sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins (original) (raw)
Abstract
Mutations that influence lin-12 activity in Caenorhabditis elegans may identify conserved factors that regulate the activity of lin-12/Notch proteins. We describe genetic evidence indicating that sel-10 is a negative regulator of _lin-12/Notch_-mediated signaling in C. elegans. Sequence analysis shows that SEL-10 is a member of the CDC4 family of proteins and has a potential human ortholog. Coimmunoprecipitation data indicate that C. elegans SEL-10 complexes with LIN-12 and with murine Notch4. We propose that SEL-10 promotes the ubiquitin-mediated turnover of LIN-12/Notch proteins, and discuss potential roles for the regulation of lin-12/Notch activity by sel-10 in cell fate decisions and tumorigenesis.
Keywords: C. elegans, lin-12, CDC4 family, SEL-10, Notch
Many cell–cell interactions that specify cell fate are mediated by receptors of the LIN-12/Notch family and ligands of the Delta/Serrate/LAG-2 (DSL) family (for review, see Artavanis-Tsakonas et al. 1995). Caenorhabditis elegans affords an opportunity to study a simple case of lateral specification involving an interaction between two cells of the hermaphrodite gonad. These cells, named Z1.ppp and Z4.aaa, are initially equivalent in their developmental potential: each has an equal chance of becoming the anchor cell (AC), a terminally differentiated cell type that is necessary for vulval development, or a ventral uterine precursor cell (VU), which contributes descendants to the ventral uterus. In any given hermaphrodite, however, only one of these cells will become the AC, whereas the other becomes a VU (Kimble and Hirsh 1979).
Laser ablation studies have shown that this process of lateral specification, the AC/VU decision, depends on interactions between Z1.ppp and Z4.aaa (Kimble 1981; Seydoux and Greenwald 1989). Furthermore, genetic studies have indicated that _lin-12_-mediated signaling controls the AC/VU decision: if lin-12 activity is inappropriately elevated, Z1.ppp and Z4.aaa become VUs, whereas if lin-12 activity is reduced, Z1.ppp and Z4.aaa become ACs (Greenwald et al. 1983). Genetic mosaic analysis (Seydoux and Greenwald 1989) and reporter gene studies (Wilkinson et al. 1994) have indicated that both Z1.ppp and Z4.aaa initially express lin-12 and lag-2, but that a stochastic small variation in ligand and/or receptor activity is subsequently amplified by a feedback mechanism that influences lin-12 and lag-2 transcription. Thus, Z1.ppp and Z4.aaa assess their relative levels of lin-12 activity as part of the decision-making process, before either cell commits to the AC or VU fates, and the feedback mechanism ensures that only one of the two cells will become an AC and the other will become a VU.
It is striking that the receptors (LIN-12/Notch proteins), ligands (DSL proteins), and at least one downstream signaling component (CBF1/Su(H)/LAG-1; for review, see Christensen et al. 1996 and references therein) that mediate lateral specification are highly conserved in animals as distantly related as C. elegans, Drosophila, and vertebrates. Furthermore, a feedback mechanism like that first described for the AC/VU decision (Seydoux and Greenwald 1989) also exists for a Notch-mediated lateral interaction in Drosophila (Heitzler and Simpson 1991) and seems likely to operate in Notch-mediated lateral interactions in vertebrates (e.g., Austin et al. 1995; Chitnis et al. 1995; Washburn et al. 1997). The identification of genes that influence lin-12 activity during the AC/VU decision may reveal other conserved factors that participate in signal transduction or regulate the activity of lin-12/Notch proteins.
Genetic screens based on suppression or enhancement of lin-12 mutations have identified a number of genes that influence lin-12 activity. Here we describe sel-10, which was first identified in a screen for suppressors of phenotypes associated with partial loss of lin-12 activity (Sundaram and Greenwald 1993). We have found that sel-10 acts as a negative regulator of lin-12 signaling, and that SEL-10 is a member of the CDC4 family of F-box/WD40 repeat-containing proteins. CDC4, the most extensively studied member of this family, is a Saccharomyces cerevisiae protein that is involved in the ubiquitin-mediated degradation of cell cycle regulators such as SIC1 (for review, see King et al. 1996). CDC4 binds to SIC1, thereby targeting the ubiquitination machinery to this substrate (Feldman et al. 1997; Skowyra et al. 1997). Similarly, we have shown that C. elegans SEL-10 can interact physically with the intracellular domains of C. elegans LIN-12 and murine Notch4 (Robbins et al. 1992; Uyttendaele et al. 1996). We propose that SEL-10 promotes ubiquitin-mediated degradation of LIN-12/Notch proteins, and discuss potential roles for LIN-12/Notch turnover in cell fate decisions and oncogenesis.
Results
Lowering sel-10 dosage elevates lin-12 activity
Two sel-10 alleles, sel-10(ar28) and sel-10(ar41), were identified in a screen for suppressors of defects caused by a partial loss-of-function allele of lin-12 (Sundaram and Greenwald 1993). These sel-10 alleles were shown to suppress multiple defects associated with loss of lin-12 activity, and to enhance defects associated with elevated lin-12 activity (Sundaram and Greenwald 1993). Here, we provide evidence that sel-10 alleles reduce sel-10 activity, indicating that sel-10 is a negative regulator of lin-12 activity.
For the genetic analysis of sel-10, we relied on its genetic interactions with mutations in lin-12. We focused on two _lin-12_-mediated decisions (for review, see Greenwald 1997). One decision is made by two cells of the hermaphrodite gonad, Z1.ppp and Z4.aaa, between the AC and VU fates; normally, only one of these two cells becomes the AC, whereas the other becomes a VU (see introductory section). Eliminating lin-12 activity causes both Z1.ppp and Z4.aaa to become ACs (the 2 AC defect), and constitutively activating LIN-12 causes both Z1.ppp and Z4.aaa to become VUs. The other decision is made by the six vulval precursor cells, between a particular vulval fate termed 2° or an alternative fate; normally, two of the six vulval precursor cells, P5.p and P7.p, adopt the 2° fate. Eliminating lin-12 activity causes all six vulval precursor cells to adopt alternative non-2° fates, and constitutively activating LIN-12 causes all six vulval precursor cells to adopt the 2° fate. Thus, mutants in which LIN-12 is constitutively active display a 0 AC Egg-laying (Egl) defect because the absence of an AC prevents normal vulval formation; they are also Multivulva (Muv), because the descendants of each vulval precursor cell that adopts the 2° fate forms a pseudovulva.
sel-10(ar41) appears to elevate lin-12 activity: sel-10(ar41) suppresses the 2 AC defect of lin-12 hypomorphs (Sundaram and Greenwald 1993) and enhances the 0 AC defect caused by elevated lin-12 activity (Table 1, cf. lines 1 and 2). Furthermore, the double mutant lin-12(n379); sel-10(ar41) displays a Muv phenotype characteristic of high lin-12 activity that is not normally seen in lin-12(n379) single mutants (Table 1, lines 4,7).
Table 1.
sel-10 gene dosage analysis
| Relevant genotype | %Egl (n) | %Muv (n) | %Ste/Let (n) |
|---|---|---|---|
| 20° | |||
| lin-12(d)/+a | 6 (93) | 0 (93) | 0 (93) |
| lin-12(d)/+;sel-10b | 91 (54) | 0 (54) | 0 (54) |
| lin-12(d)/+;sel-10/Dfc | 92 (39) | 15 (39) | 0 (39) |
| 15° | |||
| lin-12(d); +d | 86 (60) | 0 (60) | 0 (60) |
| lin-12(d);sel-10/+e | 98 (62) | 0 (62) | 0 (62) |
| lin-12(d);+/Dff | 89 (57)i | 62 (74) | 10 (63) |
| lin-12(d);sel-10g | 100 (70)i | 78 (197) | 55 (126) |
| lin-12(d);sel-10/Dfh | – | 85 (34) | 100 (34)j |
The sel-10 locus is haploinsufficient: We observed enhancement of the Muv defect (and the sterile/lethal defect) of lin-12(n379) hermaphrodites in _nDf42/_+ hermaphrodites (Table 1, lines 4,6).
The sel-10(ar41) mutation appears to reduce sel-10 activity: the enhancement of the Muv defect of lin-12(n379)/+ hermaphrodites is more pronounced when sel-10(ar41) is placed in trans to the large deficiency nDf42 (Table 1, lines 2,3). The greater enhancement seen in trans to a deficiency may mean that the sel-10(ar41) allele is a partial loss-of-function allele rather than a null allele; alternatively, nDf42 may remove another gene that interacts with or is functionally redundant with sel-10. Molecular data (see below) indicate that sel-10(ar41) would lead to a drastic truncation of the predicted SEL-10 protein, suggesting that sel-10(ar41) strongly reduces sel-10 activity.
Elevating sel-10 dosage lowers lin-12 activity
The molecular cloning of sel-10(+) (see below) enabled us to examine the effect of elevated sel-10(+) activity, because, in general, extrachromosomal arrays formed after injecting DNA at a high concentration result in higher transgene expression (Mello et al. 1991). We found that extrachromosomal arrays containing high-copy arrays of the sel-10 genomic region (see below) appear to lower lin-12 activity as assayed by their effect on the AC/VU decision. There is a dramatic decrease in the proportion of lin-12(n379) hermaphrodites displaying the 0 AC defect in the presence of the high copy number array arEx93 (Table 2A). In addition, the presence of the arEx93 array enhances the 2AC defect caused by a partial loss of lin-12 function (Table 2B). Therefore, the level of sel-10 activity can control the level of lin-12 activity, because increasing or decreasing the activity of sel-10 has reciprocal effects on lin-12 activity.
Table 2.
Increased dosage of sel-10 reduces lin-12 activity
| A. Suppression of phenotypes associated with increased lin-12 activity | |
|---|---|
| Relevant genotype | % 0AC (n) |
| lin-12(d); dpy-20;Ex[sel-10(+) dpy-20(+)] | 51 (47) |
| lin-12(d); dpy-20;Ex[dpy-20(+)] | 95 (44) |
| B. Enhancement of phenotypes associated with reduced lin-12 activity | |
| Relevant genotype | % 2AC (n) |
| lin-12(h); dpy-20;Ex[sel-10(+) dpy-20(+)] | 97 (34) |
| lin-12(h); dpy-20;Ex[dpy-20(+)] | 30 (40) |
| lin-12(+); dpy-20(e1282);Ex[sel-10(+) dpy-20(+)] | 0 (87) |
sel-10 mutants display low penetrance defects associated with constitutive activation of lin-12
Most sel-10 animals appear wild type. We have observed that ∼1% of sel-10(ar41) hermaphrodites lack an AC (data not shown). Furthermore, ∼4% of sel-10(ar41) males display a gonad Migration (Mig) defect similar to that seen in lin-12(d) mutants, where it results from a failure to form the linker cell, the male counterpart of the hermaphrodite AC (Greenwald et al. 1983). In addition, we note that ∼8% of sel-10 mutant hermaphrodites are Egl even though they have an AC, and that sel-10 males have a reduced mating efficiency that cannot be completely accounted for by the Mig defect. These additional defects may reflect the effect of increased lin-12 activity on other cell fate decisions (Greenwald et al. 1983).
Cell autonomy of the sel-10 effect on lin-12 activity
Two lines of evidence suggest that sel-10 functions cell autonomously to elevate lin-12 activity. First, we examined the effect of reducing sel-10 activity on the activity of the intracellular domain of LIN-12. Expression of lin-12(intra) causes phenotypes associated with LIN-12 activation (Struhl et al. 1993). Because LIN-12(intra) lacks the extracellular domain and, hence, is active in the absence of external signaling, an enhancement of lin-12(intra) activity by sel-10 mutations would be evidence for cell autonomy of the sel-10 effect on lin-12 activity. We used an extrachromosomal array that contains the lin-12(intra) transgene and a transformation marker (see Materials and Methods); this array results in a low-penetrance lin-12 activated phenotype (Table 3A, line 1). When this array is combined with sel-10(ar41), there is a dramatic increase in the proportion of hermaphrodites displaying the 0 AC-Egl defect and males displaying the Mig defect (Table 3A, line 2), suggesting that sel-10(+) activity normally reduces lin-12 function in the same cell.
Table 3.
Cell autonomy of sel-10 function
| A. Enhancement of lin-12(intra)a | ||
|---|---|---|
| Relevant genotype | % Egl (n) | % Mig (n) |
| sel-10(+);dr arEx[lin-12(intra)] | 16 (88)b | 10 (57) |
| sel-10(ar41);dr arEx[lin-12(intra)] | 46 (136)b | 59 (90) |
| B. Cell ablationc | ||
| Relevant genotype | % 0AC (n) | |
| unoperated | operated | |
| lin-12(n379)/+; sel-10(+) | 10 (57) | 9 (11) |
| lin-12(n379)/+; sel-10(ar41) | 97 (71) | 83 (12) |
We have also tested whether sel-10 functions in the receiving end of _lin-12_-mediated cell-cell interactions by performing cell ablation experiments to remove the signaling cell, in this case Z4.aaa (Table 3B). This experiment enables different genotypes to be compared with respect to their intrinsic level of constitutive lin-12 activity in Z1.ppp. If Z4, the precursor to Z4.aaa, is ablated in _lin-12(n379)/_+ hermaphrodites, Z1.ppp usually becomes an AC, because the level of constitutive lin-12 activity is relatively low. However, if Z4 is ablated in lin-12(n379)/+;sel-10 hermaphrodites, Z1.ppp usually becomes a VU, suggesting that the level of constitutive lin-12 activity is relatively high. These results suggest that sel-10(+) functions to reduce lin-12 activity within the same cell, because a high level of intrinsic lin-12 activity is seen when sel-10 activity is reduced, even when the signaling cell is removed.
Cloning of sel-10 by an anti-suppression assay
sel-10 was mapped previously to an interval between lin-25 and unc-76 on LGV (Sundaram and Greenwald 1993). We refined the map position to a 300-kb interval between the cloned polymorphisms arP3 and TCPAR1 (see Materials and Methods; Fig. 1). Cosmids from the region were tested for their ability to reverse the suppression of the 2AC defect of lin-12(ar170) by sel-10(ar41) (see Materials and Methods). Arrays containing the cosmid C07E11 gave rescue in this anti-suppression assay and also reversed the enhancement of lin-12(n379) by sel-10(ar41) (data not shown). This cosmid was further subcloned and the ∼8-kb fragment in pJH166 gave results similar to those seen with the entire cosmid (Fig. 1).
Figure 1.

Molecular cloning of sel-10. See Materials and Methods for details of rescue assays, plasmid constructions, and molecular analysis. Genetic markers used to map sel-10 are italicized, and two cosmids that contain sel-10 sequences are shown in the box. The cosmid C07E11 and derivatives were tested for rescue (asterisk). pJH169 is identical to pJH166 except that it contains a stop codon in the predicted coding sequence after codon 172. Restriction sites are (B) _Bam_HI; (H) _Hin_dIII; (S) _Sal_I.
Molecular analysis of sel-10
The ends of pJH166 (Fig. 1) were sequenced and compared with sequence generated by the C. elegans genome sequencing project (Waterston et al. 1997). The entire region was found on the cosmid F55B12. A fragment from the predicted open reading frame was radiolabeled and used to screen a Northern blot and to probe a cDNA library (see Materials and Methods). Northern analysis revealed a single band of ∼2.5-kb that is present in total RNA prepared from wild-type, sel-10(ar41) and sel-10(ar28) strains (data not shown). The ends of ten cDNAs from the region were sequenced and the largest cDNA was sequenced in its entirety on one strand. Verification of the 5′ end was obtained by sequencing products amplified from the cDNA library by use of trans-spliced leader sequence SL1 (Krause and Hirsh 1987) and a _sel-10_-specific sequence for primers. The splice junction of SL1 to the sel-10 coding region occurs four bases upstream of the first predicted start codon. Figure 2 summarizes the results of the sequence analysis of sel-10.
Figure 2.

cDNA sequence and predicted protein product of sel-10. Splice junctions are indicated by arrows below the DNA sequence. The first arrow indicates the SL1 splice junction. The F-Box (Kumar and Paietta 1995, Bai et al. 1996) and the WD40 repeats are overlined and labeled in the Figure. The lesions in sel-10(ar41) and sel-10(ar28) are indicated with reverse contrast letters in the nucleotide sequence and a bold asterisk above the amino acid; both are G to A transitions resulting in W to stop codon changes in the amino acid sequence at residues 323 and 511, respectively. The cDNA termination codon is marked with an asterisk. A sequence conforming to the consensus polyadenylation signal sequence is underlined, and sites of polyA attachment are marked in bold. Two independent cDNAs contained polyA 14 nucleotides downstream of this signal; two alternative sites of attachment were also observed. Sequence information has been submitted to GenBank (accession no. AF020788).
SEL-10 encodes a protein of the CDC4 family
By use of a BLAST search (Altschul et al. 1990), we found that the predicted SEL-10 protein sequence contains two previously identified amino acid sequence motifs (Fig. 3A–C). First, there is a motif (Kumar and Paietta 1995) that is now called the F-Box, after its occurrence in cyclin F (Bai et al. 1996). The F-Box motif has been implicated in protein–protein interactions, and is found in a large variety of proteins, many of which contain other recognizable motifs carboxy terminal to the F-Box (Bai et al. 1996). Second, there are seven tandem WD40 repeats, also known as β-transducin repeats, a conserved repeat of ∼40 amino acids named for the common appearance of Trp-Asp (WD) at the end of the repeat (for review, see Neer et al. 1994). The crystal structure of β-transducin reveals that the seven repeats form a β propeller structure, which most likely mediates protein–protein interactions (Gaudet et al. 1996; Lambright et al. 1996; Sondek et al. 1996). There is a great deal of functional diversity among WD40 repeat-containing proteins.
Figure 3.
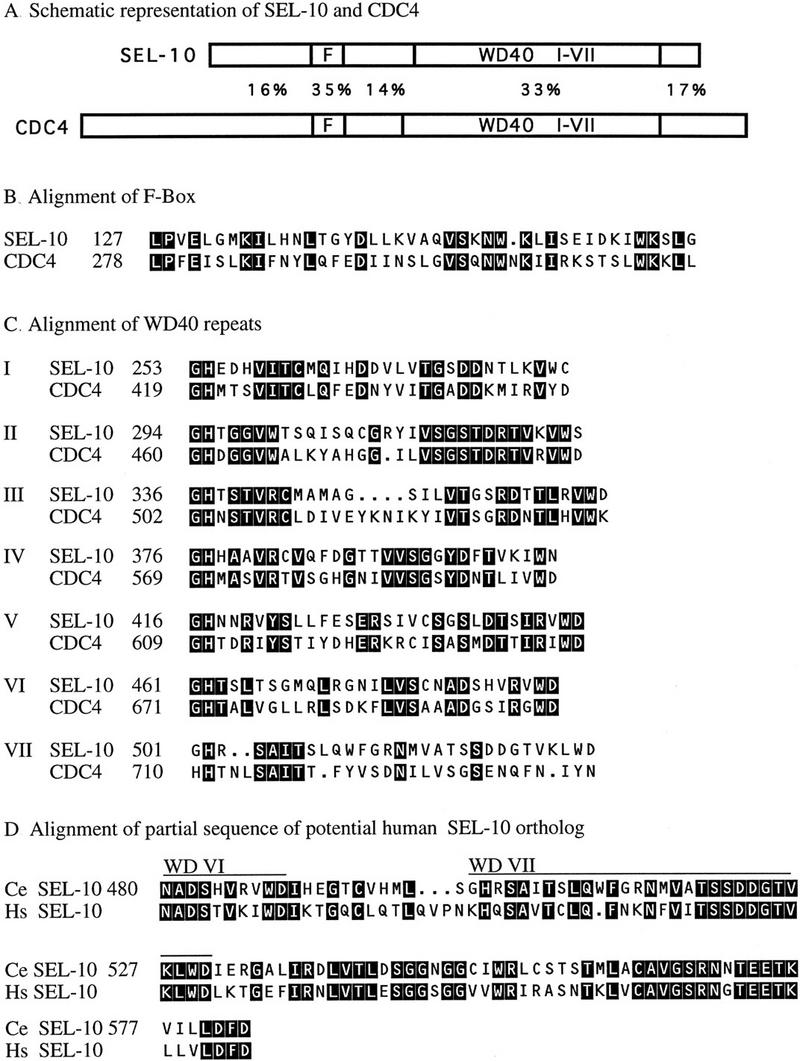
SEL-10 is a member of the CDC4 family of F-box/WD40-repeat proteins. The CDC4 sequence (accession no. X05625) is from GenBank. There are other potential CDC4 family members in the database; limited sequence data for a potential SEL-10 ortholog is given in part D. For further discussion of the CDC4 family, see Kumar and Paietta (1995) and Bai et al. (1996). Reverse contrast letters indicate amino acid identity. (A) Schematic depiction of SEL-10 and CDC4, drawn to scale. The percentage of identical amino acids in each region is indicated. Members of the CDC4 family all have this general organization, with some variability in the length and sequence of their amino and carboxyl termini. (B) Alignment of SEL-10 and CDC4 F-Boxes. (C) Alignment of WD40 repeats from SEL-10 and CDC4. (D) Alignment of SEL-10 and a potential human ortholog. Partial sequence of a human cDNA encoding a sequence highly similar to the carboxyl terminus of SEL-10 was obtained from Genbank (accession no. H22962) and was extended into the WD40 repeats by direct sequencing of clone ym50h08.s1 (G. Wu and J. Kitajewski, unpubl.). The predicted amino acid sequence encoded by the available human cDNA sequence is shown. The database also contains partial sequence information for a rat cDNA (Genbank accession no. H34371) that is predicted to encode a peptide that is 100% identical to the last 35 amino acids of the available human sequence (data not shown).
The presence of an F-box amino-terminal to a set of seven WD40 motifs is the hallmark of the CDC4 family of WD40 repeat-containing proteins, indicating that SEL-10 belongs to this family. Furthermore, separate BLAST searches with just the SEL-10 F box or the SEL-10 WD40 repeats identified members of the CDC4 family as the most similar. The F-Box motif present in proteins within the CDC4 subfamily is more conserved than among other F-Box-containing proteins (Fig. 3B), and there is more extensive homology around the F-box (Kumar and Paietta 1995). In addition, the alignment of the WD40 repeats of SEL-10 and CDC4 (Fig. 3C) reveals that a given WD40 repeat is more similar between yeast and worms than are the repeats within a given species.
The CDC4 family includes proteins in fungi and vertebrates, other predicted C. elegans proteins (E. Kipreos, S. Gohel, and E. Hedgecock, pers. comm.; E.J.A. Hubbard, unpubl.), and several mammalian proteins (Kumar and Paietta 1995; Bai et al. 1996). The best studied member of this family, Sacharomyces cerevisiae CDC4, targets SIC1 and certain G1 cyclins for degradation (for review, see King et al. 1996). Not all CDC4 family members, however, are cell cycle regulators. For example, there are proteins that negatively regulate sulfur metabolism from S. cerevisiae, Neurospora crassa, and Aspergillus nidulans (Natorff et al. 1993; Kumar and Paietta 1995; Thomas et al. 1995).
A recent database search has revealed potential human and rat SEL-10 orthologs (Fig. 3D). The limited sequence information available to date for the human gene begins in the sixth WD40 repeat and extends to the carboxyl terminus, and in this region, there is 60% sequence identity. BLAST searches of the available databases with the SEL-10 sequence carboxy-terminal to the WD40 repeats have not identified any other highly conserved sequences.
sel-10 mutations truncate the SEL-10 protein
Sequence analysis of sel-10 mutations supports the genetic evidence suggesting that they strongly reduce sel-10 activity. The sequence alterations caused by sel-10(ar41) and sel-10(ar28) were determined by direct sequencing of amplified genomic DNA products (see Materials and Methods). Both alleles are nonsense mutations at nucleotide positions 969 and 1533, respectively (see Fig. 2), resulting in truncated predicted proteins.
sel-10(ar41) removes the carboxy-terminal half of the protein, including five of the seven WD40 repeats. This observation suggests that sel-10(ar41) is likely to result in a nonfunctional SEL-10 protein. It is unlikely that the two WD40 repeats that remain in this protein are functional because there are no known WD40-repeat-containing proteins with only two repeats (Neer et al. 1994). Furthermore, the crystal structure of β transducin reveals that the seven repeats form a β propeller structure that would not be complete in the absence of five of the seven repeats (Sondek et al. 1996). Finally, comparable mutations in another C. elegans CDC4 subfamily protein, LIN-23, behave like molecular null alleles (E.T. Kipreos, S.P. Gohel, and E.M. Hedgecock, pers. comm.).
C. elegans SEL-10 physically interacts with LIN-12(intra) and murine Notch4(int3)
We probed for potential interactions between SEL-10 and the intracellular domains of LIN-12/Notch proteins, specifically LIN-12(intra), the intact intracellular domain (Struhl et al. 1993) and Notch4(int3), the intact intracellular domain with some additional sequences produced by the int3 mutation (Robbins et al. 1992; Uyttendaele et al. 1996). We initially used the yeast two-hybrid system (Fields and Song 1989) and our preliminary results suggested that SEL-10 physically interacted with the C. elegans LIN-12 intracellular domain, the C. elegans GLP-1 intracellular domain (GLP-1 is another C. elegans LIN-12/Notch protein; see Yochem and Greenwald 1989), and the mouse Notch4(int3) intracellular domain (data not shown).
To examine further whether SEL-10 binds LIN-12/Notch proteins, we carried out coimmunoprecipitation experiments by use of transfected mammalian cells (Fig. 4). 293T (Bosc23) cells (human embryonic kidney cells) were transiently transfected with hemagglutinin HA-tagged LIN-12(intra) and/or myc-tagged SEL-10 (see Materials and Methods). Transfected cells were lysed and LIN-12(intra)HA was precipitated with anti-HA antibodies (Fig. 4A) or, alternatively, SEL-10myc was immunoprecipitated with anti-myc antibodies (Fig. 4B). The immunoprecipitates were subjected to immunoblot analysis to identify bound proteins, and probed with anti-myc or anti-HA antibodies as indicated. Under both conditions, the immunoprecipitates were found to contain both LIN-12HA and SEL-10myc. This result suggests that SEL-10 and LIN-12 are able to interact physically, either directly or in a complex.
Figure 4.

Coimmunoprecipitation of C. elegans SEL-10 with either C. elegans LIN-12(intra) or murine Notch4(int3) from transfected 293T cells. (A) Samples were immunoprecipitated with anti-HA antibody and the Western blot was probed with anti-myc to visualize SEL-10myc (top) or anti-HA to visualize LIN-12(intra)HA (bottom). Arrow indicates the expected mobility of SEL-10myc. (B) Samples were immunoprecipitated with anti-myc antibody and the Western blot was probed with anti-HA (top) or anti-myc (bottom). Arrow indicates the expected mobility of LIN-12(intra)HA. (C) Samples were immunoprecipitated with anti-Notch4 antibody and the Western blot was probed with anti-HA to visualize SEL-10HA (top) or anti-Notch4 to visualize Notch4(int3) (bottom). Arrow indicates the expected mobility of SEL-10HA. (D) Samples were immunoprecipitated with anti-HA antibody and the Western blot was probed with anti-Notch4 (top) or anti-HA (bottom). Arrow indicates the expected mobility of Notch4(int3). For details, see Materials and Methods. (Lane 1) pQNClacZ; (lane_2_) pQNClin-12(intra)HA + pQNClacZ; (lane 3) pQNClacZ + pQNCsel-10myc; (lane 4) pQNClin-12(intra)HA + pQNCsel-10myc; (lane 5) mock transfected cells; (lane 6) pLNCint3 + pQNCX; (lane 7) pLNCX + pQNCsel-10HA; (lane 8) pLNCint3 + pQNCsel-10HA.
We also examined whether the C. elegans SEL-10 protein would interact with the murine Notch4(int-3) protein. Cells were transfected with Notch4(int-3) and/or SEL-10HA, and immunoprecipitation was performed with cell lysates by use of either anti-Notch4 antibodies (Fig. 4C) or anti-HA antibodies (Fig. 4D). Immunoblot analysis showed that the immunoprecipitates contained a complex of Notch4(int-3) and SEL-10 proteins; thus, SEL-10 is able to complex with Notch4(int-3) protein.
The observations that sel-10 negatively regulates lin-12 activity, that SEL-10 resembles CDC4, and that SEL-10 physically interacts with LIN-12, taken together, strongly suggest that SEL-10 functions biochemically like CDC4 to promote LIN-12 turnover. We have attempted to examine the effect of coexpressing C. elegans SEL-10 on the steady-state levels and ubiquitination of Notch4(int-3) and observed a modest decrease in the steady state level (data not shown); however, at this time the mechanism underlying this decrease is not clear. Furthermore, there appears to be polyubiquitination of Notch4(int-3) even in the absence of transfected SEL-10, perhaps caused by the activity of an endogenous mammalian _sel-10_-like gene (data not shown).
Discussion
In this paper, we have presented genetic evidence indicating that sel-10 is a negative regulator of _lin-12_-mediated signaling in C. elegans. Mutations that lower sel-10 activity, elevate lin-12 activity, and increasing sel-10 dosage lowers lin-12 activity, suggesting that the level of sel-10 activity can influence lin-12 activity. Furthermore, sel-10 appears to act in the same cell as lin-12.
We propose that the mechanism by which sel-10 affects lin-12 activity may be by controlling LIN-12/Notch protein levels. Sequence analysis indicates that SEL-10 is related to the S. cerevisiae protein CDC4, which promotes the ubiquitin-dependent degradation of cell cycle regulators (for review, see King et al. 1996). CDC4 complexes with its substrates (Feldman et al. 1997; Skowyra et al. 1997), and we have found that C. elegans SEL-10 complexes with the intracellular domain of LIN-12. Proteins related to SEL-10 exist in mammals, and C. elegans SEL-10 physically complexes with Notch4(int3), the intracellular domain of murine Notch4. These observations suggest that negative regulation of LIN-12/Notch by SEL-10 may be an evolutionarily conserved feature.
SEL-10 may target LIN-12/Notch proteins for ubiquitin-mediated degradation
The attachment of ubiquitin to substrates involves a series of protein complexes. Ubiquitin is activated by linkage to an E1 ubiquitin activating enzyme, then transferred to an E2 ubiquitin conjugating enzyme. Some ubiquitination events also require the action of a third complex, termed E3. It is thought that E3 complexes may contribute to substrate specificity (for review, see Ciechanover 1994; King et al. 1996). The S. cerevisiae protein CDC4 may function in an E3 complex. CDC4 is one of a group of genes that also includes CDC34, CDC53, and SKP1; together, the proteins encoded by these genes directly regulate the level of the cyclin-dependent kinase inhibitor SIC1, which must be destroyed for progression from G1 to S phase. CDC34 is an E2 ubiquitin conjugating enzyme (Goebl et al. 1988), and the current view is that CDC4, CDC53, and SKP1 function as an E3 complex (Bai et al. 1996; Mathias et al. 1996). On the basis of our analysis of sel-10 and the data for CDC4, we propose that SEL-10 functions as part of an E3 complex to target the intracellular domains of LIN-12/Notch proteins for ubiquitin-dependent degradation.
The carboxy-terminal region of all LIN-12/Notch proteins contain PEST sequences and one or more lysines immediately carboxy-terminal to the cdc10/SWI6 motifs. The presence of PEST sequences often indicates that ubiquitin-mediated turnover occurs, although the two are not necessarily strictly correlated (Rogers et al. 1986). With respect to the CDC34/CDC4-mediated events, it appears that the PEST sequences couple phosphorylation of the substrate to attachment of ubiquitin to lysine residues (for review, see King et al. 1996). The PEST sequence found in the carboxy-terminal region of all LIN-12/Notch proteins, however, may not be required for SEL-10-mediated degradation of LIN-12/Notch proteins, because sel-10 mutations can still enhance the gain-of-function phenotype of a glp-1 allele that is truncated prior to the PEST sequence (E.J.A. Hubbard, unpubl.).
An important issue to consider in the context of SEL-10 as a component of an E3 complex is its specificity for LIN-12/Notch proteins. The available C. elegans genetic data suggest that sel-10 is an allele-nonspecific, gene-specific suppressor of lin-12, supporting a role for SEL-10 specifically in regulating the activity of LIN-12, or perhaps a small set of proteins including LIN-12. Allele-nonspecificity is indicated by the observation that mutations in sel-10 suppress/enhance all lin-12 alleles tested (Sundaram and Greenwald 1993; this report; E.J.A. Hubbard, unpubl.). Gene-specificity is suggested by the fact that mutations in sel-10 have not been identified in numerous screens in many laboratories for suppressors of hypomorphic mutations in genes encoding proteins other than LIN-12/Notch proteins; furthermore, we have not observed suppression of various marker mutations used in routine strain constructions or of hypomorphic alleles of several other genes encoding receptor proteins (E.J.A. Hubbard, unpubl.).
The available genetic data also suggest that sel-10 activity does not regulate cell cycle progression, a possibility raised by the phenotype of cdc4 mutants. Mutations in cul-1, a C. elegans gene related to S. cerevisiae CDC53, cause hyperplasia of larval blast cells, suggesting that cul-1 regulates cell-cycle progression (Kipreos et al. 1996). We have not seen any evidence that hyperplasia occurs in sel-10(ar41) mutants (E.J.A. Hubbard, unpubl.). Because sel-10(ar41) mutants have little or no sel-10 activity (see Results), we think that it is unlikely that sel-10 is involved in cell cycle regulation per se, unless there is another functionally redundant gene that masks cell cycle involvement of sel-10. In contrast, mutations in another CDC4 related gene, lin-23, do cause hyperplasia, consistent with a role for lin-23 in the regulation of cell cycle progression (E.T. Kipreos, S.P. Gohel, and E.M. Hedgecock, pers. comm.).
Potential roles for LIN-12/Notch protein downregulation or turnover in cell fate decisions
Proteolysis of LIN-12/Notch proteins might occur in response to ligand binding or occur constitutively. For a variety of cell surface receptors, ligand-induced polyubiquitination appears to be a mechanism for down-regulation (for review, see Ciechanover and Schwartz 1994). Although we do not, at this time, have direct evidence for ligand-induced ubiquitination of LIN-12/Notch receptors, we note that LIN-12(intra), which genetically and physically interacts with SEL-10, behaves like an activated receptor (Struhl et al. 1993). Alternatively, SEL-10 may target any form of LIN-12/Notch (activated or unactivated) for degradation. Although constitutive turnover is not strictly a mechanism for controlling receptor activity per se, it would, in effect, sensitize the system to other control mechanisms such as transcriptional regulation (see below) by generally reducing the amount of LIN-12.
Constitutive turnover or ligand-induced down-regulation of LIN-12/Notch proteins may be important for cell fate decisions to occur normally. We can best illustrate potential roles for turnover or down-regulation with the AC/VU decision (Seydoux and Greenwald 1989; Wilkinson et al. 1994) as an example. Initially, Z1.ppp and Z4.aaa have equal signaling and receiving potentials; ligand (LAG-2) and receptor (LIN-12) may interact, but signaling activity is below a critical threshold. SEL-10-mediated turnover or down-regulation of LIN-12 might prevent this initial signaling from causing both cells to achieve the threshold value of effector activity. Thus, one possible role for receptor turnover or down-regulation would be to limit the output from a single ligand-receptor interaction.
Another potential role for receptor turnover or down-regulation is in enhancing differences in lin-12 activity between interacting cells. During the AC/VU decision, a small stochastic difference between the two cells is amplified by a feedback mechanism (Seydoux and Greenwald 1989). The feedback mechanism appears to involve differential transcription of ligand and receptor genes: activation of LIN-12 appears to repress transcription of lag-2 and to stimulate transcription of lin-12 (Wilkinson et al. 1994). The feedback mechanism ensures that the cell with higher lin-12 activity becomes the VU whereas the cell with lower lin-12 activity becomes the AC. Down-regulation of LIN-12 would be necessary for differences in transcription to be manifest. In the absence of down-regulation, signaling from activated receptor would persist, masking the effects of differential transcription. Indeed, this situation is analogous to the role of ubiquitin-mediated degradation of G1 cyclins (King et al. 1996).
Turnover of LIN-12/Notch proteins may play different or additional roles in other cell fate decisions. For example, in Drosophila eye development, Notch appears to be utilized for sequential cell fate decisions (Cagan and Ready 1989), which would seem to necessitate clearance of activated Notch after each decision so that a new assessment of Notch activity can be made. Furthermore, it is also conceivable that for some LIN-12/Notch mediated decisions, the cell fate adopted may depend on the intensity of signal, as has been seen for receptors for gradient morphogens (e.g., Nellen et al. 1996). If any LIN-12/Notch-mediated decisions do display such dosage sensitivity, it is likely that they would depend on rapid turnover of activated receptor complexes so that the correct threshold value is read.
The fact that most sel-10(ar41) individuals are phenotypically wild-type, with only a small proportion displaying phenotypes associated with LIN-12 activation, may be explained in this context if there is a redundant gene product or regulatory mechanism. There are other CDC4 related genes in the C. elegans genome (E.J.A. Hubbard, unpubl.). Furthermore, there may be other mechanisms for degrading LIN-12. For example, sel-1, another negative regulator of lin-12 activity, may also be involved in LIN-12 turnover (Grant and Greenwald 1997), but because SEL-1 is an extracytosolic protein, it is not likely to be directly involved in the ubiquitination of the intracellular domain of LIN-12.
Potential roles for sel-10 in oncogenesis
Mammalian tumors induced by expression of Notch4(int3) or other truncated forms of Notch largely consisting of the intact intracellular domain are thought to result from constitutive Notch activity (Ellisen et al. 1991; Robbins et al. 1992; Uyttendaele et al. 1996). Because SEL-10 down-regulates Notch activity, it may act to restrain either normal or oncogenic functions of activated Notch, and, hence, suppress cell growth. If so, loss-of-function mutations in vertebrate sel-10 could contribute to oncogenesis mediated by Notch by elevating the level of Notch protein. For instance, human T acute lymphoblastic leukemias, which in the majority of cases do not contain oncogenic Notch alterations (Drexler et al. 1995), and human breast tumors, which thus far have not been reported to contain oncogenic Notch alterations, may carry mutations in other proteins that influence Notch activity, such as sel-10 homologs.
Materials and methods
General methods and strains
General methods are described by Brenner (1974). The wild-type parent for all strains was C. elegans var. Bristol strain N2. Mapping experiments utilized the Bristol/Bergerac congenic strain GS352, in which the region between rol-4 and par-1 of Bristol was replaced with the corresponding region from the Bergerac strain BO (Tuck and Greenwald 1996). Strains were grown at 20°C unless otherwise noted. Mutations used are described in Hodgkin (1997); additional references for critical alleles are also given. Note that all genetic constructions containing sel-10 used a sel-10 chromosome that does not have the linked modifier mutation arX (Sundaram and Greenwald 1993). LGIII: dpy-17(e164), unc-36(e251), and unc-32(e189); lin-12(ar170) (Hubbard et al. 1996; E.J.A. Hubbard, unpubl.); lin-12(n379) (Greenwald et al. 1983). LGIV: dpy-20(e1282). LGV: nDf42 (M. Hengartner and H.R. Horvitz, pers. comm.), lon-3(e2175), rol-4(sc8), sel-10(ar41) (Sundaram and Greenwald 1993), him-5(e1490), unc-76(e911).
Mapping of the sel-10 locus
sel-10 had been genetically mapped between lin-25 and unc-76 V (Sundaram and Greenwald 1993) and ∼0.2 MU to the left of him-5 (data not shown). We mapped sel-10 between arP3 and TCPAR1 by identifying Rol Him non-Unc recombinants from heterozygotes of the genotype rol-4 BO unc-76/lon-3 sel-10 him-5 constructed with the strain GS352. Fifty independent recombinants were analyzed by Southern blot hybridization for the presence of arP3 and TCPAR1 (Tuck and Greenwald 1996), and each recombinant strain was tested for the presence of sel-10(ar41) by crossing into lin-12(n379) and scoring for the Muv phenotype. Mapping data can be found in ACeDB (Edgley et al. 1997).
sel-10 cloning by antisuppression assay
sel-10(ar41) partially suppresses the 2AC defect caused by lin-12(ar170): at 25°C, ∼80% of lin-12(ar170) animals have 2AC whereas ∼25% of lin-12(ar170); sel-10(ar41) animals have 2AC. We used reversal of suppression as the basis of assessing sel-10(+) activity of microinjected DNAs. Transgenic lines were generated by microinjecting the germ lines of lin-12(ar170); dpy-20(e1282);sel-10(ar41) him-5(e1490) hermaphrodites with cosmid or plasmid DNA (Mello et al. 1991) at a concentration of 5 μg/ml, along with the dpy-20(+) transformation marker DNA at 10 μg/ml (plasmid pMH86; Han and Sternberg 1991) and carrier Bluescript DNA (Stratagene) at 90 μg/ml. Synchronous populations were obtained by allowing groups of transgenic hermaphrodites to lay eggs at 20°C for 1- to 2-hr and tranferring the eggs to 25°C. The non-Dpy L3 hermaphrodites were then scored for the number of anchor cells. The injected tester DNA was considered to contain sel-10(+) sequences if >50% of the non-Dpy animals had 2AC. Typically, 60%–80% of hermaphrodites had 2AC in these rescued lines. Some arrays scored as having sel-10(+) activity were subjected to a second test, the ability to reverse the Muv phenotype of lin-12(n379);sel-10 (ar41). Initial rescue was obtained with a pool of seven overlapping cosmids from the region (each at 5 μg/ml), then with the single cosmid C07E11, and then with plasmids derived from C07E11, as shown in Figure 1.
Plasmids containing sel-10 genomic sequences
pJH151 was constructed by digesting cosmid C07E11 with _Bam_HI and ligating the 15-kb fragment to Bluescript KS+ (Stratagene). pJH166 was constructed by ligating an 8-kb _Pst_I–_Sal_I fragment from pJH151 into Bluescript KS+. The _Pst_I site was from the vector, whereas the _Sal_I site is from the genomic sequences. The ∼9-kb _Sal_I fragment was removed from pJH151 to form pJH165, and pJH167 was made by ligation of the internal _Hin_dIII fragment of pJH151 into Bluescript. To construct pJH169, pJH166 was cut with _Pme_I and a linker containing an _Nhe_I site with a stop codon in all frames (NEB 1060) was inserted, creating a stop codon after amino acid 172 in the SEL-10 sequence.
sel-10 overexpression
arEx93 was generated by microinjecting dpy-20 hermaphrodites with pJH166 [sel-10(+)] at a concentration of 100 μg/ml and pMH86 [dpy-20(+)] at 10 μg/ml. Strains carrying this array segregate sterile animals as well as fertile animals; the basis for the sterility has not been established. Many of the fertile animals display a leaky Egl phenotype similar to that observed in certain lin-12 hypomorphic mutants. Qualitatively similar results were observed with other lines at this concentration and with lines established by use of pJH166 at 50 μg/ml (data not shown).
The control array arEx149 was established by microinjecting unc-32; dpy-20 hermaphrodites with pMH86 at 10 μg/ml, and Bluescript DNA at 90 μg/ml into unc-32; dpy-20 animals.
Molecular analysis of sel-10
Standard methods were used for the manipulation of recombinant DNA (Sambrook et al. 1989). sel-10(+) cDNAs were obtained by screening ∼100,000 pfu from a phage library kindly provided by R. Barstead (Barstead and Waterston 1989). Ten positive plaques were purified by two subsequent rounds of screening with a radiolabeled fragment from pJH166 (∼8-kb _Bam_HI–_Sal_I fragment) as a probe. cDNA 1A, the longest cDNA obtained, was sequenced in its entirety on one strand and compared with genomic sequence from the genome project with GENEFINDER (Waterston et al. 1997). The sequence of the cDNA 1A differed from the GENEFINDER prediction in the location of the junction between the second and third exons and in the predicted 3′ end. Four of the cDNAs were polyadenylated at their 3′ ends (one 294, one 581, and the other two 601 bases after the predicted stop codon). Of these, only the last two were in the context of a conserved polyadenylation signal. The 5′-most cDNA end was located in codon 1 (cDNA 8 begins at G of the first ATG), but a PCR product was amplified from DNA prepared from the same cDNA library (Barstead and Waterston 1989; C. Dong, pers. comm.) contained the SL1 spliced leader at the predicted sequence 4 bases 5′ of the first ATG. The 22 base SL1 sequence and a primer straddling the 5th and 6th exons were used for the 5′ end amplification.
Sequence analysis
Standard techniques were used to obtain sequence of the 1A cDNA (Sambrook et al. 1989). The lesions associated with the sel-10(ar41) and sel-10(ar28) mutations were found by direct sequencing of two PCR products from single-stranded templates (Allard et al. 1991; Kaltenboeck et al. 1992), by use of internal primers to cover the entire region. One small segment was subcloned and sequenced (from two independent reactions each), as the sequence from this region was not easily generated by use of the direct method. Sequence comparisons and alignments were obtained by use of Blast (Altschul et al. 1990) through the NCBI web site and GCG (version 8, Devereux et al. 1984) programs.
Plasmids for cell culture experiments
Plasmids used in the transient transfection experiments were constructed in pLNCX (Miller et al. 1989) or pQNCX (Qingyou Yan and J.K., unpubl.), vectors that drive gene expression under the control of a CMV promoter. pQNClacZ contains the bacterial lacZ gene; pQNClin-12(intra)HA encodes a protein with a methionine-containing hemagglutinin epitope (Wilson et al. 1984) fused in frame amino-terminal to LIN-12(intra) at amino acid 939. pLNCint-3 contains cDNA corresponding to the Notch4 region expressed in the int3 insertion, beginning at amino acid 1411; the Notch4(int3) protein includes the entire intracellular domain of Notch4 and additional sequences (Uyttendaele et al. 1996). pQNCsel-10myc (pJH186) encodes a protein with six myc epitope tags (Roth et al. 1991) fused in frame to cDNA 1A at amino acid 13 of SEL-10. pQNCsel-10HA (pJH184) encodes a protein with a methionine-containing hemagglutinin epitope from pACT2 (Durfee et al. 1993) fused in frame (along with a short stretch of polylinker) to cDNA 1A at amino acid 13.
Transfection, immunoprecipitations, and Western blot analysis
293T (Bosc23) cells (Pear et al. 1993) were maintained in Dulbecco’s modified Eagle medium with 10% fetal bovine serum (FBS). A confluent plate of cells was split 1:3 the day prior to transfection. For one 60-mm plate of cells, 4 μg of each plasmid DNA was transfected by the calcium phosphate precipitation method. The total amount of DNA was kept constant by supplementation with vector DNA or _lacZ_-containing plasmids.
Two days after transfection, cells were harvested and lysed in TENT buffer (50 mm Tris-HCl at pH 8.0, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100) containing protease inhibitors (2 μg/ml aprotinin, 2 μg/ml of leupeptin, 2 μg/ml of pepstatin, 0.5 mm PMSF). Lysates were clarified by centrifugation at 10,000_g_ for 10 min and protein content was determined with the BioRad Protein determination kit and samples were normalized for protein content. Extracts were precleared with Sepharose CL-4B beads, incubated with antibodies (3 μl of anti-Notch4 antiserum, 50 μl of 12CA5 anti-HA supernatant, or 200 μl of 9E10 anti-myc supernatant) for 6 hr at 4°C, then incubated with 40 μl of 50% slurry of protein A–Sepharose for 1 hr at 4°C. The protein A–Sepharose beads were washed with TENT buffer three times by vortexing for 10 min, beads were boiled in 30 μl 1× protein loading buffer, and then electrophoresed on a 10% SDS–polyacrylamide gel and transferred onto nitrocellulose membrane. The blot was blocked overnight at 4°C with TBST (10 mm Tris at pH 8.0, 150 mm NaCl, 0.2% Tween 20) containing 1% bovine serum albumin (TBST–BSA). The blot was then incubated with 1° antibody diluted (1:2,000 anti-Notch4; 1:50 for 12CA5; 1:10 for 9E10) in TBST–BSA for 1 hr, washed three times for 5 min each with TBST, and incubated with 2° antibody in TBST–BSA for 1 hr. After three washes, the signal was visualized by chemiluminescence (Amersham, ECL).
The anti-Notch4 antiserum (G. Wu and J. Kitajenski, unpubl.) is directed against the carboxy-terminal region of Notch4 (residues 1788–1964) (Uyttendaele et al. 1996). 12CA5 anti-HA antibody was obtained from Berkeley Antibody Co., Richmond, CA. 9E10 anti-myc antibody was prepared from culture supernatants of the 9E10 hybridoma (Evan et al. 1985).
Acknowledgments
We are grateful to H. Uyttendaele for Notch4(int3) constructs, Q. Yan for expression vectors, and G. Struhl for plasmids and epitope tags. We also thank D. Brousseau and R. Ruiz for technical assistance; N. Adler, C. Leung, I. Temkin, and J. Yu for help with genetics; and the Caenorhabditis Genetics Center and M. Koelle for providing strains. We are indebted to E. Kipreos, M. Goebl, and R. Deshaies for sharing information and ideas prior to publication, S. Tuck and T. Schedl for helpful advice, and B. Grant, D. Levitan, A. Melendez, C. Wen, S. Hubbard, and G. Struhl for valuable discussions and comments on the manuscript. This work was supported by the Damon Runyon-Walter Winchell Cancer Research Fund (E.J.A.H.), National Institutes of Health grant GM37602 (I.G.) and the U.S. Army Medical Research and Material Command (USAMRMC) under grant DAMD17-94-J-4410 (J.K.). E.J.A.H. is a Postdoctoral Associate and I.G. is an Associate Investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL greenwald@cuccfa.ccc.columbia.edu; FAX (212) 305-1721.
References
- Allard MW, Ellsworth DL, Honeycutt RL. The production of single-stranded DNA suitable for sequencing using the polymerase chain reaction. BioTechniques. 1991;10:23–26. [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–235. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- Austin CP, Feldman DE, Ida JA, Cepko CL. Vertebrate retinal ganglion cells are selected from competent progenitors by the action of Notch. Development. 1995;121:3637–3650. doi: 10.1242/dev.121.11.3637. [DOI] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Barstead RJ, Waterston RH. The basal component of the nematode dense-body is vinculin. J Biol Chem. 1989;264:10177–10185. [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan RL, Ready DF. Notch is required for successive cell decisions in the developing Drosophila retina. Genes & Dev. 1989;3:1099–1112. doi: 10.1101/gad.3.8.1099. [DOI] [PubMed] [Google Scholar]
- Chitnis A, Henrique D, Lewis J, Ish-Horowicz D, Kintner C. Primary neurogenesis in Xenopus embryos regulated by a homolog of the Drosophila neurogenic gene Delta. Nature. 1995;375:761–766. doi: 10.1038/375761a0. [DOI] [PubMed] [Google Scholar]
- Christensen S, Kodoyianni V, Bosenberg M, Friedman L, Kimble J. lag-1, a gene required for lin-12 and glp-1 signaling in C. elegans, is homologous to human CBF1 and Drosophila Su(H) Development. 1996;122:1373–1383. doi: 10.1242/dev.122.5.1373. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: Mechanisms of recognition of the proteolytic substrate and involvement in the degradation of native cellular proteins. FASEB J. 1994;8:182–191. doi: 10.1096/fasebj.8.2.8119489. [DOI] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HG, MacLeod RAF, Borkhardt A, Janssen JWG. Recurrent chromosomal translocations and fusion genes in leukemia-lymphoma cell lines. Leukemia. 1995;9:480–500. [PubMed] [Google Scholar]
- Durfee T, Becherer K, Chen P-L, Yeh S-H, Kilburn AE, Lee W-H, Elledge SJ. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes & Dev. 1993;7:1–12. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Edgley ML, Turner CA, Riddle DL. On-line C. elegans resources. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 1059–1062. [Google Scholar]
- Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RMR, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4, Skp1, and CDC53/Cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O-k. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gaudet R, Bohm A, Sigler PB. Crystal structure at 2.4 A resolution of the complex of transducin βγ and its regulator, phosducin. Cell. 1996;87:577–568. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- Goebl MG, Yochem J, Jentsch S, McGrath JP, Varshavsky A, Byers B. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science. 1988;241:1331–1335. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- Grant B, Greenwald I. Structure, function and expression of SEL-1, a negative regulator of LIN-12 and GLP-1 in C. elegans. Development. 1997;124:637–644. doi: 10.1242/dev.124.3.637. [DOI] [PubMed] [Google Scholar]
- Greenwald I. The development of the vulva. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 519–542. [PubMed] [Google Scholar]
- Greenwald I, Seydoux G. Analysis of gain-of-function mutations of the lin-12 gene of Caenorhabditis elegans. Nature. 1990;346:197–199. doi: 10.1038/346197a0. [DOI] [PubMed] [Google Scholar]
- Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell. 1983;34:435–444. doi: 10.1016/0092-8674(83)90377-x. [DOI] [PubMed] [Google Scholar]
- Han M, Sternberg PW. Analysis of dominant-negative mutations of the Caenorhabditis elegans let-60 ras gene. Genes & Dev. 1991;5:2188–2198. doi: 10.1101/gad.5.12a.2188. [DOI] [PubMed] [Google Scholar]
- Heitzler P, Simpson P. The choice of cell fate in the epidermis of Drosophila. Cell. 1991;64:1083–1092. doi: 10.1016/0092-8674(91)90263-x. [DOI] [PubMed] [Google Scholar]
- Hodgkin J. Genetics. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 881–1048. [Google Scholar]
- Kaltenboeck B, Spatafora JW, Zhang X, Kousoulas KG, Blackwell M, Storz J. Efficient production of single-stranded DNA as long as 2 kb for sequencing of PCR-amplified DNA. BioTechniques. 1992;12:164–171. [PubMed] [Google Scholar]
- Kimble J. Alteration in cell lineage following laser ablation of cells in the somatic gonad of Caenorhabditis elegans. Dev Biol. 1981;87:286–300. doi: 10.1016/0012-1606(81)90152-4. [DOI] [PubMed] [Google Scholar]
- Kimble J, Hirsh D. The post-embryonic cell lineages of the hermaphrodites and male gonads in Caenorhabditis elegans. Dev Biol. 1979;87:396–417. doi: 10.1016/0012-1606(79)90035-6. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peteres J-M, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1658. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Paietta JV. The sulfur controller-2 negative regulatory gene of Neurospora crassa encodes a protein with beta-transducin repeats. Proc Natl Acad Sci. 1995;92:3343–3347. doi: 10.1073/pnas.92.8.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Mathias N, Johnson SL, Winey M, Adams AEM, Goetsch L, Pringle JR, Byers B, Goebl MG. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S phase transition and identifies a conserved family of proteins. Mol Cell Biol. 1996;16:6634–6643. doi: 10.1128/mcb.16.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- Natorff R, Balinska M, Paszewski A. At least four regulatory genes control sulphur metabolite repression in Aspergillus nidulans. Mol Gen Genet. 1993;238:185–192. doi: 10.1007/BF00279546. [DOI] [PubMed] [Google Scholar]
- Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- Nellen D, Burke R, Struhl G, Basler K. Direct and long range action of a DPP morphogen gradient. Cell. 1996;85:357–368. doi: 10.1016/s0092-8674(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Baltimore D. Production of high-titre helper-free retroviruses by transient transfection. Proc Natl Acad Sci. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, Blondel BJ, Gallahan D, Callahan R. Mouse mammary tumor gene int-3: A member of the Notch gene family transforms mammary epithelial cells. J Virol. 1992;66:2594–2599. doi: 10.1128/jvi.66.4.2594-2599.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- Roth MB, Zahler A, Stolk JH. A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J Cell Biol. 1991;115:587–596. doi: 10.1083/jcb.115.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Seydoux G, Greenwald I. Cell autonomy of lin-12 function in a cell fate decision in C. elegans. Cell. 1989;57:1237–1245. doi: 10.1016/0092-8674(89)90060-3. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Crystal structure of a GA protein βγ dimer at 2.1 A resolution. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- Struhl G, Fitzgerald K, Greenwald I. Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Greenwald I. Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics. 1993;135:765–783. doi: 10.1093/genetics/135.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Kuras L, Barbey R, Cherest H, Blaiseau PL, Surdin-Kerjan Y. Met30p, a yeast transcriptional inhibitor that responds to S-adenosylmethionine, is an essential protein with WD40 repeats. Mol Cell Biol. 1995;15:6526–6534. doi: 10.1128/mcb.15.12.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck S, Greenwald I. lin-25, a gene required for vulval induction in Caenorhabditis elegans. Genes & Dev. 1995;9:341–357. doi: 10.1101/gad.9.3.341. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development. 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes BJ, Cado D, Robey E. Notch activity influences the ab versus gd T cell lineage. Cell. 1997;88:833–843. doi: 10.1016/s0092-8674(00)81929-7. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Sulston JE, Coulson AR. The genome. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 23–46. [PubMed] [Google Scholar]
- Wilkinson HA, Fitzgerald K, Greenwald I. Reciprocal changes in expression of the receptor lin-12 and its ligand lag-2 prior to commitment in a C. elegans cell fate decision. Cell. 1994;79:1187–1198. doi: 10.1016/0092-8674(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Yochem J, Greenwald I. glp-1 and lin-12, genes implicated in distinct cell-cell interactions in Caenorhabditis elegans, encode similar transmembrane proteins. Cell. 1989;58:553–563. doi: 10.1016/0092-8674(89)90436-4. [DOI] [PubMed] [Google Scholar]