | none | Y | |
| 13 | 21 | N | none | Y | |
| 14 | 12 | Y | LL | Y | LL |
| 15 | 10 | N | none | Y | |
| 16 | 10 | Y | none | Y | |
| 17 | 13 | N | none | N | |
| 18 | 13 | Y | ML | N |
Figure 3.
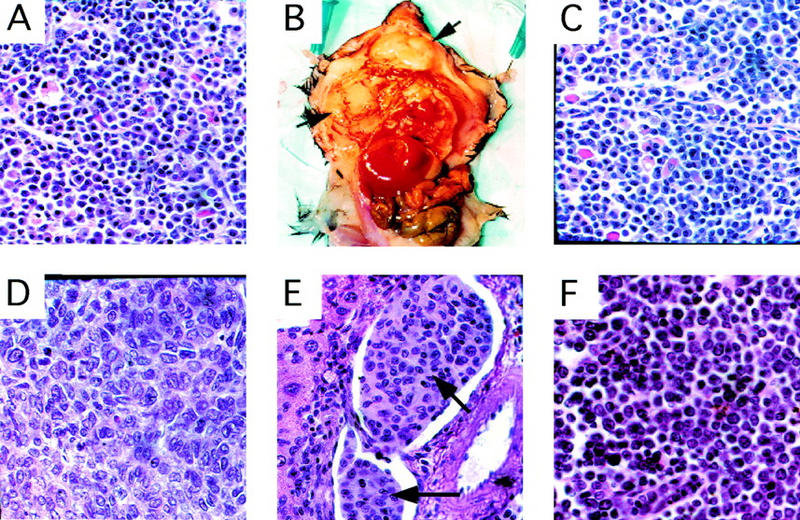
Histology of primary and transplanted tumors . (A) Primary plasma-cell infiltrated lymph node from a CBP+/− mouse (Table 2, mouse 5). Transplantation of bone marrow from this animal into WT recipient mice resulted in diffuse plasmocytomas (arrows) in recipients (B, C). (D) Primary HS in a CBP+/− mouse. (E) HS (arrow) infiltrating hepatic vasculature, arising in a WT recipient transplanted with bone marrow from a CBP+/− donor without overt tumor. (F) Infiltration of a lymph node with primary lymphocytic leukemia in a CBP+/− mouse.
In humans, myelodysplastic syndrome (MDS) is a disease characterized by pervasive defects in hematopoiesis and a high incidence of progression to leukemia. Given the highly penetrant deficiencies in hematopoietic differentiation and the presence of hematologic malignancies in some CBP+/− mice, a search was performed for covert tumorigenic cells in the CBP+/− mice from the above-noted cohort that were free of obvious tumor at the time of necropsy. Bone marrow and spleen cells from such CBP+/− mice were transplanted into sublethally irradiated WT recipients, and the latter were observed for tumor formation. Transplants from 10 CBP+/− mice without overt tumor masses resulted in the appearance of gross malignancies in recipient animals in three cases (Table 2). From a CBP heterozygote with plasma cell-infiltrated lymph nodes (Table 2, mouse 5; Fig. 3A), aggressive and diffusely invasive plasmacytomas arose in multiple transplant recipients (Fig. 3B,C). Renal amyloid deposition, monoclonal gammopathy, and plasma cell aneuploidy, all characteristics of multiple myeloma-like disease, were also found in these recipients (data not shown). Bone marrow transplants from two other CBP+/− mice without overt pathology (Table 2, mice 4 and 10) also resulted in tumors in the recipients, each identified as invasive HS (Fig. 3E). In addition, bone marrow from the above-noted CBP+/− mouse with primary lymphocytic leukemia (Table 2, mouse 14) gave rise to the same disease in multiple recipients. As expected, bone marrow or spleen cell transplants from WT littermates (n = 5) were nontumorigenic in any recipients.
Overall, at least 39% of the CBP+/− mutant mice between 10–21 months of age had overt tumors or harbored covert tumorigenic cells detected after transplantation (Table 2). In comparison, no hematologic tumors were found in WT littermates (n = 20) or p300+/− (n = 15) animals, matched for age and strain. Although the numbers of animals available for analysis were limited, the incidence of hematologic tumors in the CBP+/− cohort was significantly elevated in comparison with their wild-type littermates (P = 0.003, Fisher exact test) or to p300+/− animals (P = 0.007).
The tumors that appeared in recipient animals were uniformly of donor (CBP+/−) origin, as indicated by the presence of a neomycin resistance allele in their genomic DNA, determined both by PCR (data not shown) and/or by Southern blot analysis (see below).
Loss of heterozygosity at the CBP locus
There has been speculation that CBP may have tumor suppressor activity (Giles et al. 1998) on the basis of an increased incidence of cancer in RTS patients (Miller and Rubinstein 1995), although loss of heterozygosity (LOH) at the CBP locus has not been demonstrated in these cases. In tumors arising in our CBP+/− animals, we determined the status of the remaining WT CBP allele by Southern blot analysis. Genomic DNA derived from a mediastinal mass in a CBP+/− mouse with primary myelogenous leukemia (Table 2, mouse 18) revealed specific loss of the WT CBP allele, with retention of the mutant null allele (Fig. 4A). Southern blot analysis of the DNA from a HS (Fig. 4B) and from multiple plasmacytomas (Fig. 4C) arising in bone marrow-transplanted recipients also revealed specific loss of the WT CBP allele in tumors arising after transplantation from two separate donor animals (Table 2, mice 5 and 10). Southern blot analysis of the p300 locus revealed no diminution in signal in the tumor cells in comparison with normal tissue DNA (data not shown). Further supporting the existence of LOH in the plasmacytomas that were tested, immunoblotting of extracts of tumor tissue with CBP- and p300-specific monoclonal antibodies revealed loss of CBP protein expression, with preservation of a strong p300 protein signal (Fig. 4D).
Figure 4.
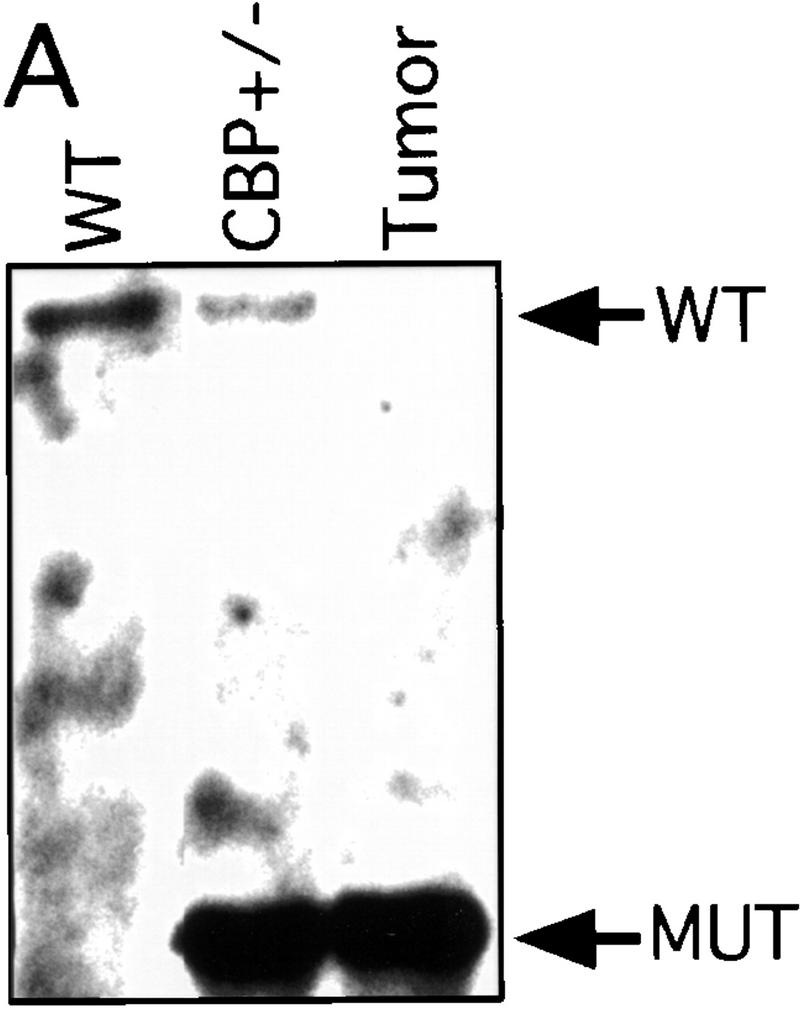



LOH at the CBP locus in tumors. Southern blot analysis of tumors isolated from a mediastinal mass in an animal with myelogenous leukemia (A) (Table 2, mouse 18); HS arising in a WT recipient transplanted with bone marrow from a CBP+/− mouse (B); and plasmacytomas arising in multiple recipients (T2, T5, T6) of bone marrow from a CBP+/− mouse (C). Comparisons are made to WT, and CBP+/− controls. Expected WT and targeted mutant (MUT) alleles are indicated by arrows. (D) p300 and CBP Western blot analysis of protein extracts from plasmacytomas arising in multiple transplant recipients (T2, T5, T6), compared with equivalent amounts of protein extract from spleens of WT and CBP+/− controls.
Discussion
CBP/p300 have been shown to interact with a number of transcription factors important at various steps of hematopoietic differentiation. It has been shown previously that embryos homozygous for a truncated CBP allele have defects in primitive hematopoiesis and vasculoangiogenesis (Oike et al. 1999b). In this work we demonstrate that inactivation of even a single CBP allele results in pervasive defects in definitive hematopoietic differentiation. In contrast, no such abnormalities were found in p300+/− mice. Thus, our results demonstrate that there is a critical requirement for a full complement of CBP, but not p300, for normal execution of this complex process.
Results presented here and reported previously demonstrate that monoallelic loss of CBP constitutes a state of haplo-insufficiency for a number of developmental processes in mice (Tanaka et al. 1997; Oike et al. 1999a), as is the case in humans with RTS (Petrij et al. 1995). We have now demonstrated that mice with monoallelic mutation of CBP also display an increased cancer risk, similar to the increased cancer risk observed in patients with RTS (Miller and Rubinstein 1995). CBP/p300 are obligate cellular targets of the E1A and large T antigen oncoproteins, and E1A must bind both the nuclear pocket proteins (pRB, p107, and p130) and CBP/p300 for the subsequent transformation of primary cultured cells (Svensson et al. 1991). Whereas pRB is a classical tumor suppressor, the function of which is inactivated by the binding of E1A, the case for CBP/p300 having tumor-suppressing function has, heretofore, been speculative. The high incidence of tumors in aging CBP hemizygous mice and the existence of LOH at the CBP locus in tumors provide the first experimental evidence that CBP possesses tumor-suppressing activity. In this regard, it should be noted that the available data do not rule out the additional possibility that the CBP+/− state represents a state of haploinsufficiency for CBP-mediated tumor suppression, akin to the situation with p27kip1 (Fero et al. 1998).
Given the multitude of transcriptional events in which CBP participates, the exact mechanism whereby loss of CBP contributes to hematologic tumorigenesis remains unclear. The relatively penetrant and pervasive antecedent hematopoietic differentiation defects in CBP+/− mice raises the possibility that defective differentiation may result in an increased pool of primitive hematopoietic cells at risk for the accumulation of additional genetic changes contributing to neoplastic transformation. CBP and/or p300 play a role in the maintenance of genomic integrity (Caporossi and Bacchetti 1990; Drews et al. 1998), and loss of CBP function could, in theory, contribute to genomic instability.
Recent data have also demonstrated that disruption of normal balance between the coactivator and corepressor function of certain transcription factors may be important in leukemogenesis, at least in the case of certain acute myelogenous leukemias (for review, see Redner et al. 1999). In these cases, translocations targeting RARα or AML1 results in fusion proteins with preferential affinity for corepressor complexes instead of CBP/p300 coactivator complexes. This molecular transformation presumably leads to an alteration in gene expression patterns in hematopoietic precursor cells, which is linked to the ensuing leukemogenesis. Conceivably, loss of a single copy of CBP may result in aberrant function of certain hematopoietic transcription factors due to an imbalance in coactivator/corepressor complex interactions, especially for transcription factors preferentially associated with CBP and not p300. Loss of both copies of CBP may result in further coactivator/corepressor imbalance, and may be functionally similar to diminished affinity for coactivator complexes due to translocation in critical hematopoietic transcription factors.
Another mechanism whereby loss of CBP may contribute to tumorigenesis is suggested by recent work demonstrating that, in Drosophila, dCBP acetylates dTCF and is a negative regulator of the Wnt-signaling pathway (Waltzer and Bienz 1998). In mammalian cells, it has been suggested that c-myc is a downstream target of this pathway (He et al. 1998). Thus, one might speculate that loss of CBP results in deregulated TCF activity and, hence, deregulated expression of c-myc, a powerful oncogene active in human (Facchini and Penn 1998) and murine tumor development (Potter 1997). Validation of these possibilities requires further investigation.
Finally, the results presented here provide an alternative hypothesis as to how certain recurrent translocations involving the CBP locus may contribute to the development of certain human leukemias and MDS. These translocations, whereby MOZ or MLL are translocated to the CBP locus (Giles et al. 1998), result in the synthesis of fusion proteins containing significant segments of the CBP protein (Borrow et al. 1996; Sobulo et al. 1997). These translocation events have been postulated previously to result in a gain of function for the fusion partners. In light of our results, one might speculate that such translocations may render the CBP locus haplo-insufficient for its hematopoietic differentiation and/or tumor suppressor functions, thereby resulting in defective hematopoiesis and leukemogenesis.
Materials and methods
Generation of CBP knockout mice
A 129 mouse genomic λ phage library (Stratagene) was screened with a probe corresponding to the CH1 region of the murine CBP cDNA (351-bp _Mfe_I fragment). The three exons spanning the CH1 region were mapped by Southern blotting and the precise exon-intron boundaries were determined by sequencing. Two of the exons were deleted by _Mfe_I and _Nco_I digestion and replaced with a PGK-neomycin cassette. PGK-TK was also inserted for negative selection. T/C ES cells (a gift of Dr. P. Leder, Harvard Medical School, Boston, MA) were electroporated with the targeting plasmid, linearized with _Not_I. From 350 ES cell colonies surviving selection with G418 and Gancyclovir, 2 contained a correctly targeted allele. One of the ES cell clones gave rise to chimeric mice with a C57BL/6 background. These mice then transmitted the targeted allele via the germ line.
PCR and Southern and Western blot analyses
Genomic DNA or cell lysates were prepared from tail sections, yolk sacs, or tumor samples by standard methods (Ausubel et al. 1988). Genomic DNA was analyzed either by Southern blot analysis or by multiplex PCR (Ausubel et al. 1988) specific for the NEO gene (reflecting the presence of the targeted allele) and one of the targeted CBP exons (reflecting the presence of an untargeted, wild-type CBP allele). Sequences of these primers and PCR conditions are available upon request. For Southern blot analysis, we utilized the 5′ probe depicted in Figure 1 and hybridized it to _Bgl_II-digested genomic DNA. Western blot analysis was performed by standard techniques (Harlow and Lane 1988), with mouse monoclonal antibodies specific for GAPDH (Biodesign International), the amino terminus of CBP (C-1, Santa Cruz Biotechnology), an internal epitope of CBP (AC26), or p300 (RW128) (Yao et al. 1998).
Hematologic analysis
Hematologic analyses was performed as described previously (Rebel et al. 1999). Briefly, PB cells were obtained by cardiac puncture after mice were anaesthetized. Cells were then treated with ammonium chloride to lyse the erythrocytes. Bone marrow cells were harvested by flushing the marrow from both femurs and tibia. Phenotypic analysis by FACS was performed by measuring cells reactive with lineage-specific antibodies (PharmMingen) as follows: 6B2 (B cells), 53-7.3 (T cells), 8C5 and M1/70 (monomyelocytic cells), and Ter119 (erythroid cells). Colony forming cells (CFCs) were assayed in methylcellulose-containing medium supplemented with specified cytokines (Stem Cell Technologies). For detection of pre-B CFCs, 5 × 104 cells per dish were plated, and, for myeloid and erythroid CFCs, 1.5 × 104 cells were plated per dish. After 7 (for pre-B CFC) and 12 days (for myeloid and erythroid CFC), colonies were counted. Myeloid colonies represent all colonies that contain granulocytes/macrophages (G/M), and include multi-lineage GEMM colonies, whereas the erythroid colonies were scored as colonies containing only erythroid and/or megakaryocytic cells.
Splenocyte and bone marrow transplantation
Wild-type C57BL/6 recipient mice were irradiated sublethally with a single dose of 425–500 cGy. Bone marrow from donor mice was prepared as described above, and spleen cells were prepared by maceration. A total of 5 × 106 bone marrow cells or 1 × 107 spleen cells were injected intravenously into the irradiated, recipient mice.
Acknowledgments
This work was supported by grants from the National Cancer Institute/NIH and Novartis Pharmaceutical Corporation (to D.M.L.), the Damon Runyon-Walter Winchill Cancer Research Fund (to T-.P.Y.), the American Society of Hematology (to V.I.R.), and the Howard Hughes Medical Institute (to A.L.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL david_livingston@dfci.harvard.edu; FAX (617)632-4381.
E-MAIL yao00001@mc.duke.edu; FAX (919) 681-8461.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: Wiley-Interscience; 1988. [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH. CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci. 1998;95:2061–2066. doi: 10.1073/pnas.95.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow J, Stanton VJ, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- Caporossi D, Bacchetti S. Definition of adenovirus type 5 functions involved in the induction of chromosomal aberrations in human cells. J Gen Virol. 1990;71:801–808. doi: 10.1099/0022-1317-71-4-801. [DOI] [PubMed] [Google Scholar]
- Drews R, Kolker M, Moran C, Sachar D, Chan V, Schnipper L. Genetic analysis of adenovirus E1A: Induction of genetic instability and altered cell morphologic and growth characteristics are segregatable functions. Mutat Res. 1998;421:9–25. doi: 10.1016/s0027-5107(98)00149-3. [DOI] [PubMed] [Google Scholar]
- Eckner R. p300 and CBP as transcriptional regulators and targets of oncogenic events. Biol Chem. 1996;377:685–688. [PubMed] [Google Scholar]
- Eckner R, Arany Z, Ewen M, Sellers W, Livingston DM. The adenovirus E1A-associated 300-kD protein exhibits properties of a transcriptional coactivator and belongs to an evolutionarily conserved family. Cold Spring Harb Symp Quant Biol. 1994;59:85–95. doi: 10.1101/sqb.1994.059.01.012. [DOI] [PubMed] [Google Scholar]
- Facchini LM, Penn LZ. The molecular role of Myc in growth and transformation: Recent discoveries lead to new insights. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith CH, Ward JM, Chandra M. The morphology, immunohistochemistry, and incidence of hematopoietic neoplasms in mice and rats. Toxicol Pathol. 1993;21:206–218. doi: 10.1177/019262339302100213. [DOI] [PubMed] [Google Scholar]
- Giles RH, Peters DJ, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998;14:178–183. doi: 10.1016/s0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995;56:112–115. doi: 10.1002/ajmg.1320560125. [DOI] [PubMed] [Google Scholar]
- Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong JM, Iwama T, Miyaki M. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, Montminy M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. Truncated CBP protein leads to classical rubinstein-taybi syndrome phenotypes in mice: Implications for a dominant-negative mechanism. Hum Mol Genet. 1999a;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, Yasue H, Araki K, Yamamura K, Suda T. Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood. 1999b;93:2771–2779. [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van OG, Goodman RH, Peters DJ, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Potter M. Experimental plasmacytomagenesis in mice. Hematol Oncol Clin North Am. 1997;11:323–347. doi: 10.1016/s0889-8588(05)70434-2. [DOI] [PubMed] [Google Scholar]
- Rebel VI, Hartnett S, Hill GR, Lazo-Kallanian SB, Ferrara JLM, Sieff CA. Essential role for the p55 tumor necrosis factor receptor in regulating hematopoiesis at a stem cell level. J Exp Med. 1999;190:1493–1505. doi: 10.1084/jem.190.10.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redner RL, Wang J, Liu JM. Chromatin remodeling and leukemia: New therapeutic paradigms. Blood. 1999;94:417–428. [PubMed] [Google Scholar]
- Shikama N, Lyon L, La Thangue NB. The p300/CBP family: Integrating signals with transcription factors and chromatin. Trends Cell Biol. 1997;7:230–236. doi: 10.1016/S0962-8924(97)01048-9. [DOI] [PubMed] [Google Scholar]
- Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, Housman D, Doggett NA, Rowley JD, Zeleznik LN. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3) Proc Natl Acad Sci. 1997;94:8732–8737. doi: 10.1073/pnas.94.16.8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson C, Bondesson M, Nyberg E, Linder S, Jones N, Akusjarvi G. Independent transformation activity by adenovirus-5 E1A-conserved regions 1 or 2 mutants. Virology. 1991;182:553–561. doi: 10.1016/0042-6822(91)90596-4. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: A partial similarity with Rubinstein-Taybi syndrome. Proc Natl Acad Sci. 1997;94:10215–10220. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L, Bienz M. Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature. 1998;395:521–525. doi: 10.1038/26785. [DOI] [PubMed] [Google Scholar]
- Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Yao TP, Ku G, Zhou N, Scully R, Livingston DM. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci. 1996;93:10626–10631. doi: 10.1073/pnas.93.20.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]