The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo (original) (raw)
. 2000 May 1;14(9):1132–1145.
Abstract
The ShcA gene products have served as a model for the analysis of phosphotyrosine-recognition domains, and for the functions of docking proteins during tyrosine kinase signaling. Here we show that ShcA is primarily expressed in the cardiovascular system during early mouse embryogenesis and regulates both heart development and establishment of mature blood vessels. Targeted mutation suggests that the ShcA adaptor is a pivotal target of tyrosine kinases that selectively potentiates activation of the MAP kinase pathway in the remodeling vasculature. Biochemical analysis of mutant cells shows that ShcA sensitizes cells to growth factor-induced MAP kinase activation, and also organizes cytoskeletal rearrangement in response to the extracellular matrix. ShcA may therefore orchestrate complex interactions within the vascular compartment by rendering cells permissive to respond to soluble and adhesive external cues.
Keywords: ShcA, phosphotyrosine adaptor, gene targeting, embryogenesis, cardiovascular system, MAP kinase activation
The ShcA proteins are phosphorylated on tyrosine in response to a range of extracellular signals, and have been widely studied for their ability to form multiple phosphotyrosine (pTyr)-dependent complexes, and thus to serve a docking function in tyrosine kinase signaling (for review, see Bonfini et al. 1996; Pawson and Scott 1997). Mammalian ShcA encodes three overlapping polypeptides of 46, 52, and 66 kD, that are produced through alternative splicing and differential use of translation initiation sites, and differ only in the length of their amino-terminal extensions (Pelicci et al. 1992; Migliaccio et al. 1997). These ShcA proteins possess two distinct pTyr recognition modules. At their carboxyl terminus they have an SH2 domain (Songyang et al. 1994), and toward the amino terminus they contain a pTyr-binding (PTB) domain, which recognizes phosphorylated motifs with the consensus sequence φXNXXY (in which φ is a large aliphatic residue) (van der Geer et al. 1995; Laminet et al. 1996). This motif is found in a number of activated receptor tyrosine kinases (RTKs), and in cytoplasmic proteins such as SH2-containing 5′-inositol phosphatase (Damen et al. 1996). The ShcA PTB domain can also bind acidic phospholipids, which may assist in the recruitment of ShcA proteins to membranes (Zhou et al. 1995; Ravichandran et al. 1997).
ShcA proteins physically associate with a variety of activated RTKs, primarily through their PTB domain, and consequently become phosphorylated at Tyr residues 239, 240, and 317 (on the basis of human 52-kD ShcA isoform) (Salcini et al. 1994; Gotoh et al. 1996; van der Geer et al. 1996). Both Tyr-239 and Tyr-317 are followed at the +2 position by an Asn residue, forming an optimal binding site for the SH2 domain of the adaptor Grb2 (Rozakis-Adcock et al. 1992; Songyang et al. 1994; van der Geer et al. 1996). The Ras guanine nucleotide exchange factors Sos1 and Sos2 are recruited into these complexes through their association with the SH3 domains of Grb2, providing a mechanism through which ShcA phosphorylation can lead to activation of the Ras-MAP kinase (MAPK) pathway (Rozakis-Adcock et al. 1992). ShcA can also associate, either directly or via Grb2, with the docking protein Gab-1, that in turn binds PI 3′ kinase (Ingham et al. 1998). Additional ShcA partners have been described, including SH3-containing proteins and adaptins (Weng et al. 1994; Matoskova et al. 1995; Okabayashi et al. 1996). Thus, ShcA appears to be a scaffold for the assembly of signaling proteins involved in the activation of the Ras–MAPK pathway, and potentially other signaling pathways. ShcA has been associated with JNK activation in B cells (Hashimoto et al. 1999), and the p66 ShcA isoform appears to have a unique role in regulating stress responses (Migliaccio et al. 1999).
ShcA is widely expressed in cultured cells and adult mouse tissues, and is phosphorylated in response to a large number of physiological stimuli (for review, see Bonfini et al. 1996). Two closely related mammalian genes, ShcB/Sck and ShcC/N-Shc, are primarily expressed in the nervous system (O'Bryan et al. 1996; Pelicci et al. 1996; Nakamura et al. 1998), and a single Shc gene (dshc) has been found in Drosophila (Lai et al. 1995), although the invertebrate gene product is significantly less complex than mammalian ShcA isoforms. To explore the biological and biochemical functions of ShcA proteins, we have introduced a mutation into the mouse ShcA gene. Unexpectedly, we find that ShcA is primarily expressed in the developing cardiovascular system and mediates pTyr signaling in mammalian heart development and angiogenic remodeling. Analysis of mutant cells indicates that ShcA has multiple functions, including sensitizing cells to small increases of growth factors and regulating cytoskeletal reorganization by adhesion receptors. Using an antibody that specifically recognizes the phosphorylated-activated Erk MAPK we detected high levels of MAPK activity in tissues such as the cardiovascular system and limb buds in wild-type embryos. Strikingly, MAPK activation is specifically impaired in regions of ShcA-deficient embryos that undergo angiogenic remodeling. ShcA therefore appears pivotal in the multiple signaling events required to establish a mature vasculature.
Results
Targeted disruption of the mouse ShcA gene
A targeting vector was designed to replace 2 kb of the ShcA gene with a PGK-neomycin resistance cassette (_neo_R) by homologous recombination in R1 mouse ES cells (Fig. 1A). The deleted genomic sequence contains the first two coding exons (exons 2 and 3), including the translational initiation sites for all three ShcA proteins and sequences encoding the amino-terminal half of the PTB domain (residues 1–80 of p52). Thus, any polypeptide expressed from the targeted (ShcAΔex2/3) locus would lack a functional PTB domain, and should therefore be defective for signaling activity. Southern blot analysis (Fig. 1B) of genomic DNA hybridized to a 5′ external probe identified multiple colonies with banding patterns expected from homologous recombination. Additional internal and 3′ external probes confirmed that the disrupted ShcA locus had the anticipated structure (data not shown). Two independent ES cell lines gave rise to highly chimeric males that transmitted the mutant ShcAΔex2/3 gene through the germ line, and subsequently gave the same homozygous mutant phenotype (see below).
Figure 1.
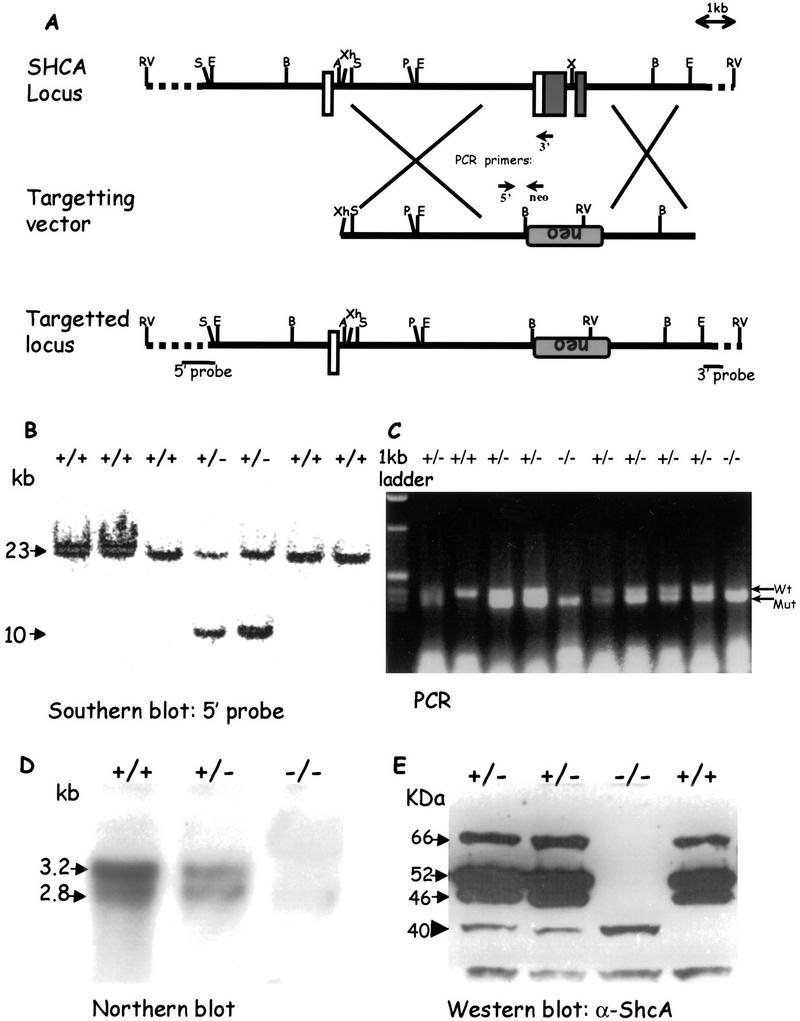
Targeted disruption of the ShcA locus. (A) Strategy for generation of the ShcAΔex2/3 mutation. A partial map of the wild-type mouse ShcA locus, including exons 1, 2, and 3 is shown. Upon homologous recombination, the targeting vector replaced a total of 2 kb of the genomic ShcA sequence with the PGK–neomycin resistance cassette in reverse orientation. Open boxes indicate noncoding exons, and shaded boxes indicate coding regions. Locations of the external 5′ and 3′ probes for Southern blotting and primers for PCR genotyping are shown. (B) Genotyping of _Eco_RV-digested ES cell-derived genomic DNA by Southern blot hybridization using a 5′ external probe as shown in A. Arrows indicate the 23-kb wild-type fragment and the 10-kb mutant allele. (C) PCR analysis of yolk sac DNA. Arrows point to the 410-bp PCR product amplified from the wild-type locus and the 347-bp band from the mutant allele. (D) Northern blot analysis of total RNA isolated from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) embryos, respectively. Total RNA (15 μg) was loaded in each lane and hybridized with a ShcA anti-sense riboprobe. The two ShcA transcripts are shown by arrows. (E) Western blot of total protein lysates isolated from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) embryos. Arrows point to the three wild-type ShcA polypeptides of 46, 52, and 66 kD and an arrowhead indicates the 40-kD truncated mutant protein. (A) _Asp_-718; (B) _Bam_HI; (E) _Eco_RI; (P) _Pvu_II; (RV) _Eco_RV; (S) _Sal_I; (X) _Xba_I; (Xh) _Xho_I; (neo) neomycin resistance cassette; (TK) thymidine kinase; (PCR) polymerase chain reaction.
Mice heterozygous for the ShcAΔex2/3 allele have not shown any detectable phenotype. However, screening of >800 offspring from heterozygous intercrosses failed to identify any viable homozygotes. To determine the point at which ShcA mutant embryos died, embryos from heterozygous intercrosses were examined. ShcAΔex2/3 allele-specific PCR analysis (Fig. 1C) of embryos collected from embryonic day (E) 9.5 to E11.5 showed that homozygotes were obtained with approximately the expected Mendelian frequency (Table 1). ShcAΔex2/3 mutant embryos had a normal morphology at E9.5 (Fig. 2). By E10.5, mutant embryos had an enlarged pericardium and abnormal cardiac contractions, but were equivalent in size to wild-type embryos. By E11.5, all ShcAΔex2/3 homozygotes appeared necrotic with pale yolk sacs, and their heart and cardiac outflow tracts were congested with blood. No homozygous mutant embryos were identified beyond E12. Examination of other areas of the embryos indicated that development of the placenta and limb buds of homozygous mutant embryos was grossly normal when compared with their littermate controls. Thus, ShcAΔex2/3 mutants die by E11.5 with evidence of a gross cardiovascular defect.
Table 1.
ShcA homozygote mutants die before E11.5 of gestation
| Developmental stage | No. of embryos | |||
|---|---|---|---|---|
| total (%) | +/+ (%) | +/− (%) | −/− (%) | |
| E8.5 | 21 (100) | 4 (19) | 12 (57) | 5 (24) |
| E9.5 | 121 (100) | 33 (27) | 60 (50) | 28 (23) |
| E10.5 | 324 (100) | 82 (25) | 171 (53) | 71 (22) |
| E11.5 | 84 (100) | 27 (29) | 50 (53) | 17a (18) |
| E13.5b | 60 (100) | 15 (25) | 31 (52) | 0 |
Figure 2.
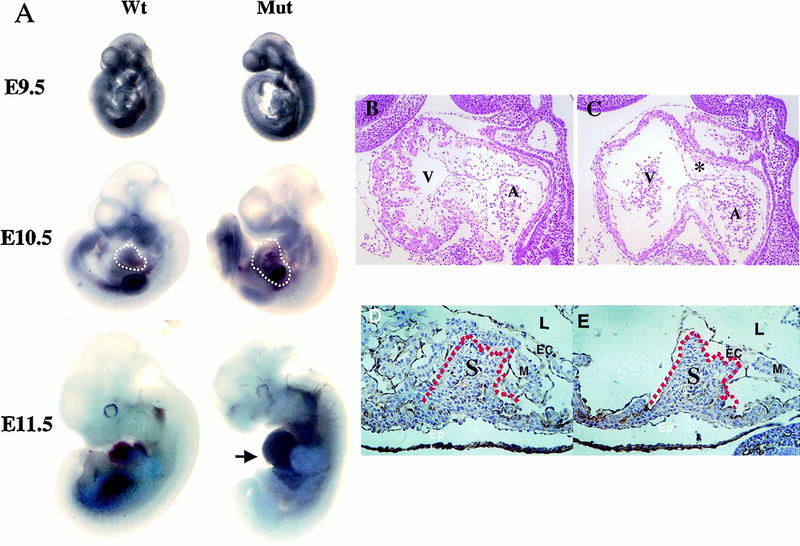
Phenotypes of homozygous ShcAΔex2/3 mutants. (A) Comparison of wild-type and mutant (Mut) embryos at E9.5, E10.5, and E11.5. The enlarged pericardium (white broken lines) is frequently displayed by ShcAΔex2/3 mutant embryos. Blood congestion (arrow) was observed in the ventral region of the mutant embryo at E11.5. (B,C) Hematoxylin and eosin stained midsagittal sections of wild-type (B) and mutant (C) E10 embryos demonstrate reduced trabeculation in the ventricle and hypoplastic endocardial cushions (asterisk) in the heart. (D,E) Higher magnification of transverse sections immunostained with anti-PECAM-1 antibodies (brownish precipitates) to highlight the endocardium, and counterstained with hematoxylin. Defects are pronounced in the myocardium of the trabeculae, but not in the intraventricular septum (red broken lines). (D) Wild type; (E) mutant. (A) Atrium; (EC) endocardium; (Epi) epicardium; (L) lumen of the heart; (M) myocardium; (S) intraventricular septum; (V) ventricle.
To analyze the effects of the Δex2/3 mutation on ShcA expression, we performed Northern and Western blot analysis. The normal 2.8 and 3.4 kb ShcA RNA transcripts and three ShcA polypeptides (46, 52, and 66 kD) could be detected as early as E9 (Fig. 1D,E), but were absent in homozygous mutants (Fig. 1D,E). However, immunoblotting with antibodies to the carboxy-terminal region of ShcA identified a small amount of a novel 40-kD protein in +/− and −/− embryos. The amount of this protein in mutant embryos was ∼2% of ShcA protein in wild-type embryos. RT–PCR analysis suggested that the mutant locus yields a transcript that initiates at the start site from the endogenous 2.8-kb mRNA, and is aberrantly spliced into the _neo_R cassette and subsequently into the fourth exon of the ShcA gene (data not shown). Coupled in vitro transcription/translation identified Met-134 as the likely initiation codon for the p40 protein. Thus, the ShcAΔex2/3 mutant allele encodes a low level of a 40-kD protein that lacks residues critical for PTB domain function.
ShcAΔex2/3 mutants show abnormal heart development
Whole mount examination of E10 mutant embryos showed that the heart had advanced to the point where normal right-sided cardiac looping was completed. Expression of cardiac markers such as cardiac actin and myosin light chains 2A and 2V was normal (data not shown), but the left ventricle was thin walled and irregular in shape, a defect that became more pronounced later in development.
To investigate this phenotype in more detail, serial transverse and sagittal sections of embryos from E8.5 to E11 were analyzed. Beginning at E9.5, a difference was observed in the normal pattern of finger-like projections formed by cardiomyocytes, known as trabeculae, and by E10.5, the trabeculae were severely reduced in the ShcAΔex2/3 mutant embryos (Fig. 2C), as compared with wild-type littermates (Fig. 2B). The thin myocardium associated with reduced trabeculation in ShcAΔex2/3 mutants likely contributes to poor heart contractility, leading to congestion of blood in the heart chamber. In addition, the endocardial cushions were poorly developed in mutant embryos (Fig. 2B,C), although the intraventricular septum appeared normal (Fig. 2D,E). Owing to the broad expression of ShcA in adult tissues and its potential involvement in many tyrosine kinase systems, this specific cardiac phenotype was unexpected. We therefore analyzed the pattern of ShcA expression during early embryonic development.
ShcA is predominantly expressed in the developing cardiovascular system
Whole-mount RNA in situ hybridization analysis of E9 wild-type embryos indicated that ShcA is primarily expressed in the developing cardiovascular system (Fig. 3). ShcA transcripts were detected at high levels in the heart (Fig. 3A), and in the region of the paired dorsal aorta. This latter staining was continuous with the branching arteries that traverse bilaterally through the branchial arches toward the aortic sac (Fig. 3B), and with the intersomitic vessels (Fig. 3C). This pattern of ShcA transcripts persists at E9.75. However, expression by this stage was more widespread, and especially pronounced in the craniofacial mesenchyme (Fig. 3D).
Figure 3.
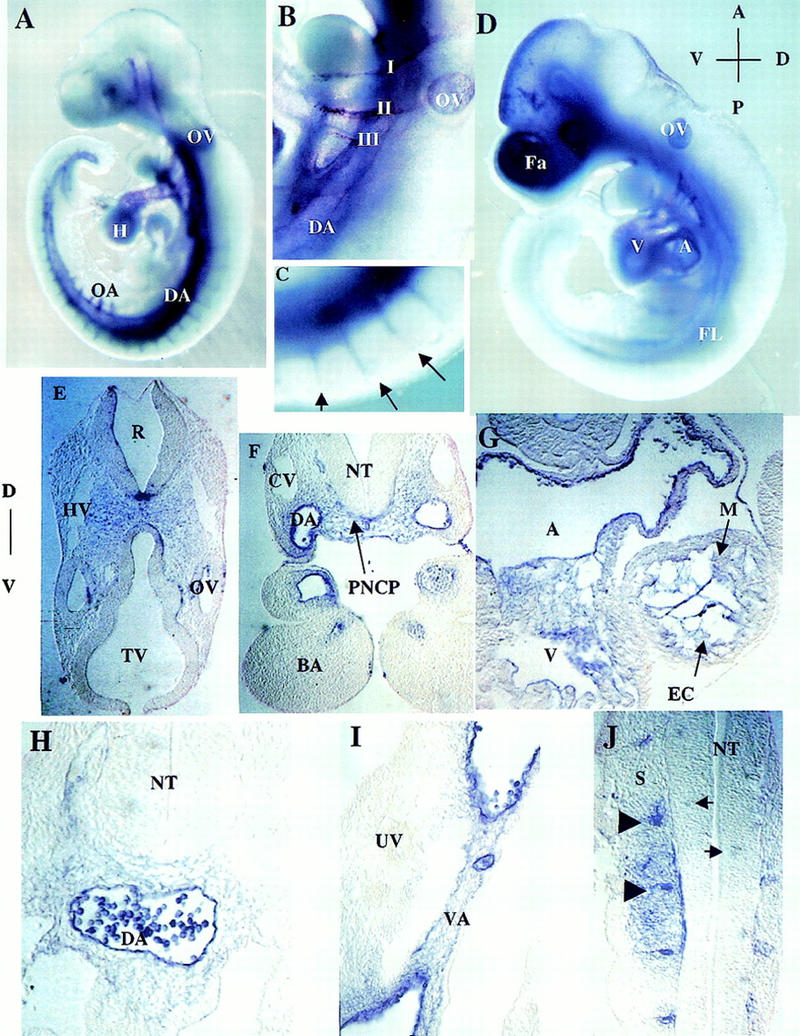
Expression pattern of ShcA RNA in wild-type embryos. (A) E9.0 whole-mount embryo. ShcA transcripts are abundant within and around the paired dorsal aortae, the contiguous omphalomesenteric artery, and the vitelline artery, as well as in the branching intersomitic arteries. Staining is clear in the ventricular chamber of the heart, and in the first, second, and third branchial arch arteries. Abundant transcripts are also evident around head vessels. (B) Higher magnification of A, branchial arch region. Note the expression of ShcA transcripts in and around aortic arteries that traverse through the branchial arches. (C) Higher magnification of the branching intersomitic vessels (arrows) as in A. (D) E9.75 whole-mount embryo. Transcripts are now detectable in the cranial mesenchyme. Scattered staining is obvious in the head. Expression in the dorsal aortae, heart, and otic vesicle remains detectable. (E_–_J) Rostral to caudal transverse sections of the E9.0 embryo shown in D. High levels of ShcA transcripts are found in the head mesenchyme in which the anterior carotid arteries develop (E); in and around the dorsal aorta (F,H,I) and branchial arch arteries (F); Transcripts are barely detectable in the anterior cardinal vein (F) and common cardinal vein (H). Prominent expression is also observed in the endocardium and surrounding mesenchymal cells within the heart (G), in the vitelline artery but not in the umbilical vein (I), in the endothelium of intersomitic vessels (arrowheads) and neighboring cells (J). Patchy staining within the neural tube is indicated by arrows (J). (I, II, III) First, second, and third branchial arch arteries; (A) aorta; (BA) branchial arch; (CV) cardinal vein; (DA) dorsal aorta; (EC) endocardium; (Fa) facial cranial mesenchyme; (FL) forelimb; (H) heart; (HV) head vein; (M) myocardium; (NT) neural tube; (OA) omphalomesenteric artery; (OV) otic vesicle; (PNCP) peri-neural capillary plexus; (R) rhombomere; (S) somites; (TV) telencephalic vesicle; (UV) umbilical vein; (V) ventricle; (VA) vitelline artery.
To obtain a more detailed analysis of ShcA expression, a histological examination of serial sections through these embryos was undertaken. ShcA RNA was identified in the head mesenchyme, in the region in which the anterior carotid arteries develop (Fig. 3E), in the endothelial lining of the branchial arch arteries, and in mesenchymal cells adjacent to the neuroepithelium (Fig. 3F). Expression was also observed in the endocardium, and in the loosely arranged mesenchyme of the endocardial cushions in the heart (Fig. 3G). More caudally, transcripts were prominent in the endothelial lining of the dorsal aorta, vitelline artery, and intersomitic vessels (ISVs), and in periendothelial mesenchymal cells (Fig. 3H–J). Patchy staining, possibly representing vessel sprouts, was also detected in the neural tube (Fig. 3J). Of interest, the signal was generally weak or absent from the venous system at this early stage, in contrast to the strong arterial expression (Fig. 3E–I). These results indicate that during early development ShcA is most prominently expressed in the endothelium and surrounding mesenchymal cells of the cardiovascular system.
ShcAΔex2/3 mutants have defects in angiogenesis and cell–cell contacts
The abundant cardiovascular expression of ShcA is consistent with the failure in heart development in mutant embryos, and raised the possibility that ShcA might have a broader role in vessel morphogenesis. We therefore analyzed wild-type and ShcAΔex2/3 mutant mice carrying a Tek–lacZ transgene, in which β-galactosidase (β-Gal) provides a specific marker for endothelial cells (ECs) (Dumont et al. 1994). Whole-mount staining with X-Gal revealed no absolute block to the early assembly of blood vessels derived by vasculogenesis or angiogenesis in ShcAΔex2/3 mutants (Fig. 4A,B). However, the β-Gal staining pattern was significantly less complex in the mutant embryos, particularly in the head. In addition, the ECs forming the endocardium in the mutant heart appeared thinner and more elongated (Fig. 4D). Defective vasculature was also apparent in cross sections of the yolk sac, which showed large and uniformly sized lumens (Fig. 4F). Discrete structures indicative of angioblast-derived blood islands appeared highly disorganized (Fig. 4F).
Figure 4.
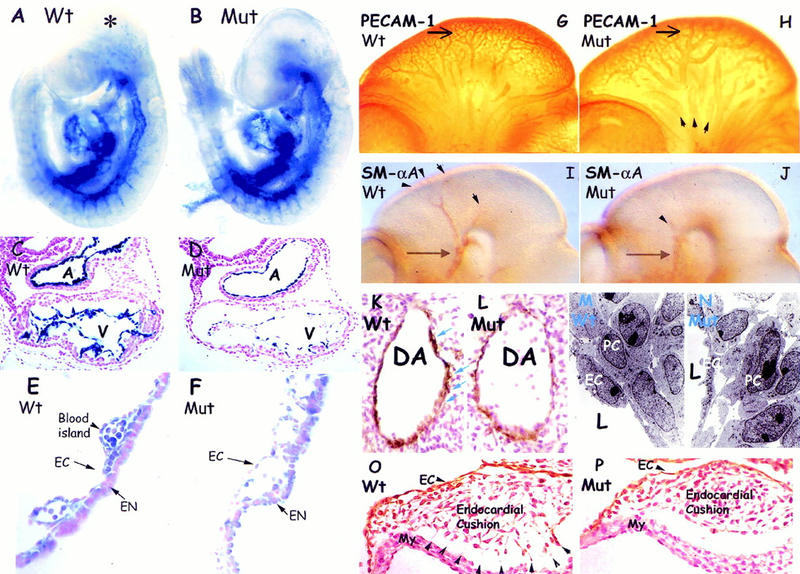
Defective angiogenic remodeling associated with abnormal cell–cell contacts in ShcA mutants. (A_–_B) Endothelial-specific Tek-lacZ stained heterozygous (A) and homozygous (B) ShcA mutant embryos at E9.5. Similar vascular networks highlighted by blue β-Gal staining were observed in mutant (Mut) and wild-type littermates, suggesting that early vasculogenic events are normal. Punctate staining (asterisk) was observed in wild-type but not in mutant embryos. (C_–_F) Hematoxylin and eosin stained transverse sections revealed an elongated endocardial lining in the mutant heart (D) compared with the more cobblestone-shaped ECs in the wild-type (C). Yolk sac cross sections demonstrated disrupted and dilated blood vessels in the mutant (F). The mesodermal angioblast-derived blood islands (E) observed in the wild-type (arrowhead) were disorganized in the mutant (F). (G_–_H) Whole-mount PECAM-1-stained wild-type (G) and ShcA mutant (H) embryos at E10.5. The anterior carotid arteries (arrows) in the wild-type (G) had matured displaying narrow vessel diameters with multiple branching vessels of various sizes. The comparable arteries in mutant embryos (H) were more simple, and dilated, with infrequent smaller branches. The primary head veins (arrowheads) were also dilated. (I,J) Anti-SMαA antibodies were utilized to mark support cells. VSMCs surrounding vessel branches (arrowheads) from anterior carotid arteries (arrows) in the head of the wild-type embryos (I) were reduced in ShcA mutant (J). (K,L) Anti-SMαA immunolabeled transverse sections. SMαA-expressing cells (brown) around the dorsal aorta in ShcA mutant (L) were discontinuous and reproducibly reduced in numbers, as compared with the wild-type (K) staining pattern showing at least two layers of tightly connected cells (arrows). (M,N) Transmission electron microscopy of comparable regions of the dorsal aorta of E10 wild-type (M) or mutant (N) embryos. There were increases in the intercellular spaces separating peri-endothelial mesenchymal cells and the overlying endothelial lining in the mutant compared with wild-type vessels. (O,P) Anti-PECAM-1-immunostained transverse sections across the heart counterstained with hematoxylin of a ShcA E10 mutant (P) demonstrated reduced cell contacts between mesenchymal cells in the endocardial cushion and the cardiac myocytes of the heart wall. Extensive cell processes (arrowheads) were obvious in wild-type (O). (A) Atrium; (DA) dorsal aorta; (EN) endoderm; (EC) endothelial cell; (L) lumen of vessel; (My) myocardium; (PC) pericytes; (V) ventricle.
We used antibodies to PECAM-1 to compare vascular development throughout the embryo. ShcAΔex2/3 mutants have a similar overall vascular patterning to age-matched wild-type embryos, but showed dilatation and decreased complexity of the primary head vessels at E10.5 (Fig. 4G,H). For example, the anterior carotid arteries in the ShcA mutant heads were expanded in size and had a more simple morphology than in wild-type embryos.
The decreased complexity of the vascular architecture and increased vessel size observed in ShcAΔex2/3 mutants indicates a perturbation in later step(s) of vascular development involving angiogenic remodeling (for review, see Risau 1997). During vessel enlargement and angiogenic sprouting, signals from ECs promote proliferation of VSMC precursors and their comigration along angiogenic sprouts; VSMCs in turn regulate EC behavior. Thus, the mature vascular network is stabilized by surrounding VSMCs, pericytes (PCs), and fibroblasts, in a fashion regulated by the extracellular matrix (ECM) and growth factors (for review, see Folkman and D'Amore 1996; Hirschi and D'Amore 1997). To test whether the defect in vessel maturation in ShcAΔex2/3 mutants was accompanied by aberrant VSMC organization, these cells were examined by immunostaining with antibodies to smooth muscle α-actin (SMαA). In mutant embryos, SMαA-positive signals were consistently reduced throughout the entire vascular system compared with wild-type. This defect was especially apparent in the head of E10.5 mutant embryos (Fig. 4I,J), which showed a relative absence of VSMC-coating branches from the anterior carotid arteries (Fig. 4J). Histological sections revealed that VSMCs were scattered and frequently reduced in the mutants (Fig. 4L; average VSMC/cross section = 37.1, n = 12) as compared with the normal staining pattern (Fig. 4K; average VSMC/cross section = 48.2, n = 12), suggesting an impaired association of mutant periendothelial VSMCs with the endothelium.
To evaluate the contacts between endothelial and surrounding cells in the dorsal aorta, ultrathin sections were prepared from comparable regions of the dorsal aorta of wild-type and mutant embryos, and were analyzed by transmission electron microscopy (TEM). These data revealed weakened cell-to-cell contacts between the abluminal EC surfaces and surrounding support cells, and also between VSMCs and their neighbors (Fig. 4M,N).
Histological sections of the heart also indicated that ShcAΔex2/3 mutants have deregulated adhesive contact of the endothelium with adjacent ECM components and support cells. Between E9 and E10, the endocardium responds to myocardial signals within the atrioventricular (AV) canal and undergoes an epithelial-mesenchymal transformation to form the endocardial cushions (for review, see Eisenberg and Markwald 1995). In wild-type embryos, the endocardium-derived mesenchymal cells invade the ECM (the cardiac jelly) of the endocardial cushions and extend processes to connect with the myocardial layer (Fig. 4O). These cell–cell contacts were substantially reduced in ShcAΔex2/3 mutant embryos (Fig. 4P). Reduced cellular adhesion was also observed in the outflow tract and yolk sac (Fig. 4F; data not shown).
Taken together, these results suggest that during angiogenic remodeling, the initial specification of VSMCs or PCs occurs independently of ShcA. However, the subsequent expansion of these progenitors and their comigration with vessel sprouts is deficient in the ShcAΔex2/3 mutants, potentially contributing to the inadequate vessel maturation observed in mutant embryos.
ShcA is phosphorylated on tyrosine and associated with signaling proteins during embryogenesis
A number of mammalian RTKs and their ligands are specifically expressed in vessel cells and are important for cardiovascular development. RTKs, including ErbB2/B3/B4 and Tie-1/Tie-2 (Tek), were expressed to normal levels in ShcAΔex2/3 mutants (data not shown). To test whether ShcA may be a direct target for activated RTKs involved in formation of the vascular system, lysates from wild-type E10 embryos were immunoprecipitated with antibodies to ShcA, a distinct adaptor protein Nck, pTyr, or control serum, and blotted with anti-pTyr antibodies (Fig. 5A). The ShcA isoforms were all strongly phosphorylated on tyrosine, and coprecipitated with a series of larger pTyr-containing proteins (∼120, 140, and 180 kD), which potentially correspond to activated RTKs. These results indicate that ShcA proteins are active in signaling at a time when they are primarily localized to cardiovascular tissue.
Figure 5.
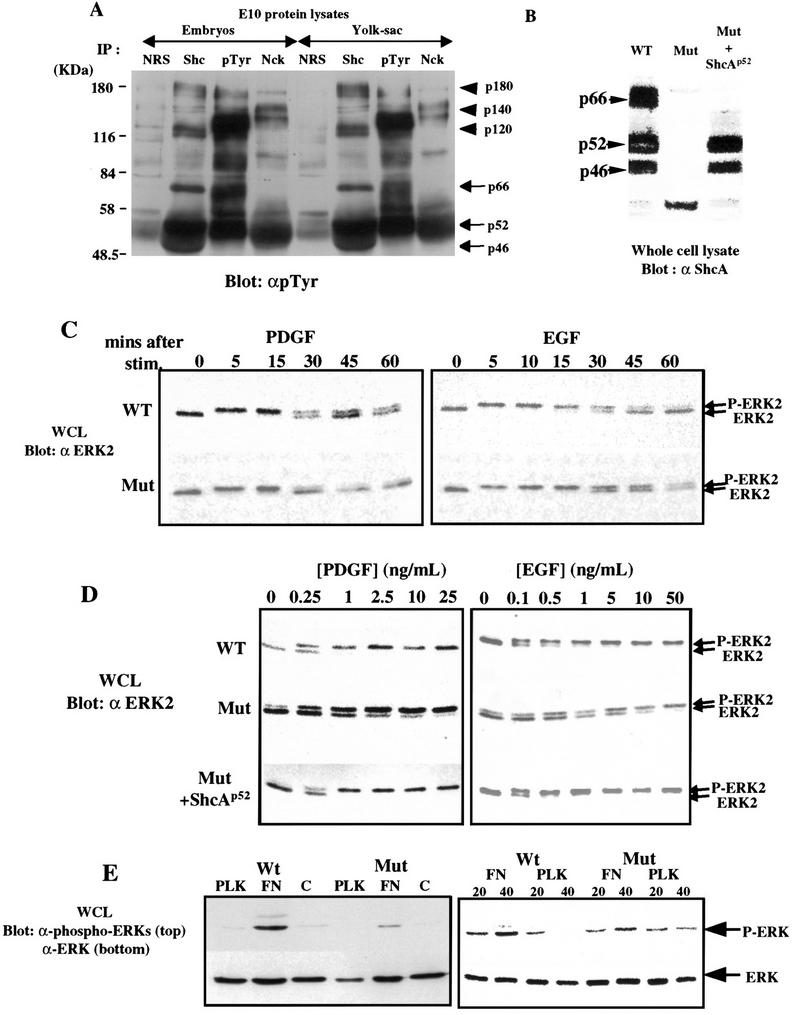
ShcA sensitizes cells for MAPK activation. (A) E10 embryos (left) or yolk sacs (right) were lysed and immunoprecipitated with normal rabbit serum (NRS), anti-ShcA antiserum (Shc), anti-pTyr antibody (pTyr), or anti-Nck antibodies. ShcA proteins of 46, 52, and 66 kD were highly phosphorylated on tyrosine (arrows). In addition, a number of higher molecular mass pTyr-containing proteins (arrowheads) were also specifically precipitated with anti-ShcA antibodies. (B) Whole cell lysates (WCL) were prepared from wild-type or ShcA (Mut) MEFs, or from ShcA cells stably transfected with a p52 ShcA cDNA (Mut + ShcAp52) and immunobloted with anti-ShcA antiserum. (C) Equal amounts of extracts from wild-type and ShcA (Mut) MEFs were prepared after stimulation of serum-deprived cells with PDGF-BB (25 ng/ml) or EGF (50 ng/ml) for the indicated time (min), electrophoresed, and subjected to Western blot analysis with anti-Erk2 antibody. The upper band is the fully phosphorylated, activated form of Erk2. (D) Lysates were obtained from wild-type, ShcA (Mut) and ShcA transfected mutant (Mut + ShcAp52) cells, which had been serum deprived overnight and then treated with the indicated concentrations of PDGF-BB or EGF for 5 min. Erk2 phosphorylation was detected using anti-Erk2 antibody as described in C. (E) Serum-starved quiescent cells were plated on fibronectin (FN), poly-l-lysine (PLK), or left in suspension for 30 min and the phosphorylation of MAPK was visualized using anti-phosphospecific Erk antibody (top,left). The same blot was probed with pan anti-Erk antibodies to show the total amount of Erk in each lane (bottom,left).The same experiment was repeated for the time (min) indicated (right). The results shown in C, D, and E are typical of three independent experiments.
ShcA mutant cells and embryos show impaired sensitivity to growth factors
Phosphorylated ShcA proteins from E10 lysates coprecipitated with Grb2 (data not shown), raising the possibility that ShcA might influence signaling to the Ras/MAPK pathway in vivo. We therefore investigated the ability of PDGF-BB and EGF to activate MAPK in mouse embryo fibroblasts (MEFs) derived from ShcA-deficient embryos. This assay may be physiologically relevant because PDGF signaling, for example, is important for the formation of vessel walls (Hellstrom et al. 1999). MEFs were serum deprived, and stimulated with PDGF-BB (25 ng/ml) or EGF (50 ng/ml). Erk2 activation was monitored by a gel-shift assay, which identifies the active phosphorylated species of this MAPK isoform. In both wild-type and ShcA mutant cells, Erk2 was activated to a similar extent and with similar kinetics. Maximal Erk2 activation was apparent within 5 min in both cell types, and in response to both growth factors (Fig. 5C). Anti-pTyr blotting of whole cell lysates revealed a similar overall response to PDGF and EGF in wild-type and mutant MEFs (data not shown).
However, mutant and wild-type cells had very different dose-response curves with respect to Erk2 activation (Fig. 5D). Addition of EGF at 0.5 ng/ml fully activated MAPK in wild-type cells, but only induced 20%–30% increase in Erk2 activation in mutant cells. Full Erk2 activation in mutant cells required at least 50-fold higher concentrations of EGF. This impaired sensitivity to EGF was observed in five different cultures of ShcA mutant fibroblasts derived from independent litters (data not shown). Similarly, MAPK was optimally activated by 1 ng/ml of PDGF-BB in wild-type cells, but required at least 25 ng/ml for strong activation in mutant cells. To test whether this decreased sensitivity to growth factor stimulation resulted solely from the loss of wild-type ShcA proteins, mutant cells were transfected with a mouse cDNA encoding the p52 and p46 ShcA isoforms. Mutant cell lines stably expressing the p52 and p46 proteins to ∼70% the level seen in wild-type cells (Fig. 5B) recovered full Erk2 activation at low growth factor concentrations. The extent of MAPK activation was confirmed in each case by measuring in vitro kinase activity (data not shown). These data indicate that wild-type p52/46-kD ShcA proteins are required for cultured cells to respond to low concentrations of growth factors.
Whole-mount immunostaining with phosphospecific Erk MAPK antibodies demonstrates selective loss of MAPK activation in the cardiovascular system of ShcA mutant embryos
The preceeding data obtained from MEFs suggest that ShcA may sensitize cells to growth factor signaling in vivo. To compare MAPK activation in wild-type and mutant embryos, we used an antibody to the doubly phosphorylated form of MAPK (dp-MAPK) in a whole-mount analysis. A similar approach has been used previously in Drosophila to correlate the dynamic pattern of MAPK activation with RTK expression (Gabay et al. 1997). E10.5 wild-type embryos showed intense dp-MAPK staining associated with head vessels and the tips of vessel sprouts that was markedly reduced in ShcA mutant embryos (Fig. 6A–D). This reduction appeared selective, as both wild-type and mutant embryos had comparable dp-MAPK staining in other regions of the head such as the mid/hindbrain junction (Fig. 6A,B). To examine whether MAPK activation might be a cause or a consequence of the cardiovascular defects in ShcA mutants, we analyzed embryos for MAPK activation at E9.5, prior to significant manifestation of phenotypic abnormalities. Compared with wild-type embryos, ShcA mutants exhibited a severe and specific reduction of dp-MAPK staining in regions that correlate with cardiovascular development and the normal pattern of ShcA expression (Fig. 6E). Reduced MAPK activation was evident in the heart ventricle (Fig. 6F,G) and in the dorsal side of the embryos around the termini of intersomitic vessels (Fig. 6H,I), although the ventral regions of the trunk and intersomitic vessels were less affected. Double labeling with dp-MAPK and PECAM-1 confirmed that vessel development at this stage was grossly normal, despite the regional reduction of MAPK activation in mutant embryos (Fig. 6J,K). The restricted expression of ShcA in cells of the vascular system is therefore required for their ability to appropriately activate the MAPK pathway. ShcA deficiency may therefore desensitize the vascular precursor cells to external stimulation required for angiogenic remodeling. Taken with the specific expression of ShcA in the cardiovascular system, these results argue that the phenotype of ShcA mutant embryos reflects a direct role for ShcA in cardiovascular development.
Figure 6.
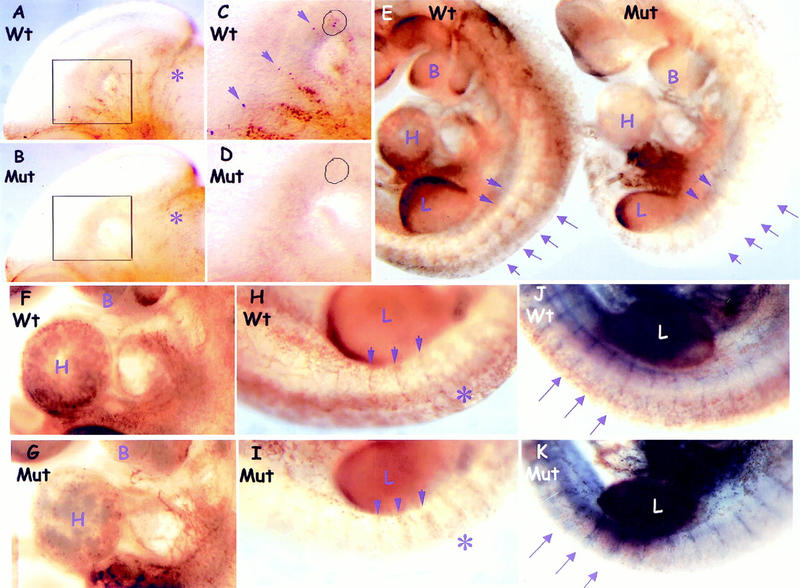
MAPK activation is selectively reduced in the developing cardiovascular system of ShcA mutant embryos. (A_–_D) Pattern of Erk activation in E10.5 heads. (C,D) Higher magnification views of the boxed areas of A and B, respectively. Intense dp-Erk staining was specifically detected along wild-type head vessels (A, arrows in C), which was much reduced in the mutant (B,D). Sites of activated MAPK (circles) that were prominent in the tips of wild-type vessel sprouts (C) were extremely weak in ShcA mutant (D). Comparable staining was observed around the mid-hindbrain region (asterisks) in both wild-type and mutant embryos. (E_–_K) Comparison of Erk activation in whole E9.5 embryos labeled with anti-dp-Erk (brown) and anti-PECAM-1 (blue, J and K only) antibodies. In mutants, dp-Erk staining was reduced in the cardiovascular system but unaffected in other regions, including the developing limbs and ventral side of the trunks (E). In mutants, dp-Erk expression was reduced in the left ventricle of the heart (wild-type, F, Mut, G) and around the dorsal side of the trunk (asterisks and arrows), although staining toward the proximal ISV was less affected (arrows) (Wt, H and J, Mut, I and K). Double labeling for dp-Erk and PECAM revealed the normal appearance of ISV at E 9.5 in both wild-type (J) and mutant (K), but with reduced MAPK activity around distal ISVs in the mutant (arrows). (B) Branchial arches; (H) heart; (L) limb.
Loss of ShcA affects fibronectin-induced MAPK activity, focal complex distribution and the actin cytoskeleton
ShcA mutant embryos exhibited adhesion defects in yolk sac (Fig. 4E,F), in the heart (Fig. 4O,P) and around dorsal aorta (Fig. 4K–N). Because activation of some integrins triggers ShcA phosphorylation and activation of the MAPK cascade (Wary et al. 1996), we compared the behavior of wild-type or mutant cells plated on fibronectin (FN). Both cell types adhered to FN-coated plates within 10 min. MAPK activation was observed in wild-type serum-starved cells plated on FN, but not on poly-l-lysine (Fig. 5E); ShcA mutant MEFs plated on FN showed a reduced level of Erk activation as measured by blotting with phosphospecific Erk antibodies (Fig. 5E), and is partially rescued by the expression of wild-type p52 ShcA (data not shown).
Compared with wild-type cells, ShcA-deficient MEFs also showed significant alterations in the organization of their focal contacts and actin stress fibres, as revealed by immunostaining with antibodies to paxillin (Fig. 7A,B) or vinculin (data not shown) and with rhodamine–phalloidin to identify F-actin (Fig. 7B). Double labeling with phalloidin (red) and anti-paxillin antibody (green) showed that wild-type cells plated on FN contained lamellipodia, with sparsely arranged, polarized actin stress fibres terminating in coarse paxillin-positive structures (Fig. 7B). In contrast, ShcA mutant MEFs had intensely staining actin bundles around the entire cell periphery, with fine paxillin-positive focal complexes that showed an atypical radial distribution. Polarized flat protrusions resembling lamellipodia were only detected in 15%–20% of the mutant cells. ShcA mutant cells were rounded, with extensive membrane blebbing. These morphological and cytoskeletal defects were suppressed by expression of the p52/46 ShcA isoforms in the mutant cells (Fig. 7A,B). Thus, ShcA potentially plays a significant role in fibroblasts in organizing cytoskeletal responses to ECM components such as FN.
Figure 7.
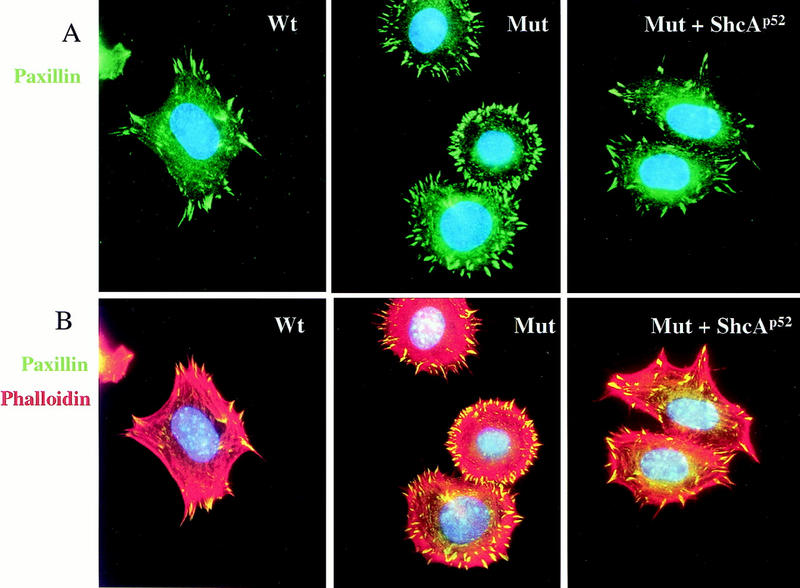
ShcA regulates cytoskeletal organization in MEFs. (A) Quiescent cells were plated on FN-coated coverslips for 40 min and stained with anti-paxillin antibody (green) to examine focal contact formation. ShcA mutant cells showed an abnormal radial distribution of focal complexes. (B) The same cells were double labeled with phalloidin (red) to identify F actin and for paxillin (green) as in A. ShcA mutant MEFs contained thick arrays of actin bundles all around the cell periphery, in contrast to the organized actin stress fibres terminating in paxillin-positive focal contacts in wild-type cells. Expression of the p52/46 ShcA isoforms in the mutant cells converted the morphology back to wild-type.
Discussion
ShcA activity is required for maturation of blood vessels and heart development
Our results indicate that during early embryogenesis ShcA is specifically expressed in cells of the developing cardiovascular system, which consequently become sensitized to signals that direct heart development and angiogenesis. Although the principal vascular progenitors are properly induced and assembled in ShcA mutants, the vessels show selective defects in their subsequent organization into a mature vascular network. This is most evident in the yolk sac and head vasculature, in which vessels are dilated and remain relatively uniform in size. ShcA mutant embryos exhibited adhesion defects in yolk sacs, in the heart, and in major blood vessels. Ultrastructural analysis of the dorsal aorta reveals a significant reduction in the contacts of ECs with periendothelial cells and the surrounding mesenchyme. Similarly, support cells around endothelial tubes are poorly organized. Thus, ShcA appears important in the reciprocal signaling between ECs and support cells required for angiogenic remodeling and heart development. Consistent with this view, ShcA proteins are highly phosphorylated on tyrosine at E10, a critical time for both angiogenesis and vessel stabilization, and are complexed with other phosphoproteins and with Grb2.
Interesting, despite early cardiovascular expression and tyrosine phosphorylation (Fig. 5a; data not shown) of the 66-kD ShcA isoform in the developing embryos, mice lacking only p66 are viable (Migliaccio et al. 1999). It is therefore probable that the 46/52-kD ShcA proteins are the isoforms essential for embryonic development. The ShcAΔex2/3 mutant allele encodes a very low level of 40-kD protein that lacks a function PTB domain. Although we cannot exclude the possibility that the 40-kD mutant protein retains some biological activity, several observations argue that this truncated protein is unlikely to cause a dominant-negative effect or to have significant signaling functions. First, heterozygotes have no apparent phenotype. In addition, quantitative analysis showed that the truncated protein is only expressed at 2% of the wild-type ShcA level. Furthermore, the 40-kD polypeptide lacks a functional PTB domain, which has been shown biochemically to be important for ShcA-signaling function. Recently, a genetic screen in Drosophila identified mutations in the dshc locus and showed that a point mutation in the PTB domain completely abolished dshc function, resulting in the same phenotype as the null mutation. This indicates that an intact PTB domain is essential for dshc activity (Luschnig et al. 2000). Most significantly, the defects in growth factor signaling and cytoskeletal organization in ShcA mutant cells were rescued by re-expression of the wild-type 46/52-kD proteins, indicating that at the cellular level the mutation causes a strong loss of function.
Mutations in ShcA, specific RTKs or their ligands cause similar cardiovascular defects
The phenotypes of embryos lacking endothelial RTKs or their ligands highlight the importance of reciprocal signaling between EC and VSMC compartments. Embryos deficient for the Tie2/Tek receptor (Dumont et al. 1994; Sato et al. 1995), its ligand Ang-1 (Suri et al. 1996) and the Flt4/VEGFR3 RTK (Dumont et al. 1998) have defects in the later stages of vascular patterning that are very similar to those observed in ShcA mutants, and are linked to a failure to recruit VSMC and PC precursors to developing vessels. The PDGF family is also important for vascular wall assembly (Soriano 1994; Schatteman et al. 1995; Lindahl et al. 1997). Analysis of PDGF-B and βPDGFR null embryos suggests that PDGF-B released by ECs drives the proliferation and migration of βPDGFR-positive VSMCs along angiogenic sprouts (Hellstrom et al. 1999). In _ShcA_-deficient embryos, VSMCs fail to expand along endothelial tubes, indicating that ShcA is required for vessel wall development.
In addition to its role in angiogenesis, ShcA is highly expressed in the embryonic heart, and ShcA mutants have reduced cardiac trabeculation and a hypoplastic endocardial cushion. Development of the myocardium and endocardium requires signaling by soluble factors such as neuregulin and Ang-1, and the cell surface protein ephrin-B2 (for review, see Yancopoulos et al. 1998). Targeted mutations that affect neuregulin (Meyer and Birchmeier 1995) or the myocardial RTKs ErbB2 (Lee et al. 1995) and ErbB4 (Gassmann et al. 1995) cause very similar phenotypes in the heart as the ShcA mutation. Mutations in EphB2/B3 (Adams et al. 1999), Ang-1 (Suri et al. 1996), and Tie-2/Tek (Dumont et al. 1994; Sato et al. 1995) also cause trabeculation defects resembling those in ShcA mutant embryos.
Taken together, these data suggest that ShcA is a target for one or more of the RTKs involved in cardiovascular development. Several of these receptors, including Flt4/VEGFR3 and ErbB2/4 have binding sites for the ShcA PTB domain (Fournier et al. 1995; Laminet et al. 1996), whereas others such as the βPDGFR associate with the ShcA SH2 domain (Yokote et al. 1994). Multiple ligand-receptor systems may therefore signal through ShcA to establish a mature vascular system.
ShcA sensitizes cells to growth-factor signaling
These observations raise the question of the biochemical functions of ShcA in the developing vasculature. ShcA has been implicated in activation of the Ras/MAPK pathway through its ability to bind the Grb2–Sos1 complex. By using mutant MEFs, we found that ShcA was dispensable for MAPK activation in response to moderate concentrations of EGF or PDGF, but was necessary for efficient MAPK activation in cells exposed to low amounts of growth factors. These results suggest that when the supply of growth factor is abundant, the ability of autophosphorylated EGF or PDGF receptors to bind Grb2 directly is sufficient to fully stimulate the MAPK pathway. However, at low concentrations of growth factors, the ShcA adaptor is required to sensitize the cellular machinery that activates the MAPK pathway. This interpretation is supported by the finding that low concentrations of EGF that fail to elicit binding of Grb2 to the receptor, nonetheless induce ShcA phosphorylation and formation of a ShcA–Grb2–Sos1 complex (Wennstrom and Downward 1999). ShcA is generally considered to be broadly expressed. However, in situ hybridization analysis indicates that ShcA expression is, in fact, highly dynamic in vivo. Thus, the ability of cells to respond to external cues during embryogenesis may in part be determined by their level of ShcA expression. A signal that increased ShcA expression could then render cells permissive to respond to small increases in growth-factor stimulation that would otherwise fail to activate the MAPK pathway.
To test whether the ability of ShcA to potentiate signaling may underlie the defects in mutant embryos, we probed the state of MAPK activation in vivo using phosphospecific MAPK antibodies. Mutant embryos show a striking and specific loss of phosphorylated MAPK in the cardiovascular system that precede the onset of phenotypic abnormalities and coincide with the pattern of ShcA expression. Because other sites of MAPK activation, such as the limb bud, were not significantly affected in mutant embryos, these data argue that ShcA is not an invariant component of pathways leading to MAPK activation, but rather plays a specific role in coupling RTKs to such pathways in specific embryonic tissues such as the heart and blood vessels. The notion that tight control of the Ras/MAPK pathway is important in cardiovascular development receives support from the phenotype of mice lacking the p120-Ras GTPase activating protein (Henkemeyer et al. 1995). Such embryos die by E10.5 with severe defects in angiogenesis, including a failure to remodel the yolk sac and thinning of the dorsal aorta.
These data suggest a functional distinction between the adaptor proteins that mediate pTyr signaling. A protein such as Grb2 is highly conserved in evolution, and is a component of a core pathway with an essentially invariant function in metazoan animals. In contrast, Shc proteins have apparently evolved as multicellular organisms became more complex, and may serve to amplify and coordinate the activities of core factors such as Grb2, in response to an increasing array of extracellular signals. Consistent with this view, genetic analysis in Drosophila has demonstrated that dshc appears to mediate a specific aspect of signaling by a subset of RTKs, as dshc mutations cause a partial reduction in the functions of the Torso receptor and DER (Luschnig et al. 2000).
ShcA regulates cellular morphology
The proliferation and migration of vascular cells require integrins (Stromblad and Cheresh 1996). Previous work has suggested that ShcA is a binding partner and substrate of integrin-activated tyrosine kinases such as FAK and Src (Schlaepfer et al. 1999), and appears to contribute to integrin-mediated MAPK activation (Wary et al. 1996; Schlaepfer and Hunter 1997). We found that Erk activation was reduced in mutant cells plated on FN, indicating that ShcA provides a significant, but not a unique route for integrin-activated kinases to recruit Grb2. Surprisingly, ShcA mutant cells plated on FN have an aberrant rounded morphology, characterized by an increased number of small focal complexes, and a disorganized, depolarized actin cytoskeleton. This function of ShcA may relate to some of the vascular defects in mutant embryos, such as the failure of mesenchymal cells in the endocardial cushion to project processes that invade the surrounding FN-enriched ECM.
Conclusion
ShcA has been viewed as a generic adaptor. In contrast, the present data suggest that ShcA has highly specific functions in mediating signals generated by complex cell–cell interactions. ShcA sensitizes cells in culture and in vivo for MAPK activation and controls cytoskeletal architecture. These combined activities suggest how ShcA may function during cardiovascular development. Consistent with a more general role for ShcA in the morphogenesis of complex structures, E10 ShcA mutant embryos show reduced branching of specific cranial ganglia (unpubl.), and transgenic expression of ShcA in mouse mammary epithelial cells induces increased tertiary branching of the virgin mammary gland (Rauh et al. 1999).
Materials and methods
Construction of the ShcA targeting vector and generation of chimeric mice
The ShcA targeting vector was constructed by inserting a 5.4-kb ShcA genomic fragment from 129Sv strain DNA into the vector pPNT. The 3′ arm, consisting of a 2.5-kb fragment, was inserted between the _neo_R and tk selection cassettes. _Not_I-linearized targeting vector (30 μg) was electroporated into R1 ES cells as described (Nagy et al. 1993; Wood et al. 1993). The frequency of homologous recombination was 1 in 33 (n = 200). Aggregation chimeras were generated with targeted ES cells and male chimeric mice were then bred with 129Sv or CD1 females to obtain germ-line transmission. Timed matings were carried out by assigning the morning of identification of vaginal plugs as day 0.5 (E0.5).
Southern blotting and PCR
Genomic DNA prepared from tail biopsies or embryonic yolk sac was subjected to PCR and/or Southern blot analysis. For Southern blot analysis, 5 μg of genomic DNA was digested with _Eco_RV, separated on 0.65% agarose gels, and then blotted onto nylon membrane filters (Genescreen, NEN Life Science). For PCR reactions, tissues were digested with proteinase K overnight at 55°C. The reaction was cycled 30 times (1 min at 94°C, 1 min at 56°C, 2 min at 72°C), which amplified a 410-bp fragment of the wild-type ShcA gene and a 347-bp fragment of the mutant allele. Oligos used were PCR-5′ (5′-TGCTCTAGCTCTTGTCTGGTAG-3′) corresponding to the 5′ flanking region, PCR-3′ (5′-GTTAAGAGAAACTCTGGACCAATC-3′) matching the deleted part of the DNA, and neo (5′-ATGCCTGCTCTTTACTGAAGGC-3′) corresponding to the _neo_R gene (Fig. 1A).
RNA preparation and Northern blot analysis
E9.5 embryos were collected in DEPC-treated PBS and stored at −70°C until they were genotyped. Total RNA from individually genotyped embryos was isolated by RNeasy mini kit (Qiagen) and 15 μg was used per embryo. Hybridization was performed as described for Southern blotting except that washing was carried out with 0.1× SSC—4% SDS solution at 75°C. A [32P]UTP-labeled antisense RNA probe was synthesized with T3 RNA polymerase by using pBSSK-p66mShcA as a template (linearized with _Stu_I).
Whole-mount RNA in situ hybridization
RNA in situ hybridization analysis was performed as described (Conlon and Rossant 1992). To make the RNA probe, a fragment (corresponding nucleotides 1–1505 of p66 cDNA) from the mouse ShcA cDNA in pBSSK vector was linearized with _Stu_I and transcribed with T3 RNA polymerase (anti-sense probe), or linearized with _Bam_HI and transcribed with T7 RNA polymerase (sense probe).
Tek–lacZ β-Gal staining
For whole-mount staining, embryos were collected and incubated for 15–30 min in fresh fixative solution (0.2% gluteraldehyde, 5 mm EGTA, 2 mm MgCl2, 4% formaldehyde). X-Gal staining of genotyped embryos was performed as described (Henkemeyer et al. 1995). Embryos were then sectioned and counterstained with hematoxylin.
Immunohistochemistry
Whole-mount embryo immunostaining with anti-CD-31 (PECAM-1, Pharmingen, 1:1000), anti-SMαA (clone 1A4, Sigma, 1:500) was performed as described (Henkemeyer et al. 1994). DAB-stained embryos were postfixed, photographed, sectioned, and counterstained with hematoxylin. For whole-mount immunostaining with anti-dpERK (Sigma, 1:250), embryos were freshly dissected, fixed in 8% paraformaldehyde for 4 hr to overnight at 4°C, washed in PBS containing 0.5% NP-40, and immunostained as described (Henkemeyer et al. 1994). For double labeling, secondary antibodies of peroxidase-conjugated AffiniPure goat anti-mouse IgG (Jackson Labs, 1:250) and alkaline phosphatase-conjugated goat anti-rat IgG (H + L, mouse serum pre-adsorbed, Bio-Rad, 1:400) were used for anti-dp-ERK and anti-CD-31 staining, and were detected with DAB and BCIP, respectively.
For immunohistochemical staining of sections with anti-SMαA (Sigma, 1 μg/ml), 7–10 μm paraffin sections were blocked with 10% normal goat serum and 0.15% Triton X-100 and incubated with primary antibody overnight at 4°C in a humidified chamber. The slides were washed at least three times in PBS and then further processed using Vectastain ABC elite detection kit (Vector laboratory).
Lysate preparation and immunoblotting
E9.5 or E10.5 embryos were homogenized in PLC-lysis buffer using a Teflon homogenizer essentially as described (Lai et al. 1995). Supernatants were collected after repeated centrifugation. For each immunoprecipitation, the equivalent of one E10.5 embryo (∼2 mg protein) was used, followed by SDS-PAGE, and filters were probed with appropriate antibodies and developed by using either an ECL kit, ECL + Plus kit or [125I]-protein A (Amersham).
Transmission electron micrographs
E10 embryos were fixed in 2% gluteraldehyde in 0.1 m sodium cacodylate buffer. Ultrathin (50 nm) transverse plastic sections of genotyped embryos were stained with uranyl acetate and lead citrate and viewed at 60 kV in a Philips CM100 Biotwin TEM.
Generation of MEFs cell lines
Dissected E10 embryos were incubated with trypsin for 15 min at 37°C, then seeded on 6-cm2 tissue culture plates in DMEM containing 15 % FBS. Yolk sacs from each embryo were genotyped by PCR. Experiments described in this study were repeated with cell lines derived from separate embryos collected from different parents.
Expression of p52/46 ShcA in ShcA mutant MEFs
A 1.6-kb _Bsg_I–_Eco_RI mouse ShcA cDNA fragment (Migliaccio et al. 1997) encoding the p52/46 ShcA polypeptides was engineered into MSCVpac vector (Markowitz et al. 1988) at _Xho_I–_Eco_RI site. Subconfluent ShcA mutant MEFs were transfected overnight with Lipofectamine (GIBCO BRL) containing 15 μg of DNA in a 150-mm cell culture dish. Cells were first cultured in DMEM with 10% FBS for 24 hr and then selected with puromycin (Sigma, 1 μg/ml). Puromycin-resistant clones were isolated and screened for expression of ShcA proteins by immunoblotting using anti-ShcA antibody (Pelicci et al. 1992). Expression level was quantified using ECL + Plus (Amersham).
Characterization of MAPK activation
Subconfluent cells were serum starved for 2 days in DMEM containing 20 mm HEPES (pH 7.2), then stimulated with EGF or PDGF-BB at 37°C. For studies with FN (Sigma, 10 μg/ml) or PLK (Sigma, 0.5 mg/ml), cells were treated essentially as described (Schlaepfer and Hunter 1997). Stimulated cells were then lysed in 2× SDS-sample buffer and boiled. Equal amounts of lysates were resolved by SDS-PAGE and analyzed by immunoblotting with the anti-Erk2 monoclonal antibody (UBI, 0.5 μg/ml), anti-phospho-MAPK antibody (NEB, 1 μg/ml), or with anti-Erk1 monoclonal antibody (Santa Cruz, 0.5 μg/ml) as indicated.
Detection of focal contacts formation and actin organization
Quiescent cells were trypsinized and plated on FN-coated glass coverslips for 40 min at 37°C, washed, fixed for 15 min in 4% paraformaldehyde with 5% sucrose, then permeabilized with 0.5% Triton X-100 for 5 min, and finally blocked in PBS containing 5% BSA and 2% normal goat serum. Following incubation with anti-paxillin mAb (transduction, 1 μg/ml) or anti-vinculin mAb (Sigma, 1 μg/ml) overnight at 4°C, cells were washed and then treated with fluorescein goat anti-mouse IgG (Molecular Probes, 1:100) and Texas Red-X phalloidin (Molecular Probes, 1:100). Cells were subsequently washed, labeled with Hoechst no. 33258 (Sigma, 1 μg/ml), mounted, and analyzed using a Leica DMRXE microscope equipped with appropriate filters and using 100× oil-immersion objectives.
Acknowledgments
We thank B.-Z. Shilo for advice, M. Puri for Tek-lacZ transgenic mice, S. Kulkarni for morula aggregation, K. Harpal for histology, D. Holmyard for electron microscopy, and T. Saxton and M. Puri for encouragement and comments on the manuscript. K.-M.V.L. held a fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC). This work was supported by a grant from the Medical Research Council of Canada (MRC), a Terry Fox Program Project grant from the National Cancer Institute of Canada, and a Howard Hughes International Research Scholar Award and an MRC Distinguished Scientist Award to T.P.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Pawson@mshri.on.ca; FAX (416) 586-8869.
References
- Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes & Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfini L, Migliaccio E, Pelicci G, Lanfrancone L, Pelicci PG. Not all Shc's roads lead to Ras. Trends Biochem Sci. 1996;21:257–261. [PubMed] [Google Scholar]
- Conlon RA, Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
- Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5- triphosphate 5-phosphatase. Proc Natl Acad Sci. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes & Dev. 1994;8:1897–1909. doi: 10.1101/gad.8.16.1897. [DOI] [PubMed] [Google Scholar]
- Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282:946–949. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Folkman J, D'Amore PA. Blood vessel formation: What is its molecular basis? Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- Fournier E, Dubreuil P, Birnbaum D, Borg JP. Mutation at tyrosine residue 1337 abrogates ligand-dependent transforming capacity of the FLT4 receptor. Oncogene. 1995;11:921–931. [PubMed] [Google Scholar]
- Gabay L, Seger R, Shilo BZ. In situ activation pattern of Drosophila EGF receptor pathway during development. Science. 1997;277:1103–1106. doi: 10.1126/science.277.5329.1103. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Tojo A, Shibuya M. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO J. 1996;15:6197–6204. [PMC free article] [PubMed] [Google Scholar]
- Hashimoto A, Kurosaki M, Gotoh N, Shibuya M, Kurosaki T. Shc regulates epidermal growth factor-induced activation of the JNK signaling pathway. J Biol Chem. 1999;274:20139–20143. doi: 10.1074/jbc.274.29.20139. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Lindahl Kal, P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Marengere LE, McGlade J, Olivier JP, Conlon RA, Letwin DP, Holmyard K, Pawson T. Immunolocalization of the Nuk receptor tyrosine kinase suggests roles in segmental patterning of the brain and axonogenesis. Oncogene. 1994;9:1001–1014. [PubMed] [Google Scholar]
- Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, D'Amore PA. Control of angiogenesis by the pericyte: Molecular mechanisms and significance. EXS. 1997;79:419–428. doi: 10.1007/978-3-0348-9006-9_18. [DOI] [PubMed] [Google Scholar]
- Ingham RJ, Holgado-Madruga M, Siu C, Wong AJ, Gold MR. The Gab1 protein is a docking site for multiple proteins involved in signaling by the B cell antigen receptor. J Biol Chem. 1998;273:30630–30637. doi: 10.1074/jbc.273.46.30630. [DOI] [PubMed] [Google Scholar]
- Lai KM, Olivier JP, Gish GD, Henkemeyer M, McGlade J, Pawson T. A Drosophila shc gene product is implicated in signaling by the DER receptor tyrosine kinase. Mol Cell Biol. 1995;15:4810–4818. doi: 10.1128/mcb.15.9.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laminet AA, Apell G, Conroy L, Kavanaugh WM. Affinity, specificity, and kinetics of the interaction of the SHC phosphotyrosine binding domain with asparagine-X-X-phosphotyrosine motifs of growth factor receptors. J Biol Chem. 1996;271:264–269. doi: 10.1074/jbc.271.1.264. [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Luschnig S, Krauss J, Bohmann K, Desjeux I, Nüsslein-Volhard C. The Drosophila SHC adaptor protein is required for signaling by a subset of receptor tyrosine kinases. Mol Cell. 2000;5:231–241. doi: 10.1016/s1097-2765(00)80419-0. [DOI] [PubMed] [Google Scholar]
- Markowitz D, Goff S, Bank A. A safe packaging line for gene transfer: separating viral genes on two different plasmids. J Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoskova B, Wong WT, Salcini AE, Pelicci PG, Di Fiore P. Constitutive phosphorylation of eps8 in tumor cell lines: Relevance to malignant transformation. Mol Cell Biol. 1995;15:3805–3812. doi: 10.1128/mcb.15.7.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore P, Lanfrancone L, Pelicci PG. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Pelicci L, Lanfrancone PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Muraoka S, Sanokawa R, Mori N. N-Shc and Sck, two neuronally expressed Shc adapter homologs. Their differential regional expression in the brain and roles in neurotrophin and Src signaling. J Biol Chem. 1998;273:6960–6967. doi: 10.1074/jbc.273.12.6960. [DOI] [PubMed] [Google Scholar]
- O'Bryan JP, Songyang Z, Cantley L, Der CJ, Pawson T. A mammalian adaptor protein with conserved Src homology 2 and phosphotyrosine-binding domains is related to Shc and is specifically expressed in the brain. Proc Natl Acad Sci. 1996;93:2729–2734. doi: 10.1073/pnas.93.7.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabayashi Y, Sugimoto Y, Totty NF, Hsuan J, Kido Y, Sakaguchi K, Gout I, Waterfield MD, Kasuga M. Interaction of Shc with adaptor protein adaptins. J Biol Chem. 1996;271:5265–5269. doi: 10.1074/jbc.271.9.5265. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Dente L, De GA, Verducci-Galletti B, Giuli S, Mele S, Vetriani C, Giorgio M, Pandolfi PP, Cesareni G, Pelicci PG. A family of Shc related proteins with conserved PTB, CH1 and SH2 regions. Oncogene. 1996;13:633–641. [PubMed] [Google Scholar]
- Rauh MJ, Blackmore V, Andrechek ER, Tortorice CG, Daly R, Lai VK, Pawson T, Cardiff RD, Siegel PM, Muller WJ. Accelerated mammary tumor development in mutant polyomavirus middle T transgenic mice expressing elevated levels of either the Shc or Grb2 adapter protein. Mol Cell Biol. 1999;19:8169–8179. doi: 10.1128/mcb.19.12.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS, Zhou MM, Pratt JC, Harlan JE, Walk SF, Fesik SW, Burakoff SJ. Evidence for a requirement for both phospholipid and phosphotyrosine binding via the Shc phosphotyrosine-binding domain in vivo. Mol Cell Biol. 1997;17:5540–5549. doi: 10.1128/mcb.17.9.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG, et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- Salcini AE, McGlade J, Pelicci G, Nicoletti I, Pawson T, Pelicci PG. Formation of Shc-Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene. 1994;9:2827–2836. [PubMed] [Google Scholar]
- Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Motley ST, Effmann EL, Bowen-Pope DF. Platelet-derived growth factor receptor alpha subunit deleted Patch mouse exhibits severe cardiovascular dysmorphogenesis. Teratology. 1995;51:351–366. doi: 10.1002/tera.1420510602. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Abnormal kidney development and hematological disorders in PDGF β receptor mutant mice. Genes & Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- Stromblad S, Cheresh DA. Cell adhesion and angiogenesis. Trends Cell Biol. 1996;6:462–468. doi: 10.1016/0962-8924(96)84942-7. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Lai VK, Olivier JP, Gish GD, Stephens R, Kaplan D, Shoelson S, Pawson T. A conserved amino-terminal Shc domain binds to phosphotyrosine motifs in activated receptors and phosphopeptides. Curr Biol. 1995;5:404–412. doi: 10.1016/s0960-9822(95)00081-9. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Gish GD, Pawson T. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol. 1996;6:1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Weng Z, Thomas SM, Rickles RJ, Taylor JA, Brauer AW, Seidel-Dugan C, Michael WM, Dreyfuss G, Brugge JS. Identification of Src, Fyn, and Lyn SH3-binding proteins: Implications for a function of SH3 domains. Mol Cell Biol. 1994;14:4509–4521. doi: 10.1128/mcb.14.7.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennstrom S, Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol. 1999;19:4279–4288. doi: 10.1128/mcb.19.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SA, Allen ND, Rossant J, Auerbach A, Nagy A. Non-injection methods for the production of embryonic stem cell-embryo chimaeras. Nature. 1993;365:87–89. doi: 10.1038/365087a0. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Klagsbrun M, Folkman J. Vasculogenesis, angiogenesis, and growth factors: Ephrins enter the fray at the border. Cell. 1998;93:661–664. doi: 10.1016/s0092-8674(00)81426-9. [DOI] [PubMed] [Google Scholar]
- Yokote K, Mori S, Hansen K, McGlade J, Pawson T, Heldin CH, Claesson-Welsh L. Direct interaction between Shc and the platelet-derived growth factor beta-receptor. J Biol Chem. 1994;269:15337–15343. [PubMed] [Google Scholar]
- Zhou MM, Ravichandran KS, Olejniczak EF, Petros AM, Meadows RP, Sattler M, Harlan JE, Wade WS, Burakoff SJ, Fesik SW. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature. 1995;378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]