Drosophila P-element transposase is a novel site-specific endonuclease (original) (raw)
Abstract
We developed in vitro assays to study the first step of the P-element transposition reaction: donor DNA cleavage. We found that P-element transposase required both 5′ and 3′ P-element termini for efficient DNA cleavage to occur, suggesting that a synaptic complex forms prior to cleavage. Transposase made a staggered cleavage at the P-element termini that is novel for all known site-specific endonucleases: the 3′ cleavage site is at the end of the P-element, whereas the 5′ cleavage site is 17 bp within the P-element 31-bp inverted repeats. The P-element termini were protected from exonucleolytic degradation following the cleavage reaction, suggesting that a stable protein complex remains bound to the element termini after cleavage. These data are consistent with a cut-and-paste mechanism for P-element transposition and may explain why P elements predominantly excise imprecisely in vivo.
Keywords: P transposon, transposition, cleavage site, recombinase
Transposons are mobile genetic elements that are present in many organisms. In general, the transposable element encodes a recombinase that mediates the DNA cleavage and strand transfer reactions during transposition. The recombinase can be a single polypeptide, like the transposase proteins encoded by the Tc elements, phage Mu, Tn_10,_ and P elements, or a combination of polypeptides, like with Tn_7._ The type of DNA cleavage made by the transposase protein, however, is common to all transposition reactions. All transposases cleave at the end of the transposon DNA to expose a 3′-hydroxyl group that is covalently joined to a new target site. The mechanism of transposition is determined by whether cleavage occurs at the 5′ ends. If the 5′ end of the element is cleaved to generate an excised transposon intermediate, the element transposes by a cut-and-paste mechanism. Tn_7_ (Bainton et al. 1991), Tn_10_ (Benjamin and Kleckner 1992), the Tc elements (van Luenen et al. 1994; Vos et al. 1996), and the P element (Kaufman and Rio 1992) all transpose by such a mechanism. The nature of the nontransferred strand cleavage, however, is variable and occurs either 3 bp outside [for Tn_7_ (Bainton et al. 1991; Gary et al. 1996)], 2 bp inside [for the Tc elements (van Luenen et al. 1994; Vos et al. 1996)], or at the transposon termini [for Tn_10_ (Benjamin and Kleckner 1992)]. If the 5′ end of the element is not cleaved, the element transposes by a replicative transposition mechanism in which the transposon remains attached to both the donor site and the target site. The intermediate is replicated by host replication proteins to produce two copies of the element in a structure called a cointegrate. The bacteriophage Mu can transpose by this type of mechanism (Craigie and Mizuuchi 1985; Mizuuchi 1992a). Analogous to Mu, retroviral integration, like HIV integration, does not require processing at the 5′ end of the reverse-transcribed genome by the integrase protein prior to insertion into the host genome (Engelman et al. 1991). A staggered target-site cleavage is made by most transposases and integrases, and DNA repair of the gaps that flank the newly inserted element generates the characteristic target-site duplications present after insertion.
The P transposable element of Drosophila melanogaster is one of the best characterized eukaryotic transposons. Full-length P elements are 2.9 kb in length and encode an 87-kD transposase protein. Transposase synthesis is restricted to the germ line by a regulated, tissue-specific pre-mRNA splicing mechanism (Laski et al. 1986). P elements transpose by a cut-and-paste mechanism and create an 8-bp target site duplication upon insertion (O’Hare and Rubin 1983; Engels et al. 1990; Kaufman and Rio 1992). The P-element transposase is unique in that GTP is required as a cofactor, in addition to magnesium, for activity (Kaufman and Rio 1992; Mul and Rio 1997). P-element transposition requires ∼150 bp of sequence at each end of the P element. These sequences include 31-bp terminal inverted repeats, internal transposase-binding sites, and internal 11-bp inverted repeats (O’Hare and Rubin 1983; Kaufman et al. 1989; Mullins et al. 1989). The terminal 31-bp inverted repeats are not bound by the transposase protein (Kaufman et al. 1989) but instead are bound site-specifically by a Drosophila_-encoded protein, the inverted repeat binding protein (IRBP) (Rio and Rubin 1988). This finding was surprising because most transposase proteins bind within the inverted repeat regions to sequences at or near the terminal nucleotides of the transposable element. For instance, the Tn_10 transposase binds to sequences within the IS_10_ terminal 23-bp inverted repeats, with the most critical contacts made at 6–13 bp (Kleckner et al. 1996). Similarly, the Tc_1_ and Tc_3_ transposases recognize sequences within 20–25 bp of either the Tc_1_ or Tc_3_ transposon terminal inverted repeats (van Luenen et al. 1993; Vos and Plasterk 1994). The sites to which transposase binds at the P-element termini are of unequal distance from the sites of cleavage (either 40 or 52 bp away) and may account for the inability of a P element with two 5′ ends to transpose in vivo (Mullins et al. 1989). During the mechanistically similar V(D)J recombination reaction (van Gent et al. 1996b), the Rag-1 and Rag-2 proteins also bind to sites of unequal distance from the site of cleavage (either 19 or 30 bp away) (Difilippantonio et al. 1996; Spanopoulou et al. 1996; Hiom and Gellert 1997).
For many transposable elements, such as Tn_10_ (Haniford et al. 1991; Sakai et al. 1995), Tn_7_ (Bainton et al. 1993; Sarnovsky et al. 1996), and Mu (Craigie and Mizuuchi 1987; Surette et al. 1987), and during V(D)J rearrangement (Eastman et al. 1996; van Gent et al. 1996a; Steen et al. 1997), the first step of the rearrangement reaction, DNA cleavage, requires synapsis of the two ends to form a stable nucleoprotein complex prior to cleavage. The subsequent breakage and joining reactions occur in the context of this synaptic complex or transpososome. For instance, during Mu transposition, a distinct series of stable nucleoprotein complexes have been observed for each step of the transposition reaction (Mizuuchi 1992b; Lavoie and Chaconas 1996). The earliest detectable complex, or LER complex, contains a transient species with the Mu left and right ends, and the internal enhancer element (Watson and Chaconas 1996). The type 0, or stable synaptic complex (SSC), contains the two Mu ends bound by a tetramer of transposase. The type 1, or cleaved donor complex (CDC), contains both transposase and the cleaved 3′ ends of the Mu DNA. The last, or strand transfer complex (STC), contains the Mu genome inserted into a new target site. Completion of the reaction involves disassembly of the transposase tetramer from the DNA by a host encoded protein, ClpX, and replication across the Mu DNA to generate a second copy of the Mu genome (Levchenko et al. 1995; Nakai and Kruklitis 1995; Kruklitis et al. 1996). Similarly, during V(D)J recombination, a stable nucleoprotein complex remains bound to the cleaved DNA intermediates following Rag-1/Rag-2 mediated cleavage (Zhu et al. 1996; Agrawal and Schatz 1997). Formation of a stable synaptic complex prior to, and after cleavage, is a way to ensure reaction fidelity through a properly positioned recombinase.
To characterize the P-element transposition reaction in more detail, we developed in vitro cleavage assays with transposase purified from Drosophila cell culture. Here, we show that P-element transposase requires both 5′ and 3′ P-element termini for efficient DNA cleavage to occur, suggesting that a synaptic complex forms on the P-element termini prior to cleavage. We mapped the cleavage sites made by transposase and show that transposase makes a staggered cleavage at the P-element termini that is novel for all known site-specific endonucleases. Similar to all transposable elements, the 3′ cleavage site occurs at the end of the P element. However, unlike any other known transposable element, the 5′ cleavage site occurs 17 bp within the P-element 31-bp inverted repeats, directly adjacent to the IRBP-binding site. The 17-nucleotide, 3′ extensions left behind at the donor site provide an explanation for the imprecise P-element excisions observed in vivo. Finally, we show that the P-element termini are protected from exonucleolytic degradation following the cleavage reaction, which suggests that a stable protein complex remains bound to the P-element termini following cleavage.
Results
Transposase only cleaves substrates containing both P-element termini
To better understand the mechanism of P-element transposition, in vitro assays were developed to study P-element transposase-mediated donor DNA cleavage activity. P-element transposase was partially purified from a stable cell line expressing transposase under the control of the metallothionein promoter (Mul and Rio 1997). This cell line differs from the previously reported transposase-producing cell line (Kaufman et al. 1989) in that 25% of the transposase amino terminus was chemically resynthesized to alter codon usage to the most frequently used codons in Drosophila (Lee et al. 1996). The partially purified transposase fraction (referred to as H0.1FT) was highly active for P-element transposition by use of a genetic-based in vitro assay (Kaufman and Rio 1992).
We used linear P-element-containing DNA fragments as substrates to study the first step of the transposition reaction, donor DNA cleavage. Linear DNA has been used as a substrate by transposase in strand transfer reactions in vitro (Kaufman and Rio 1992). The H0.1FT fraction was tested for cleavage activity by the assay described in Figure 1A. Restriction endonuclease fragments carrying internally deleted P elements of 303 or 628 bp in length were radiolabeled by end-filling. These substrates contain both left (5′) and right (3′) P-element termini that include the 31-bp terminal inverted repeats, the internal transposase-binding sites, and the internal 11-bp inverted repeat enhancer elements. In addition, a DNA fragment containing a single left (5′) P-element end was radiolabeled and tested for cleavage activity by transposase. If transposase can perform the cleavage reaction, then products corresponding to the radiolabeled cleaved flanking DNA should be observed following native gel electrophoresis and autoradiography. We found that only substrates containing both P-element termini were cleaved in the presence of the cofactor, GTP (Fig. 1B, lanes 1–4 for the 628-bp P element, lanes 9–12 for the 303-bp P element). The single left (5′) P element end-containing fragment was not used as a substrate (Fig. 1B, lanes 5–8). These data suggest that during P-element transposition, assembly of a cleavage complex requires both P-element termini and are consistent with the finding that substrates containing two left (5′) P-element ends are not active for transposition in vivo (Mullins et al. 1989).
Figure 1.




Transposase requires both 5′ and 3′ P-element termini during cleavage. (A) Schematic diagram of the assay used to study the first step in the P-element transposition reaction: donor DNA cleavage. Restriction endonuclease fragments carrying internally deleted P elements of 303 or 628 bp in length (as indicated by arrows), along with varying lengths of flanking DNA (hatched and shaded boxes), were radiolabeled at the 3′ end as indicated by an asterisk. These substrates contain both P-element termini that include the 31-bp terminal inverted repeats, the internal transposase binding sites, and the internal 11-bp inverted repeat enhancer elements. A DNA fragment containing a single 5′ P-element end was also radiolabeled and tested as a cleavage substrate for transposase. The predicted sizes for each cleavage product are as indicated. (B) Partially purified transposase (H0.1FT) was tested for double-stranded DNA cleavage activity as diagrammed in A. Reactions were performed at 27°C for 2 hr, and products analyzed by native PAGE and autoradiography. Shown is an autoradiograph from an experiment in which three different substrates were tested for cleavage activity: the 628-bp P element-containing fragment (lanes 1–4), the 175-bp single left (5′) P-element end-containing fragment (lanes 5–8), and the 303-bp P-element-containing fragment (lanes 9–12). For each substrate, two different amounts of the H0.1FT were tested for activity (either ∼2 μg, or 4 μg total protein, as indicated by + or ++). GTP-dependent cleavage products are indicated by arrows. (M) Radiolabeled pBR322 _Msp_I molecular weight markers. (C) Schematic diagram of the assay used to test whether P-element transposase requires both 5′ and 3′ P-element termini for DNA cleavage to occur. Plasmid substrates containing the 303-bp P element (P 5′3′, left), the left end only (P5′, middle), or the right end only (P3′, right) are as diagrammed. The predicted sizes for each product following transposase-mediated cleavage and restricion endonuclease digestion with the indicated enzymes are outlined at the bottom. (D) Partially purified transposase (H0.1FT) was tested for cleavage activity as diagrammed in Fig. 1C. Reactions were performed at 27°C for 2 hr, and products analyzed by DNA blot hybridization with a radiolabeled P-element probe following native PAGE. Shown is an autoradiograph from an experiment in which substrates containing both P-element termini (P 5′3′, lanes 1,2; only the 251-bp _Acc_I fragment is shown), each terminus alone (P 5′, lanes 3,4; P 3′, lanes 5,6), or a mixture containing equimolar amounts of both P 5′ and P 3′ (lanes 7 and 8) were tested for cleavage by transposase. For each substrate, ∼2 μg of the H0.1FT was tested for activity. GTP-dependent cleavage products are indicated by arrows. (M) radiolabeled pBR322 _Msp_I molecular weight markers.
To further examine the requirements for DNA cleavage, plasmid substrates containing both P-element termini on one plasmid, each terminus alone, or mixtures of each terminus on separate plasmids were tested in the cleavage assay as diagrammed in Figure 1C. If transposase requires synapsis of both a left (5′) and a right (3′) end for cleavage, then products should only be detected when substrates containing both ends are present in the reaction. DNA blot hybridization analysis of restriction endonuclease-cleaved reaction products is shown in Figure 1D. We found that efficient DNA cleavage only occurred in the presence of GTP and when both a left (5′) and right (3′) end were present in the reaction (Fig. 1D, cf. lanes 4 and 6 with lanes 2 and 8). Cleavage at the right (3′) end-containing substrate (lane 6) is approximately fourfold reduced than when both ends are present. The finding that a mixture containing a single left (5′) end-containing plasmid and a single right (3′) end-containing plasmid was active for cleavage by transposase (Fig. 1D, lane 8), but each single end-containing plasmid substrate alone was inefficiently cleaved by transposase (Fig. 1D, lanes 4 and 6), demonstrates that transposase can cleave P-element end-containing substrates both in cis (Fig. 1D, lane 2) and in trans (Fig. 1D, lane 8). These data are consistent with synapsis of both a left (5′) and right (3′) P-element end during the cleavage step of the P-element transposition reaction. The presence of both ends on the same DNA molecule, however, is not a prerequisite for cleavage. The two P-element ends differ in the spacing between the transposase-binding site and the 31-bp inverted repeat. A requirement for synapsis of both a left (5′) and right (3′) end for efficient cleavage to occur suggests that the nature of these spacing differences may be critical for synapsis (see Discussion). A similar observation has been made for cleavage substrates during V(D)J recombination (Eastman et al. 1996; van Gent et al. 1996a; Steen et al. 1997; van Gent et al. 1997).
Transposase makes a 17-bp staggered cleavage at each P-element end
To determine the exact position of the double-strand DNA breaks made by transposase, large scale in vitro cleavage reactions were performed with both the 628- and 303-bp P-element-containing plasmid substrates and the H0.1FT transposase fraction. The reaction products corresponding to the excised element and cleaved plasmid vector were isolated from gel slices following agarose gel electrophoresis. A PCR-based primer extension analysis of the purified cleavage products is shown in Figure 2. Primer extension experiments were used to determine the sites of cleavage for both the Tn_7_ transposase (Bainton et al. 1991) and the Tc_1_ transposase (Vos et al. 1996). We found that the 3′ cleavage site for either the left (5′) end (Fig. 2A) or the right (3′) end (Fig. 2B) was made precisely at the end of the transposon. This finding is consistent with the 3′ cleavage site of all known transposases and previous in vitro data by use of dideoxy-substituted substrate DNAs during P-element transposition (Kaufman and Rio 1992). Primer extension products are observed near the P-element termini in the lanes lacking transposase (Fig. 2, A, lane 2, and B, lane 4). We believe that these products are the result of secondary structures in the DNA that cause Taq polymerase to prematurely terminate extension, because the sequencing ladder around the terminal P-element nucleotide has the same background bands (Fig. 2A,B). In addition, the same strong stop near the terminal P-element nucleotide can be seen in the reactions that examine the 5′ cleavage sites (Fig. 2, C, lanes 2 and 4, and D, lanes 6 and 8).
Figure 2.
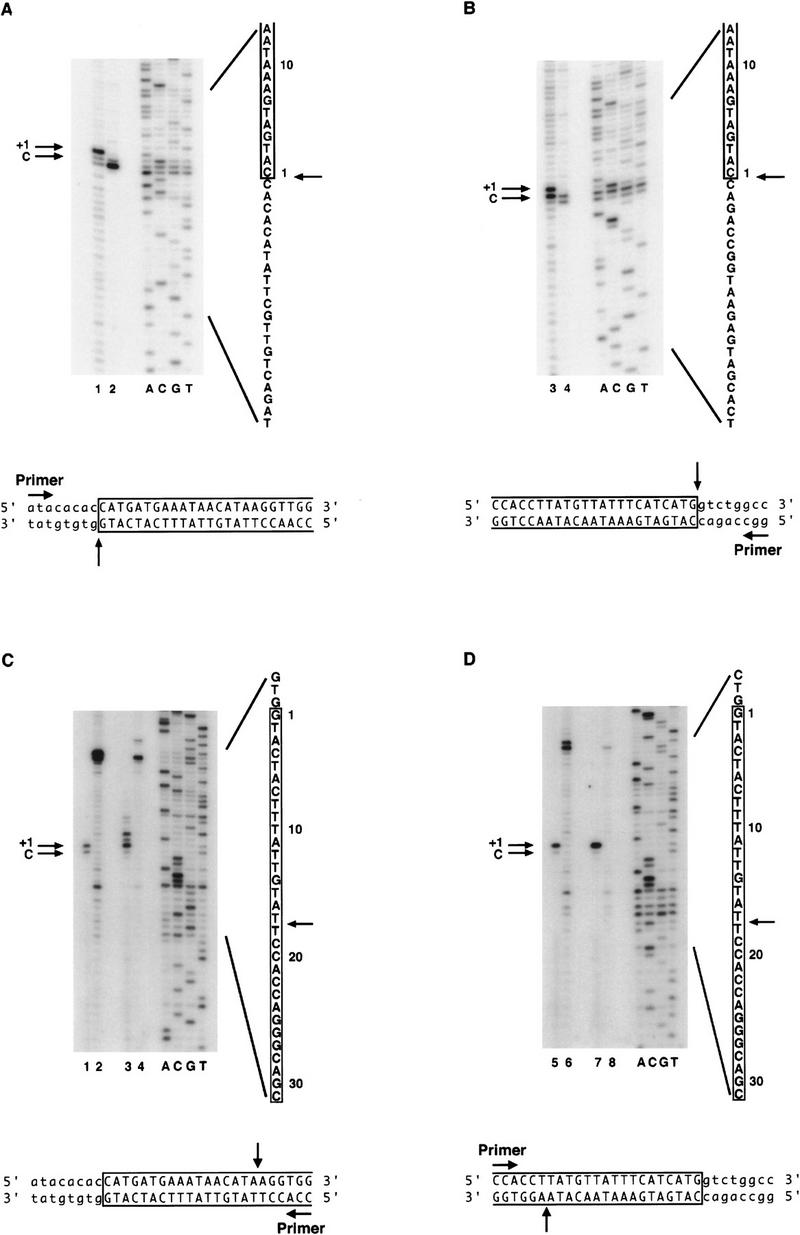
Transposase makes 17-bp staggered cleavages at the P-element termini. Large-scale cleavage reactions were performed with partially purified transposase and plasmid substrates containing either the 628-bp or 303-bp P elements. Reaction products corresponding to both the cleaved plasmid vector and the excised element were isolated from agarose gel slices. A PCR-based primer extension analysis was performed on each product in order to determine the transposase cleavage sites. Shown are autoradiographs of sequencing gels that display primer extension products from reactions in which nontemplated addition of a single nucleotide by Taq polymerase occurs (as indicated by +1 in each panel). The authentic cleavage sites are indicated by C. Sequencing reactions (ACGT) are shown as markers. The relevant sequence is indicated, with P-element-derived sequences boxed and numbered from the terminal P-element nucleotide, and the cleavage sites indicated by arrows. Schematic diagrams of the direction for primer extension and the cleavage positions for each strand are indicated below each panel. (A,B) Extension products to determine the 3′ cleavage site at the left (5′) P-element end (A) or the 3′ cleavage site at the right (3′) P-element end (B). Products were analyzed for the 303-bp P-element-derived cleavage product only. (Odd-numbered lanes) + transposase; (even-numbered lanes) − transposase. Extension yields a product that terminates exactly at the 3′ end of the P-element inverted repeat. (C,D) Extension products to determine the 5′ cleavage sites at the left (5′) P-element end (C) or right (3′) P-element end (D). (Lanes 1,2,5,6) Products derived from the 303-bp P element. (Lanes 3,4,7,8) Products derived from the 628-bp P element. (Odd-numbered lanes) + transposase, (even-numbered lanes) − transposase. Extension yields a product that terminates at nucleotide 18 of the P-element inverted repeat.
Surprisingly, we found that the 5′ cleavage site for either the left (5′) end (Fig. 2C), or the right (3′) end (Fig. 2D) occurs 17 bp within the P-element 31-bp inverted repeat sequence, directly adjacent to the IRBP-binding site (Rio and Rubin 1988). The finding that transposase makes a staggered cleavage that generates a 17-nucleotide, 3′ extension is novel. In addition, we found that at the left (5′) end only for both the 303-bp P element (E.L. Beall and D.C. Rio, unpubl.) and the 628-bp P element (Fig. 2C, lane 3), there is a clustering of primer extension products surrounding the proposed 17-bp cleavage site. It is possible that P-element transposase has reduced fidelity at the left (5′) end. The Tc_1_ transposase displays similar endonuclease activity near the site of cleavage (Vos and Plasterk 1994).
Because of the unusual nature of the 17-bp staggered cleavage made by P-element transposase, we wanted to confirm the primer extension results by an independent method. Cleavage reactions were performed with the H0.1FT transposase fraction and both the 303-bp and 628-bp P-element-containing plasmid substrates. Reaction products were analyzed by ligation-mediated PCR (LMPCR) as outlined in Figure 3A. An analogous assay has been used to identify the sites of cleavage made by Rag-1 and Rag-2 during V(D)J rearrangement (Roth et al. 1993; Schlissel et al. 1993). Reaction products were made flush with T4 DNA polymerase treatment prior to ligation of the annealed linkers and PCR by use of the primer pairs indicated in Figure 3A. LMPCR products were isolated from gel slices following agarose gel electrophoresis, inserted into a plasmid vector, and sequenced. We found that the sequence from 27 of 30 independent isolates derived from either the 5′ cleavage site at the left (5′) end or right (3′) end of the element, for both the 303-bp and 628-bp elements, began at nucleotide 18 of the P-element 31-bp inverted repeat (E.L. Beall and D.C. Rio, unpubl.). Two of the remaining DNAs began at nucleotide 19 and the third began at nucleotide 3 of the P-element 31-bp inverted repeat. These DNAs may have been derived from either exonucleolytic degradation of the terminus after cleavage, or from cleavages made by P-element transposase at another site within the 31-bp inverted repeat, similar to the alternative cleavage sites observed by the primer extension analysis (Fig. 2C, lane 3). The finding that 27/30 independent LMPCR products began at nucleotide 18 of the 31-bp inverted repeat confirms that the transposase makes a 17-bp staggered cleavage at the P-element termini during transposition.
Figure 3.



P-element termini are protected after transposase-mediated cleavage. (A) Schematic diagram of the LMPCR assay. P-element-containing plasmid substrates were incubated with partially purified transposase (H0.1FT) in the presence or absence of GTP. Products were treated with T_4_ DNA polymerase to remove the 17-bp, 3′ extension, or ligated directly to annealed linkers as shown. LMPCR was performed with the indicated primers to detect all four possible cleavage products of the sizes indicated. (Shaded boxes) P-element-derived sequences; (black boxes) 8-bp target site duplication. (B) Photograph of a native polyacrylamide gel following ethidium bromide staining. Cleavage reactions were performed in the presence (+) or absence (−) of GTP as indicated. All samples were treated with T4 DNA polymerase prior to LMPCR. (M) pBR322 _Msp_I molecular weight markers. (5′F) Amplification to detect the cleavage product at the 5′ flanking DNA end (85 bp); (3′F) amplification to detect the cleavage product at the 3′ flanking DNA end (78 bp); (5′E) amplification to detect the cleavage product at the 5′ P-element DNA end (81 bp); (3′E) amplification to detect the cleavage product at the 3′ P-element DNA end (86 bp). (C) Photograph of a native polyacrylamide gel following ethidium bromide staining. Cleavage reactions were performed in the presence of GTP (+), and either in the presence (+) or absence (−) of T4 polymerase treatment prior to LMPCR analysis. Abbreviations are as outlined in B.
The P-element termini are protected after cleavage
Efficient cleavage by Rag1 and Rag2 at the recombination signal sequences (RSS) requires a 12/23 RSS pair and occurs in a coordinated fashion during V(D)J rearrangement (Eastman et al. 1996; van Gent et al. 1996a, 1997; Steen et al. 1997). These data suggest that a synaptic complex forms at the RSS pair that allows for proper double-strand DNA break formation during the cleavage step of the reaction. Following cleavage by Rag-1 and Rag-2, the signal and coding ends are held in a stable protein complex that protects them from exonuclease degradation (Zhu et al. 1996; Agrawal and Schatz 1997).
To determine whether a stable protein complex present in the H0.1FT transposase fraction is protecting the cleaved ends, LMPCR was performed on purified cleavage products. As shown in Figure 3B, LMPCR products derived from all four possible cleaved ends are only produced in the presence of GTP (Fig. 3B, cf. lanes 1–4 with 5–8). T4 DNA polymerase treatment was required to detect P-element end-derived LMPCR products (Fig. 3C, cf. lanes 2 and 3 with 7 and 8) but not for the detection of flanking DNA end-derived products (Fig. 3C, lanes 1,2,5,6). Identical results were obtained for the 628-bp P-element-containing plasmid substrate (E.L. Beall and D.C. Rio, unpubl.). These data suggest that only the P-element termini are protected by a stable protein complex following transposase-mediated cleavage. Sequence analysis of 27 flanking DNA-derived LMPCR products showed that 33% had small deletions extending into the flanking DNA (E.L. Beall and D.C. Rio, unpubl.), which is consistent with the idea that the flanking DNA ends are not part of a stable protein complex containing the cleaved P element following excision. Similar small flanking donor DNA deletions were observed in vivo following P-element excision in embryos (Beall and Rio 1996).
Purified transposase can perform cleavage
To determine whether highly purified P-element transposase can catalyze the cleavage reaction, the H0.1FT transposase-containing fraction was further purified on a nonspecific DNA affinity column (TdT). The bound protein was eluted with increasing KCl steps. Analysis of the purified transposase fractions either by silver staining following SDS–polyacrylamide gel electrophoresis (Fig. 4A) or immunoblot analysis (Fig. 4B) is shown. Most of the purified transposase protein eluted in the first 0.6 m KCl fraction (Fig. 4, A, lane 2, and B, lane 4), and appears to be the predominant species present in this fraction. Immunoblot analysis of the TdT column fractions with anti-IRBP polyclonal antibodies revealed that most of the IRBP protein was present in the protein fraction not bound by the TdT column and the 0.3 m KCl fraction (Fig. 4C, lanes 2,3), with little or no IRBP present in the 0.6 m KCl fraction (Fig. 4C, lane 4). DNase I footprinting analysis of the TdT fractions revealed that the only site protected from DNase I digestion was the transposase-binding site (E.L. Beall and D.C. Rio, unpubl.).
Figure 4.
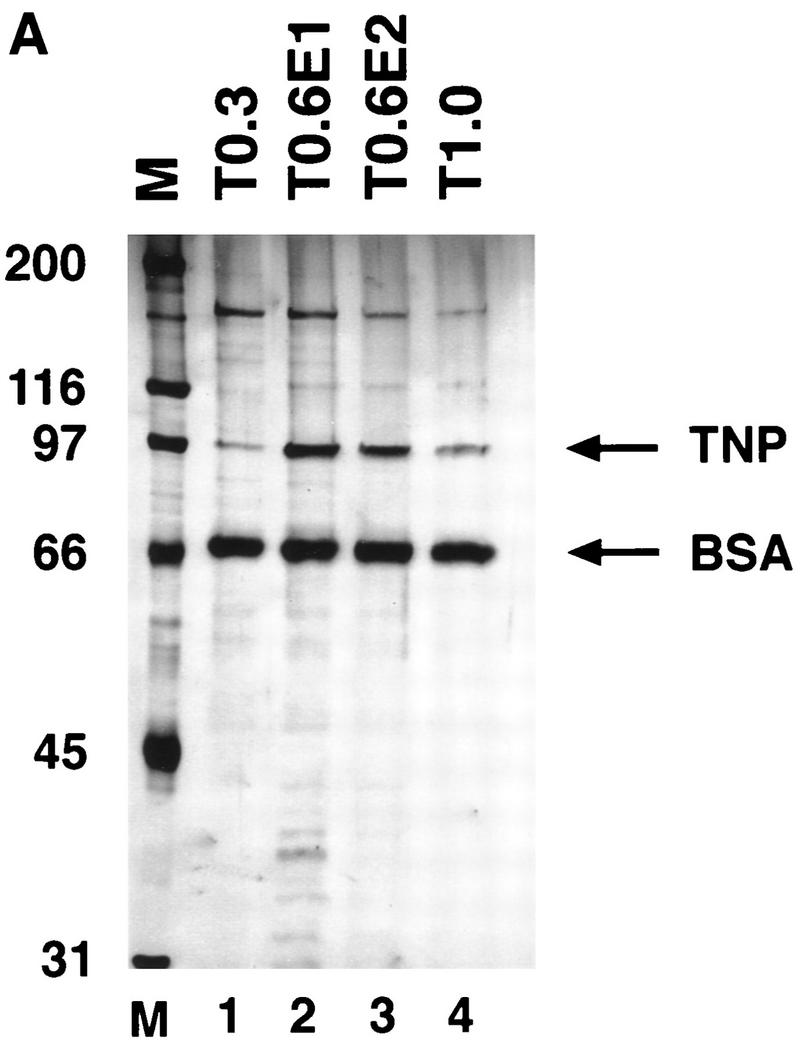



Purified P-element transposase can perform cleavage in vitro. (A) SDS-PAGE analysis of the purified transposase fractions. Samples were subjected to electrophoresis on a 7.5% acrylamide gel and stained with silver. Transposase was purified from Drosophila cell culture nuclear extracts with heparin-agarose chromatography (see Materials and Methods for details). The flowthrough (H0.1FT) fraction containing high amounts of transposase activity was chromatographed on a nonspecific DNA-affinity column (TdT), and the protein eluted with increasing KCl. (M) Molecular weight markers, with relative molecular mass in kD (left). (Lane 1) 0.3 m KCl fraction; (lane 2) 0.6 m KCl elution 1; (lane 3) 0.6 m KCl elution 2; (lane 4) 1.0 m KCl fraction. BSA was added to each fraction at 50 μg/ml. Approximately 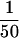 of the total fraction was loaded in each lane. (B) Immunoblot analysis of the purified transposase fractions. Samples from the TdT column were subjected to electrophoresis on a 7.5% acrylamide gel, transferred to nitrocellulose and probed with anti-transposase affinity-purified polyclonal antibodies. Molecular weight markers are described in A. (Lane 1) TdT column input; (lane 2) TdT column flowthrough; (lane 3) 0.3 m KCl fraction; (lane 4) 0.6 m KCl elution 1; (lane 5) 0.6 m KCl elution 2; (lane 6) 1.0 m KCl fraction. Approximately
of the total fraction was loaded in each lane. (B) Immunoblot analysis of the purified transposase fractions. Samples from the TdT column were subjected to electrophoresis on a 7.5% acrylamide gel, transferred to nitrocellulose and probed with anti-transposase affinity-purified polyclonal antibodies. Molecular weight markers are described in A. (Lane 1) TdT column input; (lane 2) TdT column flowthrough; (lane 3) 0.3 m KCl fraction; (lane 4) 0.6 m KCl elution 1; (lane 5) 0.6 m KCl elution 2; (lane 6) 1.0 m KCl fraction. Approximately 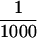 of the input and flowthrough, and
of the input and flowthrough, and 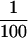 of each fraction was loaded in each lane. (C) Immunoblot analysis of the purified transposase fractions. Samples from the TdT column were subjected to electrophoresis on a 7.5% acrylamide gel, transferred to nitrocellulose and probed with anti-IRBP affinity-purified polyclonal antibodies. Molecular weight markers are described in A. (Lane 1) TdT column input; (lane 2) TdT column flowthrough; (lane 3) 0.3 m KCl fraction; (lane 4) 0.6 m KCl elution 1; (lane 5) 0.6 m KCl elution 2. Approximately
of each fraction was loaded in each lane. (C) Immunoblot analysis of the purified transposase fractions. Samples from the TdT column were subjected to electrophoresis on a 7.5% acrylamide gel, transferred to nitrocellulose and probed with anti-IRBP affinity-purified polyclonal antibodies. Molecular weight markers are described in A. (Lane 1) TdT column input; (lane 2) TdT column flowthrough; (lane 3) 0.3 m KCl fraction; (lane 4) 0.6 m KCl elution 1; (lane 5) 0.6 m KCl elution 2. Approximately 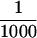 of the input and flow-through, and
of the input and flow-through, and 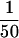 of each fraction was loaded in each lane. (D) Both partially purified (H0.1FT) and DNA affinity-purified (TdT0.6 E1) transposase-containing fractions were tested for cleavage activity with the 628-bp P-element-containing plasmid substrate. The excised element was detected by DNA blot hybridization by use of a radiolabeled P-element fragment as a probe. (M) Molecular weight markers in bp, as indicated. (−GTP) Control reactions lacking the cofactor, GTP. Reactions were performed either in the presence (+) or absence (−) of nuclear extract (NE) derived from a Drosophila somatic cell line lacking transposase (Kc cells). (NP) Reaction lacking transposase.
of each fraction was loaded in each lane. (D) Both partially purified (H0.1FT) and DNA affinity-purified (TdT0.6 E1) transposase-containing fractions were tested for cleavage activity with the 628-bp P-element-containing plasmid substrate. The excised element was detected by DNA blot hybridization by use of a radiolabeled P-element fragment as a probe. (M) Molecular weight markers in bp, as indicated. (−GTP) Control reactions lacking the cofactor, GTP. Reactions were performed either in the presence (+) or absence (−) of nuclear extract (NE) derived from a Drosophila somatic cell line lacking transposase (Kc cells). (NP) Reaction lacking transposase.
The cleavage activity of the H0.1FT and DNA affinity-purified (TdT0.6) transposase-containing fractions were compared with the 628-bp P-element-containing substrate. The excised element was detected by DNA blot hybridization with a radiolabeled P-element DNA fragment probe. Cleavage products were not detected in the absence of GTP (Fig. 4D, lanes 1,4). The purified transposase-containing fraction was able to perform the cleavage reaction (Fig. 4D, lane 5). Several products are present in the reaction containing the purified TdT0.6 fraction, however, with the predominant size corresponding to that of the excised element. The heterogeneous products are no longer detected with the DNA affinity-purified transposase fraction when the reactions were performed with the addition of Drosophila nuclear extracts lacking the transposase protein (Fig. 4D, lane 6). Preliminary analysis suggests that these heterogeneous products may represent autointegration events (E.L. Beall and D.C. Rio, unpubl.) as has been observed with the Moloney murine leukemia virus integrase (Lee and Craigie 1994) , Tn_10_ transposase (Chalmers and Kleckner 1996), and Tn_7_ transposase (M. Biery, M. Loptata, and N. L. Craig, pers. comm.). These data suggest that the purified transposase protein can perform the cleavage reaction on its own. In the absence of Drosophila DNA-binding proteins or excess nonspecific target DNA (like that present in crude nuclear extracts), it is possible that autointegration can occur.
Discussion
Here, we report the development of in vitro assays to study the cleavage step of the P-element transposition reaction. Transposase requires that both left (5′) and right (3′) P-element termini are present on DNA substrates for efficient donor DNA cleavage to occur. In addition, transposase cleaves the P-element termini to generate 17-nucleotide, 3′ extensions. By using LMPCR, we found that the P-element termini are protected from exonucleolytic degradation following donor DNA cleavage, suggesting that the termini are bound by a stable protein complex following the cleavage reaction. Finally, we found that highly purified transposase-containing fractions can perform the cleavage reaction, suggesting that the P-element transposase protein can perform the cleavage reaction in the absence of other _Drosophila_-encoded proteins.
The P-element termini are functionally distinct
Synapsis of the termini is a general prerequisite for cleavage during V(D)J recombination (Eastman et al. 1996; Steen et al. 1997; van Gent et al. 1996a, 1997) and during transposition, as has been observed for the transposons Mu (Mizuuchi et al. 1992; Surette et al. 1987) and Tn_10_ (Sakai et al. 1995). The finding that efficient transposase-mediated cleavage requires both left (5′) and right (3′) P-element termini on substrates both in vitro (Fig. 1) and in vivo (Mullins et al. 1989) suggests that synapsis of the two termini is required for efficient donor DNA cleavage to occur during P-element transposition. The experiments reported here, however, indicate that the cleavage reaction can rarely bypass the requirement for synapsis of both P-element termini. Low-level cleavage activity (about fourfold reduced) can be detected with a single right (3′) end-containing substrate (Fig. 1D). Precedents for uncoupled cleavage have been observed in other systems, such as during V(D)J recombination both in vitro when Mn2+ is used as the divalent metal ion (van Gent et al. 1995, 1996a; Eastman et al. 1996) or recently in vivo on substrates that contain both single or double RSS (Steen et al. 1997). It is noteworthy that the two P-element termini differ in the spacing between the transposase-binding site and the 31-bp inverted repeat (Fig. 6, below). Analogous to our observed low-level cleavage at the 3′ end-containing substrates (Fig. 1D, lane 6) in which the spacing between the transposase-binding site and the 31-bp inverted repeat is 9 bp, cleavage at an isolated 12-bp RSS has been observed (van Gent et al. 1995; Steen et al. 1997). It is possible that the spacing differences between the binding and cleavage sites at the two termini is critical for coupled cleavage, and that at a low frequency, the two 3′ ends or 12-bp RSS elements can be synapsed and cleaved because the longer spacer-containing substrate requires additional factor(s) for cleavage to occur. In support of this hypothesis, increased activity at a 23-bp RSS in the presence of the DNA-bending proteins HMG-1 and HMG-2 has been observed during in vitro cleavage assays (van Gent et al. 1997).
Figure 6.

Similarities between the V(D)J RSS and P-element transposon termini. Schematic diagram for synapsis of the V(D)J RSS is shown on the left. Pairing of a 12-bp spacer RSS and 23-bp spacer RSS is as indicated. Rag-1 (lightly shaded oval) is shown interacting with the nonamer element (black boxes). In the presence of Rag-2 (darkly shaded oval) and Mg2+, cleavage occurs at the heptamer (medium gray stippled boxes) to produce blunt signal ends and hairpin coding ends. (Right) A schematic diagram of the synapsed P-element termini. Pairing of the left (5′) end that contains a 21-bp spacer between the transposase-binding site (black box) and the 31-bp inverted repeat (black arrow) with the right (3′) end containing a 9-bp spacer between the two sequence elements is shown. In the presence of Mg2+ and GTP, transposase (shaded oval) makes a staggered 17-bp cleavage within the 31-bp inverted repeats, adjacent to the IRBP-binding site [(IRBP) hatched oval, p70 subunit. (Black oval) p80 subunit].
Our data also show that cleavage can occur in trans on substrates that contain a left (5′) or a right (3′) end located on separate molecules. Interestingly, a model has been proposed to explain P element-induced male recombination in Drosophila that involves synapsis of a pair of P-element ends from two different elements located on homologous chromosomes (Gray et al. 1996; Preston and Engels 1996; Preston et al. 1996). In this model, P-element transposase synapses and cleaves the 5′ terminus from an element on one homolog, and the 3′ terminus of a second element on the homologous chromosome. These two ends can be inserted into a new target site to produce the observed recombinant chromosomes. Together, these data suggest that P-element transposase requires synapsis of both left (5′) and right (3′) P-element termini for DNA cleavage to occur, but does not necessarily require that the two termini are present on the same DNA molecule. The Tn_10_ transposase has also recently been shown to synapse, cleave, and strand transfer two transposon ends derived from different DNA molecules in vitro (Chalmers and Kleckner 1996).
A model for P-element transposition
The donor DNA cleavage site has been precisely determined for several transposable elements. The 3′ cleavage site is always at the end of the transposon, whereas the 5′ cleavage site can occur at different positions surrounding the transposon end. For instance, the 5′ cleavage site occurs at the end of the Tn_10_ element to generate a flush end excised transposon intermediate (Benjamin and Kleckner 1992), 3 bp outside of the Tn_7_ element end to generate 3-nucleotide, 5′ extensions (Bainton et al. 1991; Gray et al. 1996), and 2 bp within the element for both Tc_1_ and Tc_3_ to generate 2-nucleotide, 3′ extensions (van Luenen et al. 1994; Vos et al. 1996). Cleavage at the P-element ends follows the same general rule, the 3′ site is at the transposon end, whereas the 5′ site is 17 bp within the P-element ends. However, the 17-nucleotide staggered cleavages are without precedent for all known transposase and restriction endonuclease cleavage sites determined to date. Based on the structure of the excised P-element intermediate, we propose that P-element transposition occurs by the mechanism outlined in Figure 5. In this model, P-element transposase binds to sequences within both P-element termini and initiates a double-strand DNA break at each end, as indicated. The excised element can then be inserted into a new target site. Gap repair will generate the characteristic 8-bp target site duplications as well as regenerate the P-element sequences at each end. We have recently developed in vitro strand transfer assays by use of oligonucleotide substrates that mimic the cleaved P-element termini (E.L. Beall and D.C. Rio, in prep.), which demonstrates that the excised element is an intermediate in the transposition reaction.
Figure 5.

Model for P-element transposition. A model for nonreplicative P-element transposition is diagrammed. P-element transposase binds to sequences within both P-element termini and initiates a double-stranded DNA break at each end, as indicated by arrows. The excised element, which contains 17-nt, 3′ extensions at each end, can be inserted into a new target site (left). Gap repair will generate the characteristic 8-bp target site duplications as well as regenerate the P-element sequences at each end. The 17-nt, 3′ extensions left behind at the donor site can be used for repair either from homologous P-element sequences located elsewhere in the genome by SDSA repair, or by end-joining in the absence of homologous P-sequences, (right). Imprecise repair of the double-stranded DNA break left at the donor site can produce products that contain varying lengths of P-element-derived sequences, e.g., a single repaired donor site is shown in which the cleaved termini were adjoined, extended and ligated to leave behind 34 nts of P-element sequence at the donor site. (Darkly shaded boxes) 8-bp target site duplications. (Lightly shaded boxes) P-element-derived sequences.
In the presence of homologous P-element sequences, repair of the double-strand DNA breaks at the donor site following transposition is proposed to occur by a synthesis-dependent strand annealing (SDSA) pathway in which each end initiates DNA synthesis from the template independently (Formosa and Alberts 1986; Nassif et al. 1994). In the absence of homologous sequences, Drosophila is thought to repair double-strand DNA breaks by an end-joining mechanism similar to that used by mammals. In this process, double-strand DNA breaks are repaired after direct abutting of the broken ends or by annealing short complementary sequences (Roth and Wilson 1986; Nicolas et al. 1995). We propose that P-element-derived sequences found at the donor site following excision arise from end-joining repair of the 17-nucleotide staggered breaks. In fact, a similar model was proposed to explain chromosomal excision events at the vestigial locus (Staveley et al. 1995), except that the cleavage site was proposed to be ∼16-nucleotides within the 31-bp P-element inverted repeats. Our data support and extend the previously proposed model to include the precise cleavage site made by P-element transposase: 17-nucleotides into the 31-bp P-element inverted repeats.
The donor DNA ends are not protected following cleavage (Fig. 3C) and may be substrates for exonucleolytic degradation. Consistent with this finding are several reports of varying lengths of P-element-derived sequences left behind at the donor site following excision in vivo. These studies include excision from chromosomal locations (Takasu-Ishikawa et al. 1992; Staveley et al. 1995) as well as excisions from extrachromosomal plasmid substrates (Rio et al. 1986; O’Brochta et al. 1991; Beall and Rio 1996). In the majority of cases from chromosomal P-element excision events, 15, 16, or 17 nucleotides of P-element sequences from each end of the P element (for a total of 30, 32, or 34 bp) were found at the donor site (Takasu-Ishikawa et al. 1992; Staveley et al. 1995). The most frequently observed product, 16 nucleotides derived from each end (Staveley et al. 1995), may arise through annealing the terminal two nucleotides of the 17-nucleotide extensions (TA) followed by extension and ligation, to give a product that appears to be recessed by one nucleotide but is, in fact, derived from two 17-nucleotide ends. The remaining truncated products can be explained by varying amounts of exonucleolytic degradation and/or annealing of small regions of complementary sequence followed by gap repair. The large stretches of single-stranded DNA at the donor site following excision may also explain the extremely low frequency of precise excision events in the absence of homologous sequences (Engels et al. 1990; O’Brochta et al. 1991). To generate a precise P-element excision, both 17-nucleotide extensions must be removed from each end, as well as one of the 8-bp target site duplications, prior to repair.
Some of the P-element-derived sequences found at the donor site are larger than 17 nucleotides derived from each end. At a low frequency, both 18 and 19 nucleotides from each end have been observed (Staveley et al. 1995). In addition, it was found that a polymorphism located 33 nucleotides within the donor site P element was retained following excision and template-dependent repair from ectopically located P elements, suggesting that cleavage occurred at least 33 nucleotides within the P-element ends (Keeler and Gloor 1997). We propose that these products can either arise from alternative transposase-induced cleavage products similar to those observed in our primer extension analysis (Fig. 2), or from extension of the accurately cleaved end by the repair polymerase prior to dissociation of the cleavage complex following excision. These extended ends then can be repaired directly or further extended from a second, ectopically located P element, to generate the observed products.
The prematurely terminated primer extension products that we observed near the terminal nucleotides of the P-element inverted repeat (Fig. 2C) suggest that the DNA structure at the ends of the element may be distorted. Whether the P-element termini are unwound or distorted prior to DNA cleavage has yet to be determined. The unusual structure of the cleaved termini, however, suggests that the active site of P-element transposase can perform transesterification reactions on unpaired, or single-stranded regions of DNA, at least during the strand transfer step of the reaction. The ability of single-stranded DNA to stimulate cleavage is not without precedent. Unpaired DNA sequences in model oligonucleotide substrates facilitates DNA cleavage by Mu transposase (Savilahti et al. 1995), HIV integrase (Scottoline et al. 1997), and the Rag proteins (Cuomo et al. 1996; Ramsden et al. 1996) in vitro. It is possible that local distortions in the DNA structure at both the terminal element nucleotides and the target site sequence prior to the cleavage and strand transfer reactions occurs for Mu transposase, P-element transposase, HIV integrase, and the Rag proteins during catalysis.
Roles for Drosophila-encoded proteins in transposition
It is interesting that transposase cleaves the P-element termini directly adjacent to the IRBP-binding site (Rio and Rubin 1988). IRBP is homologous to the p70 subunit of the mammalian Ku antigen (Beall et al. 1994; Jacoby and Wensink 1994). Ku, a heterodimer composed of 70- and 80-kD proteins, is the DNA-binding subunit of the DNA-dependent protein kinase (DNA–PK) (Gottlieb and Jackson 1993). DNA–PK phosphorylates a number of nuclear DNA-binding proteins (Lees-Miller et al. 1990; Anderson and Lees-Miller 1992; Boubnov and Weaver 1995), and radiosensitive mutant mammalian cell lines defective for either the 80-kD Ku subunit (xrs-6/sxi-3) or the 465-kD DNA-PKcs (V-3/scid) both display hypersensitivity to ionizing radiation and exhibit defects in immunoglobulin V(D)J recombination (Taccioli et al. 1994; Blunt et al. 1995; Boubnov et al. 1995; Kirchgessner et al. 1995; Errami et al. 1996). Similar to the mammalian Ku protein, Drosophila IRBP (Ku p70) mutants, called mus309, are hypersensitive to DNA-damaging agents and show defects in the repair of DNA breaks after P-element excision in vivo (Beall and Rio 1996).
There are several possible roles for IRBP and for DNA–PK in P-element transposition. IRBP may serve as an end-aligning factor that helps to properly position the P-element ends prior to cleavage by transposase. Host-encoded proteins have been shown to perform similar functions during Mu transposition (Mizuuchi and Mizuuchi 1989; Surette and Chaconas 1992; Lavoie and Chaconas 1993). It is possible that the Drosophila Ku protein possesses DNA helicase activity like that reported for the mammalian Ku protein (Tuteja et al. 1994). The properly positioned protein could facilitate unwinding of the 17-bp staggered ends following cleavage. Several studies have shown that the mammalian Ku protein efficiently binds to staggered double-strand DNA breaks (Mimori and Hardin 1986; Falzon et al. 1993; Morozov et al. 1994). The nature of the cleaved P-element termini suggests that the Drosophila Ku protein will remain bound to the cleaved P-element termini. In addition, a new Ku heterodimer can also bind to the cleaved donor site following excision. Repair of the DNA breaks at both the donor site, and at the new target site may be facilitated by Drosophila Ku bound at both of these sites.
The consensus phosphorylation recognition sequence for DNA–PK was determined to be a serine or threonine residue adjacent to glutamine for the substrate, p53 (Lees-Miller et al. 1992). There are eight potential DNA–PK phosphorylation sites located within the first 144 amino acids of the transposase protein (Rio et al. 1986), the portion of the protein determined to have site-specific DNA-binding activity (Lee et al. 1996). It is possible that phosphorylation by DNA–PK may regulate DNA binding by transposase, such that phosphorylation promotes disassembly of the complex following transposition. Disassembly of the cleavage complex is required for completing the Mu transposition reaction (Levchenko et al. 1995). However, the role of phosphorylation, if any, during P-element transposition has yet to be determined.
Similarities to V(D)J recombination
V(D)J rearrangement occurs in two steps: site-specific DNA cleavage that is followed by joining or repair of the cleaved double-strand DNA breaks. Efficient cleavage in vitro requires the synapsis of a pair of RSS elements by Rag-1 and Rag-2, and requires the divalent metal ion, Mg2+ (Eastman et al. 1996; van Gent et al. 1996a) (Fig. 6). The RSS consists of conserved heptamer and nonamer elements that are separated by 12 or 23 nucleotide spacer sequences. Rag-1 has been shown to recognize the nonamer (Difilippantonio et al. 1996; Spanopoulou et al. 1996; Hiom and Gellert 1997). In the presence of Rag-2 and Mg2+, cleavage occurs precisely at the heptamer to generate the two reaction intermediates: blunt-ended signal ends and hairpin-coding ends. Recombination primarily occurs between pairs of RSS that contain different spacer lengths, the so-called 12/23 rule (Eastman et al. 1996; van Gent et al. 1996a).
The arrangement of the Rag-1/Rag-2 genes in the mammalian genome (Schatz et al. 1989; Oettinger et al. 1990), the chemical mechanism of the DNA rearrangement reaction (van Gent et al. 1996b), and the similarity between the RSS nonamer and the bacterial HIN invertase-binding site, HixL (Difilippantonio et al. 1996; Spanopoulou et al. 1996), have led to the proposal that the V(D)J recombination system may have evolved from a transposon-like system (Oettinger et al. 1990; Craig 1996; Lewis and Wu 1997). In light of this idea, it is interesting to note that the overall architecture between the P-element termini and the RSS is similar (Fig. 6). The 5′ end of the P element contains a 21-nucleotide spacer that separates the internal 10-bp consensus transposase-binding site from the 31-bp terminal inverted repeat and the 3′ end of the P element contains a 9-bp spacer that separates these two sequence elements. While sequence similarities between the V(D)J RSS nonamer and the Tc_1_ transposon termini have been noted (Difilippantonio et al. 1996; Spanopoulou et al. 1996), the spacial relationship between the recombinase-binding site and the cleavage site is not similar, as can be observed for the V(D)J RSS and the P-element termini. Different spacer lengths between the recombinase-binding site and the recombination site may facilitate alignment or assembly of the termini prior to cleavage by the synaptic complex. A high degree of specificity can be achieved by requiring two termini that differ in the length of DNA that separates the binding site from the cleavage site. The 12/23 rule may simply be an important mechanism to ensure reaction fidelity in a more complex genome.
Materials and methods
Recombinant DNA
The plasmids used in the P-element cleavage assays are as follows: pISP-2/Km (Beall and Rio 1996) contains the 628-bp P element. pN/P175XTpB (Rio and Rubin 1988) contains the 5′ P-element end only. pUC18–C4 3′Δ20 (Kaufman et al. 1989) contains the 3′ P-element end only. pHSX–P5′3′ was derived from several plasmids: the 5′ P-element end-containing fragment (nucleotides 1–141) was isolated from pN/P150XTpB following _Xho_I and _Bam_HI cleavage. The 3′ P-element end-containing fragment (P-element nucleotides 2754–2907) was generated from Δ20 (Mullins et al. 1989) following _Xba_I and _Bam_HI cleavage. The 5′ and 3′ end-containing fragments were inserted into _Xho_I and _Xba_I cleaved pHSX to generate the 303-bp-containing plasmid, pHSX–P5′3′.
The substrates used in the cleavage assay described in Figure 1A were generated as follows: 25 μg of pISP-2/Km was cleaved with _Eco_RI and _Eag_I to generate a 749-bp P-element-containing fragment. 25 μg of pHSX–P5′3′ was cleaved with _Xba_I and _Xho_I to generate a 436-bp P element-containing fragment. 25 μg of pN/P175XTpB was cleaved with _Xho_I and _Kpn_I to generate a 225-bp 5′ P-element end-containing fragment. All fragments were isolated from agarose gel slices according to the manufacturer (Qiagen Gel Extraction Kit). Fragments were eluted in dH2O, and stored at −20°C.
Protein purification and immunoblot analysis
P-element transposase was purified from the Drosophila Schneider L2 stable cell line, pUChygMT-Tnp, following CuSO4 induction as described (Mul and Rio 1997). This cell line differs from the previously reported transposase-producing cell line (Kaufman et al. 1989) in that 25% of the amino terminus of transposase was chemically resynthesized to alter codon usage to the most frequently used codons in Drosophila (Lee et al. 1996). Briefly, nuclear extracts were precipitated with ammonium sulfate and fractions containing transposase pooled, dialyzed, and chromatographed on heparin-agarose (Kaufman et al. 1989). All buffers contained 50 mm NaF to inhibit phosphatase activity. The flow through (H0.1FT) contained highly active transposase, as determined by a genetic-based plasmid assay (Kaufman and Rio 1992). The H0.1FT transposase-containing fraction was chromatographed on a nonspecific DNA affinity resin (TdT) essentially as described (Kaufman et al. 1989), with the following modifications: Two oligonucleotides carrying three TdT sites were synthesized, one containing a biotin moiety at the 5′ end. These two DNAs were annealed and bound to streptavidin–agarose (Pierce) as described (Chen et al. 1994). Transposase was eluted, batchwise, with increasing amounts of KCl. The 0.6 m KCl fraction (TdT0.6) contained peak amounts of transposase, at ∼10 ng/μl as judged by silver-stained SDS–polyacrylamide gels containing known amounts of bovine serum albumin.
For the immunoblot analysis in Figure 4B, 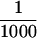 of each TdT fraction and
of each TdT fraction and 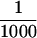 of the input and flow-through fractions were analyzed on a 7.5% SDS-polyacrylamide gel and transferred to nitrocellulose. Immunoblot analysis was performed with rabbit anti-KP affinity-purified polyclonal antibodies (kindly provided by S. Roche, University of California, Berkeley) prepared from the amino-terminal transposase KP fragment (amino acids 1–209) expressed in Escherichia coli as described (Harlow and Lane 1989; Lee et al. 1996). For the immunoblot analysis in Figure 4C,
of the input and flow-through fractions were analyzed on a 7.5% SDS-polyacrylamide gel and transferred to nitrocellulose. Immunoblot analysis was performed with rabbit anti-KP affinity-purified polyclonal antibodies (kindly provided by S. Roche, University of California, Berkeley) prepared from the amino-terminal transposase KP fragment (amino acids 1–209) expressed in Escherichia coli as described (Harlow and Lane 1989; Lee et al. 1996). For the immunoblot analysis in Figure 4C, 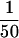 of each TdT fraction and
of each TdT fraction and 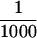 of the input and flow-through fractions were analyzed on a 7.5% SDS-polyacrylamide gel and transferred to nitrocellulose. Immunoblot analysis was performed with rabbit anti-IRBP affinity-purified polyclonal antibodies as described (Beall et al. 1994).
of the input and flow-through fractions were analyzed on a 7.5% SDS-polyacrylamide gel and transferred to nitrocellulose. Immunoblot analysis was performed with rabbit anti-IRBP affinity-purified polyclonal antibodies as described (Beall et al. 1994).
In vitro cleavage assays
The cleavage substrates described in Figure 1A were prepared as follows: 200 ng of the restriction endonuclease fragments pHSX–P5′3′ _Xba_I–_Xho_I fragment, or pISP-2/Km _Eco_RI–_Eag_I fragment, and 125 ng of pN/P175XTpB _Xho_I–_Kpn_I fragment, were radiolabeled by end-filling with the Klenow fragment of DNA polymerase I and [α-32P]dCTP. Radiolabeled fragments were extracted with 25:24:1 phenol-chloroform-isoamyl alcohol and ethanol precipitated. The pellets were resuspended to 10 fmoles/μl in dH2O. Reaction conditions for the cleavage assay were as follows: 10 fmoles of radiolabeled restriction fragment was incubated with ∼2 μg of total protein H0.1FT transposase-containing fraction in a volume of 6 μl in chromatography buffer (HGKED: 20 mm HEPES-KOH at pH 7.6, 20% glycerol, 100 mm KCl, 0.5 mm EGTA, 0.5 mm EDTA, 1 mm DTT, 0.2 mm PMSF, with the addition of 100 μg/ml of bovine serum albumin). Binding was carried out on ice for 15 min. The reaction was initiated by addition of 0.35 × HGKED (0m KCl), 10 mm MgCl2, and 2 mm GTP to make a total volume of 20 μl, and [KCl]⩽ 35 mm. Reactions were performed at 27°C for 2 hr and terminated by the addition of 125 μl of 50 mm Tris-HCl at pH 7.5, 10 mm EDTA, 0.3 m NaCl, 1% SDS, 250 μg/ml of yeast RNA, 0.1 mg/ml of proteinase K, and incubated at 37°C for 30 min. The reactions were extracted with 25:24:1 phenol/chloroform/isoamyl alcohol and ethanol precipitated. The pellets were resuspended in 10 μl of TE containing 100 μg/ml of RNase A and analyzed by native acrylamide gel electrophoresis and autoradiography.
For the DNA blot hybridization in Figure 1D, the standard cleavage assay was performed with 100 ng of circular plasmid (pHSX–P5′3′, pN/P175XTpB, or pUC18–C4 3′Δ20) as the substrate, and ∼2 μg H0.1FT transposase-containing fractions. Half of the reaction products were cleaved with either _Acc_I (pHSX– P5′3′) or _Bam_HI (pN/P175XTpB or pUC18–C4 3′Δ20) and analyzed by native gel electrophoresis and standard DNA blot hybridization following electrophoretic transfer to Hybond N+ membrane (Amersham). Products were detected with a 32P-random hexamer-labeled _Eco_RI/_Eag_I pISP-2/Km restriction fragment.
For the DNA blot hybridization in Figure 4, the standard cleavage assay was performed with 35 fmole of circular plasmid pISP-2/Km as the substrate, and ∼2 μg of H0.1FT or ∼10 ng of TdT0.6 transposase-containing fractions. Half of the reaction products were analyzed by agarose gel electrophoresis and standard DNA blot hybridization following capillary transfer to Hybond N+ membrane (Amersham). Products were detected with a 32P-random hexamer-labeled _Eco_RI/_Eag_I pISP-2/Km restriction fragment and quantitated using a PhosphorImager (Fuji).
Oligonucleotide primers and primer extension analysis
The primers used for both sequencing and primer extension analysis are as follows: to detect the 5′ cleavage site at the left (5′) end of the element for both pISP-2/Km and pHSX–P5′3′, the primer 5′IN was used, which is complementary to nts 56–79 of the 2.9-kb P-element sequence (O’Hare and Rubin 1983). To detect the 3′ cleavage site at the right (3′) end of the element for both pISP-2/Km and pHSX–P5′3′, the primer 3′IN was used, which corresponds to nts 2937–2857 of the 2.9-kb P-element sequence (O’Hare and Rubin 1983). To detect the 3′ cleavage site at the left (5′) end of the element for pHSX–P5′3′, the primer 5′EX was used, with the sequence 5′-GGTCGATAGATAGGTAGTATTG. To detect the 3′ cleavage site at the right (3′) end of the element for pHSX–P5′3′, the primer 3′EX was used, with the sequence 5′-GAAGTCTTAGAGCCAGATATGCG.
Oligonucleotides were gel purified and radiolabeled at their 5′ ends with T4 polynucleotide kinase and [γ-32P]ATP (6000 Ci/mmole, ICN radionucleotides) according to the manufacturer’s instructions (GIBCO BRL). Large scale cleavage reactions (fivefold increase) were performed with either pISP-2/Km or pHSX– P5′3′ plasmid substrates and the H0.1FT transposase-containing fraction, and both the excised P element or cleaved plasmid donor were extracted from gel slices according to the manufacturer’s instructions (Qiagen Gel Extraction Kit). Extension reactions contained 0.5 pmole of radiolabeled oligonucleotide, one-sixth of the total purified fragment, 2.5 units of Taq polymerase (Perkin Elmer Cetus), 200 mm dNTPs, and PCR buffer (10 mm Tris-HCl at pH8.3, 50 mm KCl, 2.0 mm MgCl2, 0.01% gelatin). Under these conditions, we found that Taq polymerase adds one nontemplated additional nucleotide to the 3′ ends of the amplified products, as previously observed (Bainton et al. 1991; Vos et al. 1996). Linear PCR amplification was performed in a total volume of 20 μl for 20 cycles: 30 sec at 95°C, 30 sec at 50°C, 1 min at 72°C followed by a final extension of 5 min at 72°C. DNA sequencing reactions were performed with circular plasmid templates and radiolabeled primer as described by the manufacturer (GIBCO BRL). Both extension products and sequencing reactions were analyzed on 6% polyacrylamide-urea sequencing gels.
LMPCR analysis
Large scale cleavage reactions (fivefold increase) were performed with either pHSX–P5′3′ or pISP-2/Km circular plasmid substrates and the dried DNA pellets resuspended in 30 μl TE. 5 μl (1/6 of the total cleaved substrate) was treated with T4 DNA polymerase to create flush termini. The T4-treated samples were extracted with 25:24:1 phenol-chloroform-isoamyl alcohol and ethanol precipitated with 10 μg of glycogen. The pellets were resuspended in 5 μl of dH2O and ligated to 50 pmoles of annealed linkers (FM25-2: 5′-GCGGTGACTCGGGAGATCTGAGATG, and FM11-2: 5′-CATCTCAGATC) overnight at 14°C with 1 unit of T4 DNA ligase. Ligation reactions were extracted with 25:24:1 phenol-chloroform-isoamyl alcohol and ethanol precipitated with 10 μg of glycogen. The pellets were resuspended in 30 μl of dH2O. PCR was performed on 5 μl of the treated DNA (∼15 ng, or 5 fmoles) with Taq polymerase and 25 cycles: 30 sec at 95°C, 30 sec at 50°C, 1 min at 72°C followed by a final extension of 5 min at 72°C with the following primer pairs: FM25-2 and either 5′ IN or 3′ IN to detect the cleavage products corresponding to the left or right P-element ends, respectively for the pHSX–P5′3′ plasmid substrate; FM25-2 and either 5′ EX or 3′ EX to detect the cleavage products corresponding to the left or right flanking DNA ends, respectively for the pHSX–P5′3′ plasmid substrate. PCR products were analyzed by native acrylamide gel electrophoresis and ethidium bromide staining.
PCR products were purified from agarose gel slices and cloned into _Eco_RV cleaved, T-tailed pBSKS(+) as described (Marchuk et al. 1991). Standard sequencing reactions were performed with the standard T3 sequencing primer and Sequenase 2.0 as described by the manufacturer (U.S. Biochemical) on plasmids containing LMPCR product inserts.
Acknowledgments
We thank Paul Kaufman for initiating the work that led to the development of the assays used in this paper. We also thank Charles Lee for help with the transposase protein purification, and Matt Mahoney for help with sequencing the LMPCR products. We thank G. Barnes, M. Botchan, K. Collins, A. Copeland, P. Kaufman, J. Sekelsky, and members of the Rio lab for many helpful discussions and suggestions on the manuscript. This work was supported by a grant from the National Institutes of Health (R01GM48862).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL don_rio@mendel.berkeley.edu; FAX (510) 642-6062.
References
- Agrawal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- Anderson CW, Lees-Miller SP. The nuclear serine/threonine protein kinase DNA-PK. Crit Rev Eukaryot Gene Expression. 1992;2:283–314. [PubMed] [Google Scholar]
- Bainton R, Gamas P, Craig NL. Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell. 1991;65:805–816. doi: 10.1016/0092-8674(91)90388-f. [DOI] [PubMed] [Google Scholar]
- Bainton RJ, Kubo KM, Feng JN, Craig NL. Tn7 transposition: Target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell. 1993;72:931–943. doi: 10.1016/0092-8674(93)90581-a. [DOI] [PubMed] [Google Scholar]
- Beall EL, Rio DC. Drosophila IRBP/Ku p70 corresponds to the mutagen-sensitive mus309 gene and is involved in P-element excision in vivo. Genes & Dev. 1996;10:921–933. doi: 10.1101/gad.10.8.921. [DOI] [PubMed] [Google Scholar]
- Beall EL, Admon A, Rio DC. A Drosophila protein homologous to the human p70 Ku autoimmune antigen interacts with the P transposable element inverted repeats. Proc Natl Acad Sci. 1994;91:12681–12685. doi: 10.1073/pnas.91.26.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin HW, Kleckner N. Excision of Tn10 from the donor site during transposition occurs by flush double-strand cleavages at the transposon termini. Proc Natl Acad Sci. 1992;89:4648–4652. doi: 10.1073/pnas.89.10.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunt T, Finnie NJ, Taccioli GE, Smith GC, Demengeot J, Gottlieb TM, Mizuta R, Varghese AJ, Alt FW, Jeggo PA, et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- Boubnov NV, Weaver DT. scid cells are deficient in Ku and replication protein A phosphorylation by the DN8A-dependent protein kinase. Mol Cell Biol. 1995;15:5700–5706. doi: 10.1128/mcb.15.10.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubnov NV, Hall KT, Wills Z, Lee SE, He DM, Benjamin DM, Pulaski CR, Band H, Reeves W, Hendrickson EA, et al. Complementation of the ionizing radiation sensitivity, DNA end binding, and V(D)J recombination defects of double-strand break repair mutants by the p86 Ku autoantigen. Proc Natl Acad Sci. 1995;92:890–894. doi: 10.1073/pnas.92.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers RM, Kleckner N. IS10/Tn10 transposition efficiently accommodates diverse transposon end configurations. EMBO J. 1996;15:5112–5122. [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Attardi LD, Verrijzer CP, Yokomori K, Tjian R. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- Craig NL. V(D)J recombination and transposition: Closer than expected. Science. 1996;271:1512. doi: 10.1126/science.271.5255.1512. [DOI] [PubMed] [Google Scholar]
- Craigie R, Mizuuchi K. Mechanism of transposition of bacteriophage Mu: Structure of a transposition intermediate. Cell. 1985;41:867–876. doi: 10.1016/s0092-8674(85)80067-2. [DOI] [PubMed] [Google Scholar]
- ————— Transposition of Mu DNA: Joining of Mu to target DNA can be uncoupled from cleavage at the ends of Mu. Cell. 1987;51:493–501. doi: 10.1016/0092-8674(87)90645-3. [DOI] [PubMed] [Google Scholar]
- Cuomo CA, Mundy CL, Oettinger MA. DNA sequence and structure requirements for cleavage of V(D)J recombination signal sequences. Mol Cell Biol. 1996;16:5683–5690. doi: 10.1128/mcb.16.10.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio MJ, McMahan CJ, Eastman QM, Spanopoulou E, Schatz DG. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell. 1996;87:253–262. doi: 10.1016/s0092-8674(00)81343-4. [DOI] [PubMed] [Google Scholar]
- Eastman QM, Leu TM, Schatz DG. Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature. 1996;380:85–88. doi: 10.1038/380085a0. [DOI] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- Engels WR, Johnson-Schlitz DM, Eggleston WB, Sved J. High-frequency P element loss in Drosophila is homolog dependent. Cell. 1990;62:515–525. doi: 10.1016/0092-8674(90)90016-8. [DOI] [PubMed] [Google Scholar]
- Errami A, Smider V, Rathmell WK, He DM, Hendrickson EA, Zdzienicka MZ, Chu G. Ku86 defines the genetic defect and restores X-ray resistance and V(D)J recombination to complementation group 5 hamster cell mutants. Mol Cell Biol. 1996;16:1519–1526. doi: 10.1128/mcb.16.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzon M, Fewell JW, Kuff EL. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J Biol Chem. 1993;268:10546–10552. [PubMed] [Google Scholar]
- Formosa T, Alberts BM. DNA synthesis dependent on genetic recombination: Characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell. 1986;47:793–806. doi: 10.1016/0092-8674(86)90522-2. [DOI] [PubMed] [Google Scholar]
- Gary PA, Biery MC, Bainton RJ, Craig NL. Multiple DNA processing reactions underlie Tn7 transposition. J Mol Biol. 1996;257:301–316. doi: 10.1006/jmbi.1996.0164. [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- Gray YHM, Tanaka MM, Sved JA. P element-induced recombination in Drosophila melanogaster: Hybrid element insertion. Genetics. 1996;144:1601–1610. doi: 10.1093/genetics/144.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniford DB, Benjamin HW, Kleckner N. Kinetic and structural analysis of a cleaved donor intermediate and a strand transfer intermediate in Tn10 transposition. Cell. 1991;64:171–179. doi: 10.1016/0092-8674(91)90218-n. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Hiom K, Gellert M. A stable RAG1-RAG2-DNA complex that is active in V(D)J cleavage. Cell. 1997;88:65–72. doi: 10.1016/s0092-8674(00)81859-0. [DOI] [PubMed] [Google Scholar]
- Jacoby DB, Wensink PC. Yolk protein factor 1 is a Drosophila homolog of Ku, the DNA-binding subunit of a DNA-dependent protein kinase from humans. J Biol Chem. 1994;269:11484–11491. [PubMed] [Google Scholar]
- Kaufman PD, Rio DC. P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell. 1992;69:27–39. doi: 10.1016/0092-8674(92)90116-t. [DOI] [PubMed] [Google Scholar]
- Kaufman PD, Doll RF, Rio DC. Drosophila P element transposase recognizes internal P element DNA sequences. Cell. 1989;59:359–371. doi: 10.1016/0092-8674(89)90297-3. [DOI] [PubMed] [Google Scholar]
- Keeler KJ, Gloor GB. Efficient gap repair in Drosophila melanogaster requires a maximum of 31 nucleotides of homologous sequence at the searching ends. Mol Cell Biol. 1997;17:627–634. doi: 10.1128/mcb.17.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown JM. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- Kleckner N, Chalmers RM, Kwon D, Sakai J, Bolland S. Tn10 and IS10 transposition and chromosome rearrangements: Mechanism and regulation in vivo and in vitro. Curr Top Microbiol Immunol. 1996;204:49–82. doi: 10.1007/978-3-642-79795-8_3. [DOI] [PubMed] [Google Scholar]
- Kruklitis R, Welty DJ, Nakai H. ClpX protein of Escherichia coli activates bacteriophage Mu transposase in the strand transfer complex for initiation of Mu DNA synthesis. EMBO J. 1996;15:935–944. [PMC free article] [PubMed] [Google Scholar]
- Laski FA, Rio DC, Rubin GM. Tissue specificity of Drosophila P element transposition is regulated at the level of mRNA splicing. Cell. 1986;44:7–19. doi: 10.1016/0092-8674(86)90480-0. [DOI] [PubMed] [Google Scholar]
- Lavoie BD, Chaconas G. Site-specific HU binding in the Mu transpososome: Conversion of a sequence-independent DNA-binding protein into a chemical nuclease. Genes & Dev. 1993;7:2510–2519. doi: 10.1101/gad.7.12b.2510. [DOI] [PubMed] [Google Scholar]
- ————— Transposition of phage Mu DNA. Curr Top Microbiol Immunol. 1996;204:83–102. doi: 10.1007/978-3-642-79795-8_4. [DOI] [PubMed] [Google Scholar]
- Lee CC, Mul YM, Rio DC. The Drosophila P-element KP repressor protein dimerizes and interacts with multiple sites on P-element DNA. Mol Cell Biol. 1996;16:5616–5622. doi: 10.1128/mcb.16.10.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Craigie R. Protection of retroviral DNA from autointegration: Involvement of a cellular factor. Proc Natl Acad Sci. 1994;91:9823–9827. doi: 10.1073/pnas.91.21.9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol Cell Biol. 1992;12:5041–5049. doi: 10.1128/mcb.12.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Luo L, Baker TA. Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes & Dev. 1995;9:2399–2408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- Lewis SM, Wu GE. The origins of V(D)J recombination. Cell. 1997;88:159–162. doi: 10.1016/s0092-8674(00)81833-4. [DOI] [PubMed] [Google Scholar]
- Marchuk D, Drumm M, Saulino A, Collins FS. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res. 1991;19:1154. doi: 10.1093/nar/19.5.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- Mizuuchi K. Polynucleotidyl transfer reactions in transpositional DNA recombination. J Biol Chem. 1992a;267:21273–21276. [PubMed] [Google Scholar]
- ————— Transpositional recombination: Mechanistic insights from studies of mu and other elements. Annu Rev Biochem. 1992b;61:1011–1051. doi: 10.1146/annurev.bi.61.070192.005051. [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Mizuuchi K. Efficient Mu transposition requires interaction of transposase with a DNA sequence at the Mu operator: Implications for regulation. Cell. 1989;58:399–408. doi: 10.1016/0092-8674(89)90854-4. [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Baker TA, Mizuuchi K. Assembly of the active form of the transposase-Mu DNA complex: A critical control point in Mu transposition. Cell. 1992;70:303–311. doi: 10.1016/0092-8674(92)90104-k. [DOI] [PubMed] [Google Scholar]
- Morozov VE, Falzon M, Anderson CW, Kuff EL. DNA-dependent protein kinase is activated by nicks and larger single-stranded gaps. J Biol Chem. 1994;269:16684–16688. [PubMed] [Google Scholar]
- Mul, Y.M. and D.C. Rio. 1997. Reprogramming the purine nucleotide cofactor requirement of Drosophila P element transposase in vivo. EMBO J. (in press). [DOI] [PMC free article] [PubMed]
- Mullins MC, Rio DC, Rubin GM. cis-acting DNA sequence requirements for P-element transposition. Genes & Dev. 1989;3:729–738. doi: 10.1101/gad.3.5.729. [DOI] [PubMed] [Google Scholar]
- Nakai H, Kruklitis R. Disassembly of the bacteriophage Mu transposase for the initiation of Mu DNA replication. J Biol Chem. 1995;270:19591–19598. doi: 10.1074/jbc.270.33.19591. [DOI] [PubMed] [Google Scholar]
- Nassif N, Penney J, Pal S, Engels WR, Gloor GB. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol. 1994;14:1613–1625. doi: 10.1128/mcb.14.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas AL, Munz PL, Young CSH. A modified single-strand annealing model best explains the joining of DNA double-strand breaks in mammalian cells and cell extracts. Nucleic Acids Res. 1995;23:1036–1043. doi: 10.1093/nar/23.6.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brochta DA, Gomez SP, Handler AM. P element excision in Drosophila melanogaster and related drosophilids. Mol Gen Genet. 1991;225:387–394. doi: 10.1007/BF00261678. [DOI] [PubMed] [Google Scholar]
- O’Hare K, Rubin GM. Structures of P transposable elements and their sites of insertion and excision in the Drosophila melanogaster genome. Cell. 1983;34:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- Preston CR, Engels WR. P-element-induced male recombination and gene conversion in Drosophila. Genetics. 1996;144:1611–1622. doi: 10.1093/genetics/144.4.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CR, Sved JA, Engels WR. Flanking duplications and deletions associated with P-induced male recombination in Drosophila. Genetics. 1996;144:1623–1638. doi: 10.1093/genetics/144.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden DA, McBlane JF, van Gent DC, Gellert M. Distinct DNA sequence and structure requirements for the two steps of V(D)J recombination signal cleavage. EMBO J. 1996;15:3197–3206. [PMC free article] [PubMed] [Google Scholar]
- Rio DC, Rubin GM. Identification and purification of a Drosophila protein that binds to the terminal 31-base-pair inverted repeats of the P transposable element. Proc Natl Acad Sci. 1988;85:8929–8933. doi: 10.1073/pnas.85.23.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC, Laski FA, Rubin GM. Identification and immunochemical analysis of biologically active Drosophila P element transposase. Cell. 1986;44:21–32. doi: 10.1016/0092-8674(86)90481-2. [DOI] [PubMed] [Google Scholar]
- Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: Role for short sequence homologies in the joining reaction. Mol Cell Biol. 1986;6:4295–4304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Zhu C, Gellert M. Characterization of broken DNA molecules associated with V(D)J recombination. Proc Natl Acad Sci. 1993;90:10788–10792. doi: 10.1073/pnas.90.22.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Chalmers RH, Kleckner N. Identification and characterization of a pre-cleavage synaptic complex that is an early intermediate in Tn10 transposition. EMBO J. 1995;14:4374–4383. doi: 10.1002/j.1460-2075.1995.tb00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW, Craig NL. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Savilahti H, Rice PA, Mizuuchi K. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J. 1995;14:4893–4903. doi: 10.1002/j.1460-2075.1995.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Schlissel M, Constantinescu A, Morrow T, Baxter M, Peng A. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes & Dev. 1993;7:2520–2532. doi: 10.1101/gad.7.12b.2520. [DOI] [PubMed] [Google Scholar]
- Scottoline BP, Chow S, Ellison V, Brown PO. Disruption of the terminal base pairs of retroviral DNA during integration. Genes & Dev. 1997;11:371–382. doi: 10.1101/gad.11.3.371. [DOI] [PubMed] [Google Scholar]
- Spanopoulou E, Zaitseva F, Wang FH, Santagata S, Baltimore D, Panayotou G. The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. 1996;87:263–276. doi: 10.1016/s0092-8674(00)81344-6. [DOI] [PubMed] [Google Scholar]
- Staveley BE, Heslip TR, Hodgetts RB, Bell JB. Protected P-element termini suggest a role for inverted-repeat-binding protein in transposase-induced gap repair in Drosophila melanogaster. Genetics. 1995;139:1321–1329. doi: 10.1093/genetics/139.3.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen SB, Gomelsky L, Speidel SL, Roth DB. Initiation of V(D)J recombination in vivo: Role of recombination signal sequences in formation of single and paired double-strand breaks. EMBO J. 1997;16:2656–2664. doi: 10.1093/emboj/16.10.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette MG, Chaconas G. The Mu transpositional enhancer can function in trans: Requirement of the enhancer for synapsis but not strand cleavage. Cell. 1992;68:1101–1108. doi: 10.1016/0092-8674(92)90081-m. [DOI] [PubMed] [Google Scholar]
- Surette MG, Buch SJ, Chaconas G. Transpososomes: Stable protein-DNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell. 1987;49:253–262. doi: 10.1016/0092-8674(87)90566-6. [DOI] [PubMed] [Google Scholar]
- Taccioli GE, Gottlieb TM, Blunt T, Priestley A, Demengeot J, Mizuta R, Lehmann AR, Alt FW, Jackson SP, Jeggo PA. Ku80: Product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 1994;265:1442–1445. doi: 10.1126/science.8073286. [DOI] [PubMed] [Google Scholar]
- Takasu-Ishikawa E, Yoshihara M, Hotta Y. Extra sequences found at P element excision sites in Drosophila melanogaster. Mol Gen Genet. 1992;232:17–23. doi: 10.1007/BF00299132. [DOI] [PubMed] [Google Scholar]
- Tuteja N, Tuteja R, Ochem A, Taneja P, Huang NW, Simoncsits A, Susic S, Rahman K, Marusic L, Chen J, et al. Human DNA helicase II: A novel DNA unwinding enzyme identified as the Ku autoantigen. EMBO J. 1994;13:4991–5001. doi: 10.1002/j.1460-2075.1994.tb06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, McBlane JF, Ramsden DA, Sadofsky MJ, Hesse JE, Gellert M. Initiation of V(D)J recombination in a cell-free system. Cell. 1995;81:925–934. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Ramsden DA, Gellert M. The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell. 1996a;85:107–113. doi: 10.1016/s0092-8674(00)81086-7. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996b;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Hiom K, Paull TT, Gellert M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997;16:2665–26670. doi: 10.1093/emboj/16.10.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luenen HG, Colloms SD, Plasterk RH. Mobilization of quiet, endogenous Tc3 transposons of Caenorhabditis elegans by forced expression of Tc3 transposase. EMBO J. 1993;12:2513–2520. doi: 10.1002/j.1460-2075.1993.tb05906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The mechanism of transposition of Tc3 in C. elegans. Cell. 1994;79:293–301. doi: 10.1016/0092-8674(94)90198-8. [DOI] [PubMed] [Google Scholar]
- Vos JC, Plasterk RH. Tc1 transposase of Caenorhabditis elegans is an endonuclease with a bipartite DNA binding domain. EMBO J. 1994;13:6125–6132. doi: 10.1002/j.1460-2075.1994.tb06959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos JC, De Baere I, Plasterk RH. Transposase is the only nematode protein required for in vitro transposition of Tc1. Genes & Dev. 1996;10:755–761. doi: 10.1101/gad.10.6.755. [DOI] [PubMed] [Google Scholar]
- Watson MA, Chaconas G. Three-site synapsis during Mu DNA transposition: A critical intermediate preceding engagement of the active site. Cell. 1996;85:435–445. doi: 10.1016/s0092-8674(00)81121-6. [DOI] [PubMed] [Google Scholar]
- Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]