The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2 (original) (raw)
Abstract
The high risk human papillomaviruses (HPVs) are associated etiologically with the majority of human cervical carcinomas. These HPVs encode two viral oncoproteins, E6 and E7, which are expressed consistently in cervical cancers. The function of these viral oncoproteins during a productive infection is to ensure viral replication in cells that have normally withdrawn from the cell division cycle and are committed to terminal differentiation. Expression of the E7 oncoprotein has been shown to lead to the abrogation of various negative growth regulatory signals, including a p53-mediated G1 growth arrest, TGFβ-mediated growth inhibition, and quiescence of suprabasal keratinocytes. Here we describe a novel mechanism by which E7 can uncouple cellular proliferation and differentiation. In contrast to normal, differentiating keratinocytes, HPV-16 E7-expressing keratinocytes show delayed cellular differentiation and elevated cdk2 kinase activity despite high levels of p21Cip1 and association of p21Cip1 with cdk2. We show that the HPV E7 protein can interact with p21Cip1 and abrogate p21Cip1-mediated inhibition of cyclin A and E-associated kinase activities. Based on these findings, we propose that this capacity of the HPV E7 oncoprotein to overcome p21Cip1-mediated inhibition of cdk2 activity during keratinocyte differentiation contributes to the ability of E7 to allow for cellular DNA synthesis in differentiated keratinocytes.
Keywords: Human papillomavirus, cyclin dependent kinase inhibitor, cellular differentiation, cell division cycle
The papillomaviruses are small DNA viruses that have a pronounced tropism for epithelial cells where they cause benign hyperplasias or warts. Human papillomaviruses (HPVs) can be subdivided into two groups based on the propensity of the lesions to progress to malignancy. Low risk HPVs are associated with lesions with a very low frequency of malignant progression, whereas the lesions caused by the high risk HPVs can undergo carcinogenic progression (for review, see zur Hausen 1996), with more than 95% of all cervical carcinomas harboring high risk HPVs (Bosch et al. 1995). Although HPVs infect the dividing, basal cells of host epithelia, their replication is intimately tied to the cellular differentiation machinery of these cells. Expression of the viral capsid proteins and productive viral replication can only occur in the differentiated layers of the skin that are composed of cells that have withdrawn from the cell division cycle. Limited by their small genome size, with the exception of the viral E1 and E2 proteins, papillomaviruses do not encode the enzymes that are rate limiting for DNA replication. Rather, these viruses have developed multiple strategies to subvert key regulatory circuits that control host cell replication (for review, see Howley 1996). The precise mechanisms by which HPVs are able to reactivate the dormant replication machinery in these cells to replicate their own genomes are unknown. The E6 and E7 proteins of the high-risk HPVs interact with and functionally abrogate the p53 and retinoblastoma tumor suppressor (pRB) proteins, respectively. In a nonproductive infection, integration of the viral DNA into the host genome causes dysregulated expression of E6 and E7, which contributes to malignant progression of a high-risk HPV-associated lesion (for review, see Vousden 1995). Therefore, the elucidation of the transforming principles of these viral proteins also yields important insights into the replication strategy of the high-risk HPVs and vice versa.
Because of the intricate link between the arrest of cell division and the onset of terminal differentiation, the ability of papillomaviruses to reinitiate or maintain cellular replication implies an underlying capacity to alter the cellular differentiation program. Initial studies investigating the effects of E6 and E7 expression on cellular replication were performed by expressing E6 and E7 using recombinant retroviruses to infect epithelial raft cultures (Blanton et al. 1992; Halbert et al. 1992; Woodworth et al. 1992; Cheng et al. 1995). These studies showed that high-risk and to a lesser extent also low-risk HPV E7 proteins are capable of inducing expression of proliferating cell nuclear antigen (PCNA) and reactivating cellular DNA synthesis in the suprabasal, differentiated cells of epithelial raft cultures. Interestingly, the differentiation program was not altered markedly in these rafts. Histologically, HPV E7-expressing raft cultures were reminiscent of high-risk HPV-positive squamous intraepithelial lesions (SIL) where there is an expansion of poorly differentiated suprabasal cells but differentiation is still observed in the upper strata of the lesion (Blanton et al. 1992; Cheng et al. 1995). Although these studies suggested that the E7 protein is capable of reactivating the cellular replication machinery, the underlying molecular mechanism remained unknown. The ability of HPV-16 E7-expressing cells to override a variety of negative growth regulatory signals has been well documented. This includes the abrogation of transforming growth factor β (TGFβ)-mediated growth inhibition, p53-mediated G1 growth arrest, and cell cycle withdrawal during terminal differentiation (Vousden et al. 1993; Demers et al. 1994, 1996; Hickman et al. 1994; Slebos et al. 1994).
Whereas the differentiation-specific expression of structural proteins, such as the keratins and cornified envelope proteins, has been studied extensively in squamous epithelial cells (for review, see Fuchs 1993), the basic mechanisms that drive the differentiation program in keratinocytes are less well understood. Recent studies with murine keratinocytes have focused on the elucidation of mechanisms that couple cellular differentiation to the control of the cell division cycle. These studies have demonstrated that the cyclin-dependent kinase inhibitors (CKIs) p21Cip1 and p27Kip1 each have a role in this process. The levels of these CKIs increase during cellular differentiation resulting in a cell cycle arrest. The induction of p21Cip1 is through a p53-independent pathway and involves p300 (Missero et al. 1995, 1996). Similar results were obtained in differentiating human keratinocytes and the major initial kinase target of the CKIs induced during keratinocyte differentiation is cyclin-dependent kinase 2 (cdk2) (R.M. Alani, J. Hasskarl, and K. Münger, in prep.).
In this study we propose a mechanism whereby HPV-16 E7 is able to uncouple cellular proliferation and differentiation in human keratinocytes by targeting p21Cip1. HPV-16 E7-expressing keratinocytes show delayed cellular differentiation and elevated cdk2 kinase activity despite high levels of p21Cip1 and association of p21Cip1 with cdk2. We demonstrate that HPV-16 E7 can abrogate p21Cip1-mediated cdk2 inhibition and can exist in complexes that contain p21Cip1. Because induction of p21Cip1 during keratinocyte differentiation is a p53-independent process (Missero et al. 1995), it is not targeted by the HPV E6 protein. Based on these results we suggest a model whereby abrogation of p21Cip1-mediated inhibition of cdk2 activity by the HPV E7 protein is a vital step that leads to the uncoupling of the processes of cellular differentiation and proliferation in HPV-infected keratinocytes. This function of HPV E7 may allow HPV replication in cells that have normally withdrawn from the cell division cycle.
Results
Cellular proliferation and differentiation are uncoupled in HPV-16 E7-expressing human keratinocytes
Primary cultures of human foreskin keratinocytes (HFKs) were infected with an HPV-16 E7-expressing LXSN-based retrovirus (Halbert et al. 1991) or the parental LXSN virus (Miller and Rosman 1989) as a control. Infected cells were obtained by G418 selection in low-calcium-containing, serum-free growth medium and subsequently treated with medium containing 2 mm calcium and 10% fetal bovine serum. Under these conditions, monolayer cultures of keratinocytes undergo a differentiation program that is similar to that observed in stratified squamous epithelia in vivo (Pittelkow et al. 1986; Schlegel et al. 1988). Although this system does not recapitulate the three dimensional architecture of the skin, it is possible to biochemically analyze cells at distinct phases of keratinocyte differentiation by standard methods, which is not feasible with raft cultures. Following 72 hr of treatment with differentiation medium, cells infected with the control LXSN virus displayed features typical of differentiated keratinocytes including cellular flattening, enlargement, and increased desmosomal contacts (Fig. 1A,B). In contrast, focal areas of morphologically poorly differentiated, small angular cells were observed frequently in cultures of HPV-16 E7-expressing keratinocytes under differentiation conditions (Fig. 1C,D). These phenotypic differences indicated that cellular differentiation was reduced in HPV-16 E7-expressing HFKs compared with normal cells. To determine whether these conclusions could be confirmed on a molecular basis, cell extracts were prepared from undifferentiated and differentiated HFKs expressing HPV-16 E7 and compared with matched control cells that were infected with the parental virus. Expression of keratin 10 and the cornified envelope protein involucrin was analyzed to assess keratinocyte differentiation. In normal cells, increased expression of involucrin and keratin 10 was observed at 24 and 72 hr after induction of differentiation. In the HPV-16 E7-expressing keratinocytes, the induction of these markers was delayed (Fig. 2A). We also examined the expression of differentiation markers in HPV-16 E6- and HPV-16 E6/E7-expressing keratinocytes. We found that the rate of induction of differentiation markers in HPV-16 E6-expressing keratinocytes was identical to that of matched control cells, whereas E6/E7-expressing cells exhibited a delayed expression of differentiation markers similar to HPV-16 E7-expressing keratinocytes (data not shown). We next studied the expression of proteins that are expressed typically in dividing cells such as PCNA and hyperphosphorylated forms of pRB. As shown in Figure 2A, the levels of PCNA expression were higher in E7-expressing keratinocytes than in normal keratinocytes at all time points. In agreement with the ability of HPV E7 to destabilize pRB, the overall levels of this protein were lower in E7-expressing cells than in the control cells; however, the relative levels of hyperphosphorylated pRB was higher in E7-expressing cells than in the parental cells at 72 hr after induction of keratinocyte differentiation (Fig. 2A). These results support the notion that expression of HPV-16 E7 in human keratinocytes results in sustained elevated proliferation in differentiating keratinocytes.
Figure 1.
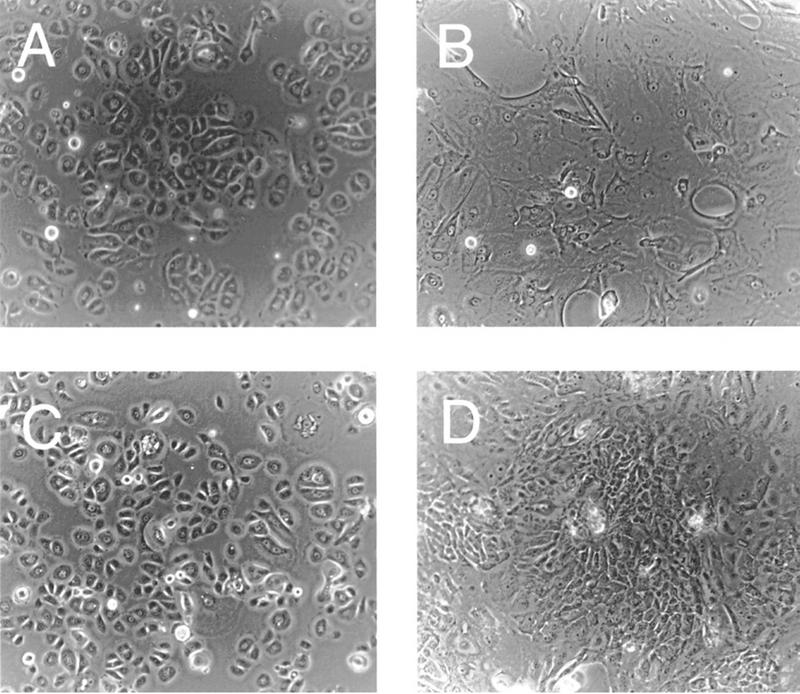
Photomicrographs of undifferentiated (A) and differentiated (B) human keratinocytes, and undifferentiated (C) and differentiated (D) HPV-16 E7-expressing keratinocytes.
Figure 2.

(A) Immunoblot analysis of markers of cellular differentiation (Involucrin; Keratin 10) and proliferation (proliferating cellular nuclear antigen, PCNA; retinoblastoma tumor suppressor protein, pRB) in keratinocytes infected with a control virus (LXSN) or an HPV-16 E7-expressing retrovirus (E7) at 0, 1, 6, 24, and 72 hr after induction of differentiation. (B) Analysis of p21Cip1, expression, p21Cip1 association with cdk2, and histone H1 (HH1) kinase activity associated with cdk2 at 0, 24, and 72 hr after induction of differentiation. To allow for quantitative comparisons, the different panels of LXSN and E7-expressing keratinocytes shown in this figure are derived from identical exposures of the same gel.
cdk2 activity is high in E7-expressing, differentiating keratinocytes despite association with p21Cip1
Previous studies have shown that expression of multiple CKIs, including p21Cip1 and p27Kip1, is induced during keratinocyte differentiation (Missero et al. 1995, 1996; R.M Alani, J. Hasskarl, and K. Münger, in prep.). The levels of p21Cip1 were considerably higher in HPV-16 E7-expressing keratinocytes compared with matched control cells at 24 and 72 hr after induction of differentiation (Fig. 2B). The induction of p27Kip1 was unaltered in E7-expressing cells compared with matched control cells (data not shown). Because previous studies had implicated cdk2 as the primary, initial target for inhibition by CKIs induced during cellular differentiation (Missero et al. 1995; R.M Alani, J. Hasskarl, and K. Münger, in prep.), we analyzed the association of p21Cip1 with cdk2. High levels of p21Cip1 in association with cdk2 were detected in E7-expressing keratinocytes at 24 and 72 hr after keratinocyte differentiation. Association of p21Cip1 with cdk2 was also detected in matched control keratinocytes but the levels were much lower (Fig. 2B). Next we compared the modulation of cdk2 activity during differentiation of control keratinocytes and HPV-16 E7-expressing keratinocytes. The activity of cdk2 decreased during differentiation in control keratinocytes and was very low at 72 hr after induction of differentiation. Surprisingly, however, cdk2 remained active in HPV-16 E7-expressing keratinocytes despite association with p21Cip1 (Fig. 2B). This was observed in multiple experiments. Induction of p21Cip1 was also detected in E6/E7-expressing keratinocytes and, like in E7-expressing cells, the activity of cdk2 during differentiation remained high in these cells (data not shown). The levels of cyclin E and cdk2 were approximately twofold increased in E7-expressing cells compared with matched control keratinocytes (data not shown).
HPV-16 E7 abrogates p21Cip1-mediated inhibition of cyclin A- and cyclin E-associated kinase activity
It has been reported that HPV-16 E7 can interact with cyclins A and E and a histone H1 kinase activity (Dyson et al. 1992; Davies et al. 1993; Tommasino et al. 1993). Although not conclusive, there is evidence to suggest that this interaction is mediated through the ability of E7 to associate with the pRB family member, p107 (McIntyre et al. 1996). Therefore, we wanted to determine whether the presence of E7 in a complex with these cyclins might alter the associated kinase activities or the susceptibility of these kinases to inhibition by p21Cip1. Cyclin A was immunoprecipitated from the human myeloid leukemia cell line, ML-1, and associated kinase activity was assayed using histone H1 as a substrate. When recombinant HPV-16 E7 fused to glutathione-_S_-transferase (GST–16E7) was titrated into the kinase assay, no effect was seen on cyclin A-associated kinase activity (Fig. 3A, lanes 1,3,4,5,6), whereas addition of GST p21Cip1 led to substantial inhibition of cyclin A-associated kinase activity (Fig. 3A, lane 2). When GST–16E7 was titrated into the kinase assay in the presence of p21Cip1, concentration-dependent recovery of kinase activity was observed (Fig. 3A, lanes 2,6,7,8,9). Approximately 80% of the initial kinase activity was recovered by addition of HPV-16 E7. No recovery of kinase activity was detected when unfused GST was incubated with cyclin A-associated kinases in the presence of p21Cip1 (Fig. 3A, lanes 2,11,12,13). Because the ability of E7 to associate with kinase complexes is thought to be attributable, in part, to its ability to associate with p107 (McIntyre et al. 1996), we also tested the ability of a pRB- and p107-binding deficient, transformation negative mutant of E7 (HPV-16 E7 Δ21-24) for its ability to abrogate the inhibitory effects of p21Cip1 in this assay. This mutant was reduced in its ability to abrogate the inhibitory effects of p21Cip1 on cyclin A-associated kinase activity (Fig. 3B).
Figure 3.

HPV-16 E7 can overcome p21Cip1-mediated inhibition of cyclin A- and cyclin E-associated histone H1 kinase activity in vitro. Cyclin A-associated (A,B) and cyclin E-associated (C) protein kinases were immunoprecipitated from the human myeloid leukemia cell line ML-1 and kinase assays were performed in the absence (−) or presence (+) of 2 μg of purified GST–p21Cip1 using histone H1 as a substrate. Increasing amounts of purified GST–16E7 (0.4, 1.1, 4.4, and 6.6 μg) (A,C) or the pRB-binding deficient mutant GST–16E7Δ21-24 (0.4, 1.1, 4.4, and 6.6 μg) (B) were titrated into the reaction. As a control, purified unfused GST (1.1, 4.4, and 6.6 μg) was added. Quantitations are shown underneath each assay.
The results presented in Figure 2B showed that cdk2 activity remained high in E7-expressing, differentiating HFKs despite increased levels of p21Cip1 and association of the CKI with cdk2. cdk2 associates with multiple cyclin partners, including cyclins A and E, both of which are important for the initiation of and progression through the synthesis phase of the cell division cycle (for review, see Pines 1993). Therefore, we wanted to determine if HPV-16 E7 was capable of counteracting p21Cip1-mediated inhibition of cyclin E-associated kinase activity as well. As was seen for cyclin A-associated kinase activity, addition of GST–16E7 had no effect on cyclin E-associated kinase activity (Fig. 3C, lanes 1,3,4,5,6). However, addition of GST–16E7 to a cyclin E-associated kinase assay in the presence of p21Cip1 led to a significant abrogation of the ability of p21Cip1 to inhibit kinase activity. (Fig. 3C, lanes 2,7,8,9,10). As before, addition of GST alone had no effect on kinase activity (Fig. 3C, lanes 2,11,12,13). The ability of E7 to abrogate p21Cip1 activity was consistently higher when cyclin E-associated kinase activity was assayed, and up to 100% of the initial kinase activity was recovered by addition of HPV-16 E7. These results demonstrate that HPV-16 E7 can counter the inhibitory activity of p21Cip1 on cyclin A- and cyclin E-associated kinases in vitro.
HPV-16 E7 can associate with p21Cip1
Because E7 can abrogate the inhibitory effects of p21Cip1, we wanted to ascertain whether or not E7 was capable of existing in complexes with p21Cip1. Insect cells were infected with recombinant baculoviruses expressing wild-type HPV-16 E7 or mutant E7 proteins (Fig. 4A). The HPV-16 E7 Δ6–10 mutant contains a deletion in the adenovirus (Ad) conserved region 1 homology domain within the amino terminus of E7. This mutant of E7 is transformation deficient, yet is still capable of interacting with pRB family members (Phelps et al. 1992). The pRB-binding-deficient HPV-16 E7 Δ21–24 mutant analyzed in the cyclin A-associated kinase assay (Fig. 3B) was described above. The HPV-16 E7 Δ35–37 mutant has a deletion in an acidic casein kinase II consensus phosphorylation site adjacent to the pRB-binding domain. This mutant is transformation competent and retains wild-type pRB-binding properties (Phelps et al. 1992). Equal expression of the HPV-16 E7 proteins were verified by immunoblot analysis (data not shown). Insect cells infected with these recombinant baculoviruses were lysed, and protein extracts were incubated with in vitro synthesized, radioactively labeled p21Cip1. Immunoprecipitations with an E7-specific monoclonal antibody showed that p21Cip1 was coimmunoprecipitated efficiently with wild-type HPV-16 E7, HPV-16 E7 Δ6–10, and HPV-16 E7 Δ35–37 and less efficiently with HPV-16 E7 Δ21–24 (Fig. 4B). This correlates with the reduced ability of this mutant of E7 to abrogate the inhibitory effects of p21Cip1 toward cyclin A- and cyclin E-associated kinases.
Figure 4.
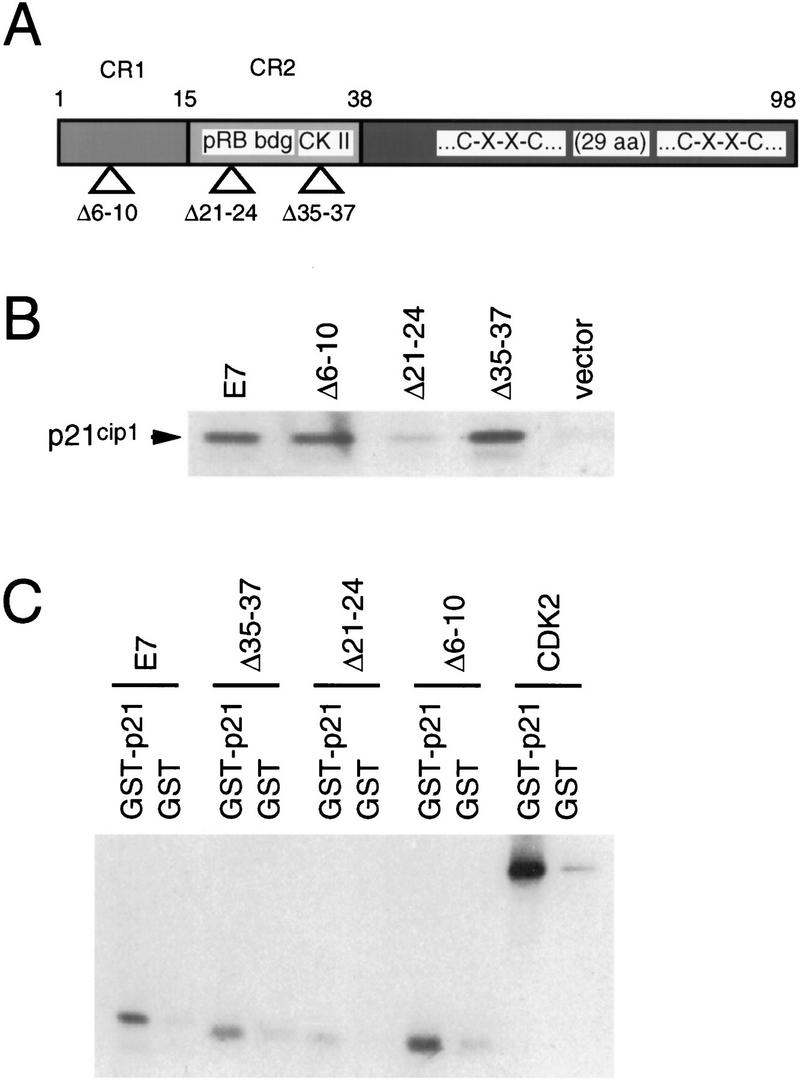
Complex formation of HPV-16 E7 and p21Cip1 in vitro. (A) Schematic representation of HPV-16 E7 and the mutants used for these experiments. (B) Radioactively labeled p21Cip1 protein was synthesized by in vitro transcription/in vitro translation in the presence of 35S-methionine and mixed with insect cell lysates containing the wild-type HPV-16 E7 protein (E7), or each of three mutant E7 proteins (Δ6–10; Δ21–24; Δ35–37). Extracts from cells infected with wild-type baculovirus (vector) were used as a negative control. HPV-16 E7/p21Cip1 complexes were purified by immunoprecipitation with an E7-specific monoclonal antibody that recognizes an epitope outside of the mutated domains of E7 (clone 7F3; obtained from Ciba-Corning Diagnostics) and analyzed by SDS-PAGE and fluorography. The signal of p21Cip1 in complex with wild-type E7 corresponds to ∼5% of the input protein. (C) HPV E7 or each of three mutant E7 proteins (Δ6–10; Δ21–24; Δ35–37) were synthesized by in vitro transcription/in vitro translation and mixed with GST–p21 or GST. Complexes were purified by affinity chromatography with glutathione–Sepharose followed by SDS-PAGE and bound E7 was detected by fluorography. The signal of wild-type E7 in complex with GST–p21 corresponds to ∼5% of the input protein. cdk2 was used as a positive control and bound to p21Cip1 at an efficiency of ∼20%.
The results of these binding experiments were confirmed using bacterially produced, purified GST fusion proteins and equal amounts of radioactively labeled proteins generated by in vitro transcription/in vitro translation. Approximately 5% of the input 35S-labeled HPV-16 E7 protein was seen to bind GST-p21Cip1 (Fig. 4C). The binding of HPV-16 E7 Δ6–10 and HPV-16 E7 Δ35–37 to p21Cip1 was similar, whereas the HPV-16 E7 Δ21–24 mutant of E7 bound to p21Cip1 with reduced efficiency (Fig. 4C). In comparison, ∼20% of the input 35S-labeled cdk2 bound to GST–p21Cip1 (Fig. 4C). In the reverse experiment, 35S-labeled p21Cip1 was found to bind GST–HPV-16 E7, whereas the HPV-16 E7 Δ21–24 mutant of E7 bound to p21Cip1 with reduced efficiency (data not shown). Although binding of HPV-16 E7 Δ21–24 to p21Cip1 was reduced, when this mutant was used in binding experiments with pRB, it was completely negative (Münger et al. 1989). Because all of the amino-terminal mutants tested here retain some ability to bind to p21Cip1, the data suggest that the carboxyl terminus of E7 as well as sequences in the pRB-binding site may be important for interacting with p21Cip1.
Low-risk HPV-6 E7 has a decreased ability to associate with p21Cip1 and abrogate p21Cip1-mediated cdk2 inhibition
Because low-risk HPVs are faced with many of the same biological challenges as the high-risk HPVs with regards to viral replication, we wanted to determine if the low-risk HPV-6 E7 protein could also interact with p21Cip1. Using a bacterially produced, purified GST–HPV-6 E7 fusion protein and in vitro transcribed/translated p21Cip1, it was seen that HPV-6 E7 could interact with p21Cip1 albeit with a significantly reduced efficiency when compared with the high-risk HPV-16 E7 (Fig. 5A). Addition of GST–HPV-6 E7 to a cyclin E-associated kinase assay in the presence of p21Cip1 led to concentration-dependent recovery of kinase activity. Although up to 95% of the initial kinase activity was recovered by HPV-6 E7, approximately fivefold higher concentrations of HPV-6 E7 were required (Fig. 5B) in comparison to the amount of HPV-16 E7 necessary for recovery of maximal activity (Fig. 3A). These results suggest that HPV-6 E7 has a decreased ability to abrogate the inhibitory effects of p21Cip1.
Figure 5.

The low-risk HPV-6 E7 protein can bind to p21Cip1 and overcome p21Cip1-mediated inhibition of cyclin E-associated histone H1 kinase activity. (A) Radioactively labeled p21Cip1 protein was synthesized by in vitro transcription/in vitro translation in the presence of [35S]-methionine and mixed with purified GST-16 E7, GST-6 E7 or unfused GST as a control. Complexes were analyzed by glutathione–sepharose affinity chromatography, SDS-PAGE, and fluorography. The signal of p21Cip1 in complex with wild-type HPV-16 E7 corresponds to ∼5% of the input protein. (B) Cyclin E-associated protein kinases were immunoprecipitated from the human myeloid leukemia cell line ML-1 and kinase assays were performed in the absence (−) or presence (+) of 2 μg of purified GST–p21Cip1 using histone H1 as a substrate. Increasing amounts of purified GST–6 E7 (0.4, 1.1, 4.4, and 6.6 μg) were titrated into the reaction. As a control, purified unfused GST (1.1, 4.4, and 6.6 μg). Quantitation is shown underneath.
The HPV-16 E7 protein associates with p21Cip1 in E7-expressing human keratinocytes
To ascertain whether or not the association between HPV-16 E7 and p21Cip1 detected in vitro could be physiologically relevant, we wanted to determine if such complexes could be detected in the primary host cells of HPVs, human keratinocytes. Because we had shown previously that HPV E7-expressing, differentiating human keratinocytes possess active cdk2 bound to p21Cip1 (Fig. 2B), we performed coupled immunoprecipitation/immunoblot experiments using lysates from such cells. Lysates prepared from matched sets of vector-infected keratinocytes were used as controls. HPV-16 E7 was coprecipitated with a p21Cip1-specific antibody (Fig. 6A). In addition, p21Cip1 was coprecipitated with an E7 monoclonal antibody specifically from HPV-16 E7 expressing keratinocytes (Fig. 6B). These results show that HPV-16 E7 and p21Cip1 can be in a complex in differentiating human keratinocytes.
Figure 6.

Coimmunoprecipitation of HPV-16 E7 and p21Cip1 in E7-expressing keratinocytes. (A) Immunoprecipitations were performed with E7-specific (left) and p21Cip1-specific (right) antibodies, resolved by SDS-PAGE and followed by immunoblot analysis with an E7-specific monoclonal antibody. (B) Immunoprecipitation was performed with a p21Cip1-specific (left) or an E7-specific (right) antibody, resolved by SDS-PAGE and followed by immunoblot analysis with a p21Cip1-specific antibody.
Discussion
As a consequence of their restricted genome size, the small DNA tumor viruses do not encode many enzymes that are rate limiting for viral DNA replication and, therefore, have to appropriate host cellular components to replicate their own genomes. This constitutes a particular challenge for the HPVs whose life cycle is intimately linked to the differentiation status of the host cell. HPVs initially infect the replicative basal cells of the epithelium but full vegetative replication, including expression of the viral capsid proteins, can only occur in the terminally differentiated strata of the infected epithelium (for review, see Howley 1996). These cells have normally withdrawn from the cell cycle. Therefore, HPVs need to reactivate cellular DNA synthesis and/or maintain the infected host cell in a replication-competent state without blocking the actual process of keratinocyte differentiation. To uncouple cellular proliferation and differentiation, viral proteins target molecules that link these two processes. Previous studies with epithelial raft cultures have demonstrated that HPV E7 but not E6 is able to reactivate cellular proliferation in suprabasal keratinocytes. Despite a dramatic increase in cellular DNA synthesis, the pattern of keratinocyte differentiation was not significantly altered in E7-expressing keratinocytes (Blanton et al. 1992; Cheng et al. 1995). Therefore, HPV E7 but not E6 is able to uncouple cellular differentiation and proliferation in keratinocytes. We used a simple monolayer system of human keratinocyte differentiation (Pittelkow et al. 1986; Schlegel et al. 1988) (Fig. 1) to investigate the molecular changes that occur when HPV E7 is expressed in differentiating keratinocytes (Fig. 2). Our goal was to identify cellular molecules that are targeted by the E7 protein to derail the tight coupling of cellular proliferation and differentiation in keratinocytes. It has been shown previously that the CKI p21Cip1 has an important role in linking differentiation and proliferation in multiple cell types (Jiang et al. 1994; Steinman et al. 1994; Guo et al. 1995; Halevy et al. 1995; Macleod et al. 1995; Parker et al. 1995; Matsumura et al. 1997), including keratinocytes (Missero et al. 1995, 1996). Induction of p21Cip1 is an early event during keratinocyte differentiation, is independent of p53-induction, and results in the specific inhibition of cdk2 activity (Missero et al. 1995, 1996; R.M. Alani, J. Hasskarl, and K. Münger, in prep.). HPV-16 E7-expressing keratinocytes express relatively high levels of p21Cip1, and the activity of cdk2 is high despite association with p21Cip1 (Fig. 2B). This is not only observed in differentiating human keratinocytes but is a general characteristic of E7-expressing cells (data not shown). Our results demonstrate that the HPV E7 protein can associate with p21Cip1 (Figs. 4 and 6) and abrogate its ability to inhibit the activities of cyclin/cdk complexes (Fig. 3). A mutant of HPV-16 E7, which is unable to bind to pRB or p107 (HPV-16 E7 Δ21–24) (Münger et al. 1989; E. Blom and K. Münger, unpubl.), is decreased in its ability to associate with and abrogate the inhibitory activity of p21Cip1, suggesting that this region of E7 encompasses the p21Cip1-association domain. However, because all of the HPV-16 E7 mutants had an intact carboxyl terminus and retained at least some partial binding activity, this suggests that the interaction between E7 and p21Cip1 may involve multiple regions. The low-risk HPV-6 E7 protein, which has a reduced ability to bind to pRB family members (Münger et al. 1989; Ciccolini et al. 1994), also had a reduced ability to interact with and reactivate p21Cip1-inhibited cyclin E-associated kinase activity (Fig. 5). This is consistent with the greatly reduced ability of HPV-6 E7 to reactivate DNA synthesis in suprabasal keratinocyte in raft cultures (Halbert et al. 1992; Demers et al. 1996).
Based on these results we propose a model where interaction with the CKI p21Cip1 and its functional abrogation contribute to the ability of HPV E7 to reestablish and/or sustain elevated cdk2 activity in differentiating keratinocytes (Fig. 2) to allow for viral replication. The interaction of viral oncoproteins with CKIs has some precedent. The tax protein of the human T-cell leukemia virus type 1 (HTLV1) can bind to the CKI p16Ink4A (Suzuki et al. 1996; Low et al. 1997) and both Ad E1A and HPV-16 E7 can interact with p27Kip1 (Mal et al. 1996; Zerfass-Thome et al. 1996). Because p27Kip1 is involved in mediating cell cycle inhibition in response to growth inhibitory molecules such as TGFβ (for review, see Polyak 1996), targeting p27Kip1 may be important for the ability of these viral oncoproteins to overcome TGFβ-mediated growth inhibition (Laiho et al. 1990; Pietenpol et al. 1990; Demers et al. 1996). Moreover, p27Kip1 is also induced during keratinocyte differentiation (Missero et al. 1996), and it is conceivable that the ability of E7 to interact with p27Kip1 in addition to p21Cip1, contributes to the ability of HPV E7 to reactivate cellular DNA synthesis in differentiated keratinocytes.
The tumor suppressor p53 can directly activate transcription of p21Cip1 (El-Deiry et al. 1993; Harper et al. 1993; Xiong et al. 1993) and, therefore CKI is an important mediator of p53-associated G1 growth arrest. The high-risk HPV E6 proteins can interact with and induce the proteasome-mediated degradation of p53 (Scheffner et al. 1990; Werness et al. 1990), resulting in the abrogation of the p21Cip1-mediated growth arrest signal sent by p53 (Kessis et al. 1993; Foster et al. 1994; Gu et al. 1994). Because E6 and E7 are generally coexpressed in a viral infection or during HPV-associated carcinogenic progression, the ability of high-risk HPV E7 to overcome a p21Cip1-mediated growth inhibition may be less significant in those cases, such as DNA damage, where significant increases of p21Cip1 are triggered by p53 as the high-risk HPV E6 protein can induce p53 degradation and dampen the p53 response. During keratinocyte differentiation, p21Cip1 is induced by a p53-independent pathway (Missero et al. 1995), and the induction of p21Cip1 is not abrogated by HPV E6 (data not shown).
We have analyzed recently the actinomycin D-induced, p53-mediated, DNA damage signal transduction pathway in E7-expressing clones of the RKO colon carcinoma cell line. We found that p21Cip1 was induced rapidly in both parental and E7-expressing RKO cells and that cyclin A- and cyclin E-associated kinases were inhibited in both normal and E7-expressing cells (Jones and Münger 1997). This may seem to be at odds with the model we propose here, namely that HPV E7 can bind to p21Cip1 and abrogate its inhibitory activity. However, it has been noted that although E7 can bind to cyclin A, cyclin E, and a histone H1 kinase activity, the antibodies used for the kinase assays in this study did not detect E7-containing kinase complexes (McIntyre et al. 1996; Jones and Münger 1997). Consequently, these analyses failed to include E7-containing kinase complexes. In support of our current model we found, in agreement with a recent study (Hickman et al. 1997), that HPV E7-associated kinase activity did not markedly change and remained high after DNA damage (Jones and Münger 1997). These findings further support the current model that the subpopulation of E7-containing kinases is less susceptible to inhibition by p21Cip1.
The small DNA tumor viruses have evolved multiple strategies for sustaining cellular proliferation in the presence of anti-proliferative signals, which reflects the requirements of these viruses to stimulate cellular DNA synthesis to replicate their own genomes. One common mechanism used by these viruses is targeting the tumor suppressor proteins pRB and p53. But while the SV40 and adenovirus oncoproteins interact with and sequester the p53 and pRB tumor suppressors, the high-risk HPV-16 E6 and E7 oncoproteins employ nonstoichiometric mechanisms and can destabilize p53 and pRB, respectively (Scheffner et al. 1990; Demers et al. 1994; Boyer et al. 1996; Jones and Münger 1997). Furthermore, each of these viruses has also evolved additional, unique ways of abrogating cellular negative growth regulatory pathways. Most notably, both Ad E1A and SV40 TAg, but not the HPV oncoproteins, can target p300 and CBP (Eckner et al. 1994, 1996; Arany et al. 1995), a family of transcriptional coactivators with histone acetyltransferase activities (Bannister and Kouzarides 1996; Ogryzko et al. 1996; Yang et al. 1996), which has an important role in differentiation (for review, see Eckner 1996). Because p300 has a role in the transcriptional induction of p21Cip1, accumulation of p21Cip1 during cellular differentiation is abrogated by the Ad E1A protein (Missero et al. 1995; Billon et al. 1996). Similarly, the Ad E1A protein can block transcriptional induction of the CKIs p21Cip1 and p15Ink4B in response to TGFβ (Datto et al. 1997). This activity also depends on the integrity of the p300-binding site on Ad E1A (Missero et al. 1995). Therefore, whereas earlier work highlighted the similarities in the mechanism of action of the oncoproteins of DNA tumor viruses, more recent studies have shown that these proteins have evolved to employ distinct strategies to interfere with the normal functions of their cellular targets. These differences may provide insights as to why lesions caused by high-risk HPVs have a much higher incidence of malignant progression in comparison with those caused by adenoviruses or polyomaviruses.
In conclusion, we believe that the ability of HPV E7 to interact with and interfere with p21Cip1-mediated inhibition of cdk complexes has a vital role in maintaining a replication competent milieu in suprabasal keratinocytes that would have normally withdrawn from the cell division cycle. As a recent study showed that multiple domains of E7 are required for the induction of suprabasal DNA replication in keratinocytes (Demers et al. 1996), it is likely that targeting of p21Cip1 is not the only mechanism that contributes to this activity of E7. In addition, levels of cyclin E, cyclin A, and cdk2 are somewhat increased, and the steady-state levels of pRB are dramatically reduced in E7-expressing cells. Future studies will be focused on delineating the contributions of each of these molecular targets for viral replication and HPV-associated carcinogenesis.
Materials and methods
Cell culture
Monolayer cultures of primary HFKs were prepared from a pool of neonatal foreskins obtained from routine circumcisions (Rheinwald and Beckett 1981). Keratinocytes were isolated using dispase incubation of foreskin tissue to allow for dermal–epidermal separation. Epidermal specimens were trypsinized and plated on standard tissue culture dishes. Cells were maintained in serum-free keratinocyte growth medium (keratinocyte–SFM, GIBCO BRL). Differentiation was induced by allowing for keratinocyte growth in high-calcium-containing medium (2.0 mm calcium) consisting of Dulbecco’s modified Eagle medium (DMEM; GIBCO BRL) supplemented with 10% fetal bovine serum as described previously (Schlegel et al. 1988). Confluence of cultures under growth and differentiating conditions was regulated strictly to ensure differentiation effects were attributable to the supplemented media provided as opposed to confluence of cells, as this has been observed previously (Poumay and Pittelkow 1995).
Packaging cell lines (PA317) producing recombinant retroviruses LXSN, HPV-16 E6, HPV-16 E7, and HPV-16 E6/E7 were kindly provided by Dr. D. Galloway (Halbert et al. 1991). These cell lines were maintained in DMEM supplemented with 10% fetal bovine serum, 50 units/ml of penicillin, and 50 μg/ml of streptomycin, and recombinant retroviruses were prepared using standard methods (Miller and Rosman 1989). Retroviral infections of primary HFKs were performed as described previously (Halbert et al. 1991; Jones and Münger 1997)
The myeloid leukemia cell line, ML-1, was a kind gift of E. Harlow (Massachusetts General Hospital Cancer Center, Charlestown, MA). The cells were grown in DMEM containing 10% fetal calf serum.
Hi5 and Sf9 insect cells were grown at 25°C in Grace’s Insect cell media (GIBCO BRL).
Antibodies and immunological methods
Monoclonal antibodies against human p21Cip1 (CP-36, CP-68) were gifts from B. Dynlacht (Zhu et al. 1995), and involucrin antibodies were kindly provided by J. Kvedar (Baden and Kvedar 1993). Cdk2 (sc-163), cyclin A (BF683; sc-239, sc-293-AC), cyclin E (HE-12; HE-111, sc-248 AC), PCNA (sc-56), and p27 antibodies (sc-528) were purchased from Santa Cruz Biotechnologies. Keratin 10 antibodies (DE-K13) were obtained from Neomarkers. Monoclonal antibodies against p53 (Ab-6) and pRB (Ab-5) were purchased from Oncogene Science. The E7 monoclonal antibody 7F3 was kindly provided by Ciba-Corning Diagnostics (7F3); the other E7 antibodies have been described previously (Jones and Münger 1997) and were obtained from Santa Cruz Biotechnologies. Horseradish peroxidase-linked secondary antibodies were purchased from Amersham.
Human keratinocytes were lysed in 250 mm NaCl, 50 mm Tris-HCl (pH 7.5), 5 mm EDTA, 0.1% NP-40 (pH 7.5), 1 mm dithiothreitol (DTT), 0.5 mm PMSF, 1 μg/ml of aprotinin and leupeptin, 2 mm NaF, 0.5 mm Na Vanadate for 30 min at 0°C. Protein samples (100 μg) were analyzed by SDS-PAGE and immunoblot analyses using PVDF membranes (Immobilon P, Millipore). Antigen–antibody complexes were detected by enhanced chemiluminescence (ECL; Amersham).
For immunoprecipitation experiments, immunocomplexes were isolated by adding protein A–Sepharose beads (GIBCO BRL) at 4°C for 90 min. Immunocomplexes were washed three times in lysis buffer then evaluated as above. Experiments were repeated several times with independent sets of keratinocyte populations.
ML-1 cells were lysed in a 0.1% NP-40 lysis buffer (100 mm Tris at pH 7.4, 120 mm NaCl, 0.5 mm PMSF, 1 μg/ml of aprotinin and leupeptin) at 0°C for 30 min.
Kinase assays
cdk2 kinase assays were performed with 100 μg of keratinocyte lysate. Immunocomplexes were bound to protein A–Sepharose (Pharmacia), washed three times in lysis buffer and twice in kinase buffer (50 mm HEPES, 5 mm MnCl2, 10 mm MgCl2, 1 mm DTT at pH 7.4). Samples were then resuspended in 10 μl of kinase buffer containing 10 μCi of [γ-32P]ATP and 1 μm ATP (Pharmacia) and incubated for 30 min at 37°C in the presence of 1 μg of histone H1 (Boehringer Mannheim). Samples were subsequently analyzed by SDS-PAGE and autoradiography.
Cyclin A and E kinase assays were performed using immunocomplexes prepared from 1 mg of protein extract from ML-1 cells using monoclonal antibodies conjugated to agarose beads. Immunoprecipitations were performed for 3 hr at 0°C. The immunocomplexes were washed three times in 0.1% NP-40 lysis buffer and twice in 2× kinase buffer (50 mm Tris HCl at pH 7.4, 20 mm MgCl2) and then resuspended in 2× kinase buffer supplemented with 1 mm DTT and 100 μm ATP (Boehringer Mannheim). The beads were resuspended in a volume to yield 10 μl/sample. The exogenous proteins, GST–p21Cip1 and GST–E7, were then added at the desired concentrations to the immunocomplexes. The total volume of added exogenous proteins was held constant at 10 μl. The mixtures were incubated at 0°C for 10 min. One microgram of histone H1 (Boehringer Mannheim) and 10 μCi of [γ-32P]ATP were added to initiate the kinase assay. Samples were incubated for 30 min at 37°C and analyzed by SDS-PAGE and autoradiography. Quantitation was performed using NIH image software (v. 1.56).
Expression plasmids
GST–p21Cip1 was obtained from S. Elledge (Harper et al. 1993) and GST-16 E7 was described previously (Wu et al. 1993). GST–HPV-6 E7 was constructed by PCR cloning an _Eco_RI–_Bam_HI fragment containing HPV-6 E7 into pGEX–2TK (Pharmacia). His6-tagged versions of HPV-16 E7, HPV-16 E7 Δ6–10, HPV-16 E7 Δ21–24, and HPV-16 E7 Δ35–37 were cloned into pVL1393 (Invitrogen). HPV-16 E7 in vitro transcription/translation plasmids have been described previously (Münger et al. 1989). Plasmids for in vitro transcription/in vitro translation of cdk2 and p21Cip1 were obtained from P. Hinds (Harvard Medical School, Boston, MA) and R. Metema (Whitehead Institute, Cambridge, MA), respectively.
Protein expression and purification
Recombinant baculoviruses were produced by transfecting Sf9 insect cells using a commercially available kit (Baculogold kit; PharMingen). Virus was amplified in Hi5 cells, and viral stocks were maintained by harvesting media from infected Hi5 cells. Protein extracts were prepared by lysing infected cells in 0.1% NP-40 lysis buffer as described above.
Bacterial expression of GST fusion proteins was performed in Escherichia coli strain DH5α. Protein induction using IPTG (GIBCO BRL), cell lysis, and purification of proteins using glutathione–Sepharose beads (Pharmacia) were done according to standard methods, described previously (Wu et al. 1993). Purified GST–fusion proteins were quantitated using the Bradford assay (Bio-Rad) and were analyzed by SDS-PAGE before use.
Protein expression by coupled in vitro transcription/in vitro translation was performed using the TNT-coupled rabbit reticulocyte lysate kit (Promega).
Interaction experiments
In vitro transcribed/in vitro translated, 35S-labeled p21Cip1 (10 μl) was mixed with 1 mg of protein extract from Hi5 insect cells that had been infected with recombinant baculoviruses expressing either wild-type HPV-16 E7 or various mutants. Mixings were performed in 67.5 mm Tris HCl, 75 mm NaCl, 0.5% NP-40 at pH 7.8 at 4°C for 1 hr. After preclearing with normal rabbit serum, the monoclonal E7 antibody 7F3 was added. After an additional incubation of 1 hr at 4°C, immunocomplexes were collected using a rabbit anti-mouse secondary antibody preabsorbed to protein A-Sepharose. The complexes were washed and analyzed by SDS-PAGE and fluorography.
For GST-binding experiments, 1 mg of purified fusion protein was incubated with 10 μl of in vitro transcribed/in vitro translated, 35S-labeled protein. Mixings were performed in 150 mm NaCl, 50 mm Tris HCl, 0.5% NP-40 at pH 7.4 for 2 hr at 4°C. After the incubation, glutathione–Sepharose was added and the mixture was incubated for an additional 30 min at 4°C. The glutathione beads were washed with mixing buffer before being analyzed by SDS-PAGE and fluorography.
For immunoprecipitation/immunoblot analyses, 1 mg of cell extracts were used for immunoprecipitations with p21Cip1 or E7-specific monoclonal antibodies followed by immunoblot analyses with E7 or p21Cip1-specific antibodies.
Acknowledgments
We thank Drs. Steven Elledge, James DeCaprio, Brian Dynlacht, Joseph Kvedar, Anindya Dutta, René Metema, and Phil Hinds for generously sharing plasmid DNAs and antibodies; Dr. Denise Galloway for the E7- and E6-expressing retroviral vectors; Dr. Ed Harlow for the ML-1 cell line; Ciba-Corning Diagnostics for their kind gift of the E7-specific monoclonal antibody 7F3; Jennifer L. Yoerkie for constructing the recombinant baculovirus clones; Ann Hwang and Rani Dhavan for performing binding assays; Eric Blom for testing the binding of E7 Δ21–24 to p107; Margaret Dale and Andrew Lasser for advice; and Miranda Grace for expert technical assistance. We also thank Dr. Yang Shi, John Daniel, and members of the Münger laboratory for support, suggestions, and critical comments on the manuscript and Dr. Denise Galloway for sharing results before publication. This work was supported by grants from the National Institutes of Health T32 AR07098-21 and K08 AR01975-01A1 (R.M.A.) and CA66980 (K.M.). K.M. is supported by a Junior Faculty Research Award (JFRA-597) from the American Cancer Society.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kmunger@warren.med.harvard.edu; FAX (619) 432-0426.
References
- Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- Baden HP, Kvedar JC. Epithelial cornified envelope precursors are in the hair follicle and nail. J Invest Dermatol. 1993;101:72S–74S. doi: 10.1111/1523-1747.ep12362869. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Billon N, van Grunsven LA, Rudkin BB. The CDK inhibitor p21WAF1/Cip1 is induced through a p300-dependent mechanism during NGF-mediated neuronal differentiation of PC12 cells. Oncogene. 1996;13:2047–2054. [PubMed] [Google Scholar]
- Blanton RA, Coltrera MD, Gown AM, Halbert CL, McDougall JK. Expression of the HPV16 E7 gene generates proliferation in stratified squamous cell cultures which is independent of endogenous p53 levels. Cell Growth Diff. 1992;3:791–802. [PubMed] [Google Scholar]
- Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV, Alihonou E, Bayo S, Mokhtar HC, Chicareon S, Daudt A, Delosrios E, Ghadirian P, Kitinya JN, Koulibaly M, Ngelangel C, Tintore LMP, Riosdalenz JL, Sarjadi-Schneider A, Tafur L, Teyssie AR, Rolon PA, Torroella M, Tapia AV, Wabinga HR, Zatonski W, Sylla B, Vizcaino P, Magnin D, Kaldor J, Greer C, Wheeler C. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. J Nat Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes & Dev. 1995;9:2335–2349. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- Ciccolini F, Dipasquale G, Carlotti F, Crawford L, Tommasino M. Functional studies of E7 proteins from different HPV types. Oncogene. 1994;9:2633–2638. [PubMed] [Google Scholar]
- Datto MB, Hu PP, Kowalik TF, Yingling J, Wang XF. The viral oncoprotein E1A blocks transforming growth factor beta-mediated induction of p21/WAF1/Cip1 and p15/INK4B. Mol Cell Biol. 1997;17:2030–2037. doi: 10.1128/mcb.17.4.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Foster SA, Halbert CL, Galloway DA. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci. 1994;91:4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Espling E, Harry JB, Etscheid BG, Galloway DA. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Guida P, Münger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66:6893–6902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R. p300 and CBP as transcriptional regulators and targets of oncogenic events. Biol Chem. 1996;377:685–688. [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes & Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, Decaprio JA, Livingston DM, Morgan JA. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Foster SA, Demers GW, Etscheid BG, Galloway DA. The ability of human papillomavirus E6 proteins to target p53 for degradation in vivo correlates with their ability to abrogate actinomycin D-induced growth arrest. J Virol. 1994;68:5698–5705. doi: 10.1128/jvi.68.9.5698-5705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, E. 1993. Epidermal differentiation and keratin gene expression. J. Cell Sci. (Suppl.) 17: 197–208. [DOI] [PubMed]
- Gu ZM, Pim D, Labrecque S, Banks L, Matlashewski G. DNA damage induced p53 mediated transcription is inhibited by human papillomavirus type 18 E6. Oncogene. 1994;9:629–633. [PubMed] [Google Scholar]
- Guo K, Wang JA, Andres V, Smith RC, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers W, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activty in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 CDK-interacting protein cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hickman ES, Picksley SM, Vousden KH. Cells expressing HPV16 E7 continue cell cycle progression following DNA damage induced p53 activation. Oncogene. 1994;9:2177–2181. [PubMed] [Google Scholar]
- Hickman ES, Bates S, Vousden KH. Perturbation of the p53 response by human papillomavirus type 16 E7. J Virol. 1997;71:3710–3718. doi: 10.1128/jvi.71.5.3710-3718.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM. Papillomavirinae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fundamental virology. Philadelphia, PA: Lippincott-Raven Publishers; 1996. pp. 947–978. [Google Scholar]
- Jiang HP, Lin J, Su ZZ, Collart FR, Huberman E, Fisher PB. Induction of differentiation in human promyelocytic HL-60 leukemia cells activates p21, WAF1/CIP1, expression in the absence of p53. Oncogene. 1994;9:3397–3406. [PubMed] [Google Scholar]
- Jones DL, Münger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71:2905–2912. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessis TD, Slebos RJ, Nelson WG, Kastan MB, Plunkett BS, Han SM, Lorincz AT, Hedrick L, Cho KR. Human papillomavirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proc Natl Acad Sci. 1993;90:3988–3992. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiho M, DeCaprio JA, Ludlow JW, Livingston DM, Massagué J. Growth inhibition by TGF-β linked to suppression of retinoblastoma protein phosphorylation. Cell. 1990;62:175–185. doi: 10.1016/0092-8674(90)90251-9. [DOI] [PubMed] [Google Scholar]
- Low KG, Dorner LF, Fernando DB, Grossman J, Jeang KT, Comb MJ. Human T-cell leukemia virus type 1 Tax releases cell cycle arrest induced by p16INK4a. J Virol. 1997;71:1956–1962. doi: 10.1128/jvi.71.3.1956-1962.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes & Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Mal A, Poon RYC, Howe PH, Toyoshima H, Hunter T, Harter ML. Inactivation of p27Kip1 by the viral E1A oncoprotein in TGFβ-treated cells. Nature. 1996;380:262–265. doi: 10.1038/380262a0. [DOI] [PubMed] [Google Scholar]
- Matsumura I, Ishikawa J, Nakajima K, Oritani K, Tomiyama Y, Miyagawa J, Kato T, Miyazaki H, Matsuzawa Y, Kanakura Y. Thrombopoietin-induced differentiation of a human megakaryoblastic leukemia cell line, CMK, involves transcriptional activation of p21(WAF1/Cip1) by STAT5. Mol Cell Biol. 1997;17:2933–2943. doi: 10.1128/mcb.17.5.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre MC, Ruesch MN, Laimins LA. Human papillomavirus E7 oncoproteins bind a single form of cyclin E in a complex with cdk2 and p107. Virology. 1996;215:73–82. doi: 10.1006/viro.1996.0008. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–982. [PMC free article] [PubMed] [Google Scholar]
- Missero C, Calautti E, Eckner R, Chin J, Tsai LH, Livingston DM, Dotto GP. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc Natl Acad Sci. 1995;92:5451–5455. doi: 10.1073/pnas.92.12.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missero C, Di Cunto F, Kiyokawa H, Koff A, Dotto GP. The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes & Dev. 1996;10:3065–3075. doi: 10.1101/gad.10.23.3065. [DOI] [PubMed] [Google Scholar]
- Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Parker SB, Eichele G, Zhang PM, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. P53-independent expression of p21(Cip)1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- Phelps WC, Münger K, Yee CL, Barnes JA, Howley PM. Structure-function analysis of the human papillomavirus E7 oncoprotein. J Virol. 1992;66:2418–2427. doi: 10.1128/jvi.66.4.2418-2427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Münger K, Howley PM, Moses HL. TGFb1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990;61:777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- Pines J. Cyclins and cyclin-dependent kinases—Take your partners. Trends Biochem Sci. 1993;18:195–197. doi: 10.1016/0968-0004(93)90185-p. [DOI] [PubMed] [Google Scholar]
- Pittelkow MR, Wille JJ, Scott RE. Two functionally distinct classes of growth arrest states in human prokeratinocytes that regulate clonogenic potential. J Invest Dermatol. 1986;86:410–417. doi: 10.1111/1523-1747.ep12285684. [DOI] [PubMed] [Google Scholar]
- Polyak K. Negative regulation of cell growth by TGF beta. Biochim Biophys Acta. 1996;1242:185–199. doi: 10.1016/0304-419x(95)00009-5. [DOI] [PubMed] [Google Scholar]
- Poumay K, Pittelkow M. Cell density and culture factors regulate keratinocyte commitment to differentiation and expression of suprabasal K1/K10 keratins. J Invest Dermatol. 1995;104:271–276. doi: 10.1111/1523-1747.ep12612810. [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res. 1981;41:1657–1663. [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Schlegel R, Phelps WC, Zhang Y-L, Barbosa M. Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. EMBO J. 1988;7:3181–3187. doi: 10.1002/j.1460-2075.1988.tb03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebos RJC, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB, Cho KR. p53-dependent G(1) arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RA, Hoffman B, Iro A, Guillouf C, Liebermann DA, Elhouseini ME. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene. 1994;9:3389–3396. [PubMed] [Google Scholar]
- Suzuki T, Kitao S, Matsushime H, Yoshida M. HTLV-1 Tax protein interacts with cyclin-dependent kinase inhibitor p16INK4A and counteracts its inhibitory activity towards CDK4. EMBO J. 1996;15:1607–1614. [PMC free article] [PubMed] [Google Scholar]
- Tommasino M, Adamczewski JP, Carlotti F, Barth CF, Manetti R, Contorni M, Cavalieri F, Hunt T, Crawford L. HPV16 E7 protein associates with the protein kinase p33CDK2 and cyclin A. Oncogene. 1993;8:195–202. [PubMed] [Google Scholar]
- Vousden KH. Regulation of the cell cycle by viral oncoproteins. Sem Cancer Biol. 1995;6:109–116. doi: 10.1006/scbi.1995.0014. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Vojtesek B, Fisher C, Lane D. HPV-16 E7 or adenovirus E1A can overcome the growth arrest of cells immortalized with a temperature-sensitive p53. Oncogene. 1993;8:1697–1702. [PubMed] [Google Scholar]
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Woodworth CD, Cheng S, Simpson S, Hamacher L, Chow LT, Broker TR, DiPaolo JA. Recombinant retroviruses encoding human papillomavirus type 18 E6 and E7 genes stimulate proliferation and delay differentiation of human keratinocytes early after infection. Oncogene. 1992;7:619–626. [PubMed] [Google Scholar]
- Wu EW, Clemens KE, Heck DV, Münger K. The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J Virol. 1993;67:2402–2407. doi: 10.1128/jvi.67.4.2402-2407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- Zhu L, Harlow E, Dynlacht BD. p107 uses a p21CIP1-related domain to bind cyclin/cdk2 and regulate interactions with E2F. Genes & Dev. 1995;9:1740–1752. doi: 10.1101/gad.9.14.1740. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections–a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–F78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]