Translation Initiator EIF4G1 Mutations in Familial Parkinson Disease (original) (raw)
Abstract
Genome-wide analysis of a multi-incident family with autosomal-dominant parkinsonism has implicated a locus on chromosomal region 3q26-q28. Linkage and disease segregation is explained by a missense mutation c.3614G>A (p.Arg1205His) in eukaryotic translation initiation factor 4-gamma (EIF4G1). Subsequent sequence and genotype analysis identified EIF4G1 c.1505C>T (p.Ala502Val), c.2056G>T (p.Gly686Cys), c.3490A>C (p.Ser1164Arg), c.3589C>T (p.Arg1197Trp) and c.3614G>A (p.Arg1205His) substitutions in affected subjects with familial parkinsonism and idiopathic Lewy body disease but not in control subjects. Despite different countries of origin, persons with EIF4G1 c.1505C>T (p.Ala502Val) or c.3614G>A (p.Arg1205His) mutations appear to share haplotypes consistent with ancestral founders. eIF4G1 p.Ala502Val and p.Arg1205His disrupt eIF4E or eIF3e binding, although the wild-type protein does not, and render mutant cells more vulnerable to reactive oxidative species. EIF4G1 mutations implicate mRNA translation initiation in familial parkinsonism and highlight a convergent pathway for monogenic, toxin and perhaps virally-induced Parkinson disease.
Introduction
Parkinson disease (PD [MIM 168600]) is characterized clinically by asymmetric resting tremor, bradykinesia, muscle rigidity, and postural instability.1 Dopaminergic loss and Lewy bodies in surviving neurons of the substantia nigra support a pathologic diagnosis.2 Although considered a sporadic illness, 10%–30% of individuals with PD report a first-degree relative with parkinsonism.3 Linkage and sequence analyses performed in multi-incident families with parkinsonism have discovered deleterious mutations in α-synuclein (SNCA [MIM 163890]), leucine-rich repeat kinase 2 (LRRK2 [MIM 609007]), vesicular protein sorting 35 (VPS35 (MIM 601501]), Parkin (PARK2 [MIM 602544]), PTEN induced putative kinase 1 (PINK1 [MIM 608309]), DJ-1 (PARK7 [MIM 602533]) and ATP13A2 (PARK9 [MIM 610515]).4,5 Although familial parkinsonism attributed to mutant genes is uncommon, the molecular etiology discovered might be generalizable to idiopathic PD. For instance, antibodies to α-synuclein robustly stain Lewy bodies and neuritic pathology,6 whereas mutations in Parkin and PINK1 have emphasized the role of stress-induced mitochondrial dysfunction in PD.7
We have performed genome-wide linkage analysis of a multi-incident northern French family (P30; Figure 1A) with autosomal-dominant late-onset parkinsonism for which known genetic causes of parkinsonism were excluded (unpublished data).8 Linkage analysis identified two regions with suggestive two-point LOD scores > 2 (θ = 0), a 31 cM interval between D3S1763 and D3S1580, and a 15 cM interval between D5S2055 and D5S393. After saturation of both loci with short tandem repeat (STR) markers, significant linkage was obtained only for chromosomal region 3q26-q28. Genomic analysis subsequently led to the identification of eukaryotic translation initiation factor 4-gamma 1 (EIF4G1) mutations (protein eIF4G1 [MIM 600495]) in affected subjects with familial parkinsonism and idiopathic Lewy body disease. eIF4G1 is a component of the translation initiation complex, eIF4F.9,10 We demonstrate that the two most frequent mutations impair complex formation, consistent with a dominant-negative loss of function, and are associated with mitochondrial dysfunction when cells are stressed.
Figure 1.

Pedigrees with EIF4G1 Mutations and Parkinsonism
Individual pedigrees are numbered and their country of origin is indicated. Only pedigrees with an (A) EIF4G1 c.3614G>A (p.Arg1205His), (B) c.1505C>T (p.Ala502Val), (C) c.2056G>T (p.Gly686Cys), (D) c.2056G>T (p.Gly686Cys) and c.3589C>T (p.Arg1197Trp) and (E) c.3490A>C (p.Ser1164Arg) mutations are illustrated. Filled symbols indicate affected individuals with parkinsonism; the age of symptom onset is shown in parentheses. Quarter-filled symbols indicate individuals with dementia. To protect confidentiality, the pedigrees do not show some individuals and siblings and/or gender is sometimes disguised with a diamond. All samples genotyped are indicted as heterozygous mutations (M) or as wild-type (wt). The asterisk (∗) indicate that this individual did not fulfill the criteria for PD, but presented resting tremor and akinesia.
Subjects and Methods
Subjects Ascertainment
The institutional review boards of all participating institutions approved the protocol (institutional review boards France CPP 94/07), and informed consent was obtained from all affected and control subjects. Participating individuals were examined by neurologists specializing in movement disorders. A full history, including a family history and a neurological examination, was completed for each patient. A clinical diagnosis of PD was determined by the presence of at least two of three cardinal signs (resting tremor, bradykinesia, and rigidity), improvement through adequate dopaminergic therapy (when tried), and the absence of atypical features or other causes of parkinsonism. Clinical criteria for probable and possible PD were consistent with former classifications.1,2 Familial history is defined herein as one or more affected relatives within two degrees of relationship.
Linkage Analysis
Peripheral blood samples were collected and genomic DNA was extracted with standard techniques. Tri- and tetra-nucleotide repeat genome-wide genotyping was completed by the mammalian genotyping service of Marshfield in family P30, for 403 markers at approximately 10 cM resolution. Linkage analysis employed MLINK with a dominant model for two-point LOD scores and SIMWALK2 for nonparametric statistics and haplotype analysis.11,12 The frequency of the deleterious allele was set at 0.0001; marker-allele frequencies were from CEPH or determined empirically. The map positions for each marker were taken from Marshfield and Rutgers combined linkage physical map.13 For tightly linked loci with no observed recombinants the intermarker genetic distances were assigned as 0.01 cM.
Sequencing Analysis and Mutation Screening
Gene sequencing of all coding exons in the 3q26-q28 interval was performed for the three affected members of family P30 (individuals with symptom onset of 61, 62, and 56 years [Figure 1A, III-2, III-3, III-5]) with an ABI 3730 sequencer with SeqScape v2.5 analysis software (Applied Biosystems). Electrophoregrams were compared with CEPH 1331-01 and -02 control subjects obtained from Genethon and the human reference sequence from the UCSC database. Primers designed for amplification of all exons including exon-intron junctions of EIF4G1 are provided in Table S1, available online. Mutations segregating with the disease were further assessed in the other P30 members (n = 23), in control subjects of the northern French (n = 146), and in other subjects of European descent (n = 370). RefSeq accessions NM_198241.2 and NP_937884.1 were used to number all variants within the EIF4G1 gene and protein.
Further sequencing of all EIF4G1 coding exons was then performed to identify other mutations in additional subjects affected with autosomal-dominant parkinsonism (n = 95) or neuropathologically confirmed Lewy body disease (n = 130) and ethnically matched controls (n = 185).
In order to test whether EIF4G1 missense mutations might be found in idiopathic PD, we genotyped a large case-control series consisting of 4430 affected and 3671 control subjects of European descent (North America: 2092 PD [130 Lewy body disease], 1666 controls [provided by Z.K.W., R.J.U., D.W.D., R.F., D.M.M., PROGENI]; Norway: 775 PD, 614 controls [J.O.A.]; France: 574 PD, 469 controls [M.-C.C.-H., A.D.]; Ireland: 400 PD, 460 controls [D.G., T.L., M.H.]; Poland: 397 PD, 366 controls [A.K.-W., G.O.]; Italy: 192 PD, 96 controls [A.R.B., E.M.V.]) and 278 affected and 379 control subjects of Arab-Berber ethnicity from Tunisia [F.H., R.A.G.]. Genotyping was performed with the Sequenom MassArray iPLEX platform (San Diego, CA). All primer sequences are available on Table S1. All mutations discovered were confirmed by direct sequencing.
Chromosome 3 Haplotype Analysis
STR and SNP markers were chosen to span the EIF4G1 locus, including D3S3037, Chr3_179.425, D3S3730, D3S3699, Chr3_182.108, D3S2312, D3S2314, Chr3_184.176, Chr3_184.543, Chr3_184.806, D3S1571, Chr3_185.212, Chr3_185.320, Chr3_185.398, Chr3_185.425, Chr3_185.464, D3S3609, Chr3_185.526, D3S3578, Chr3_185.619, Chr3_185.633, Chr3_185.653, Chr3_185.692, D3S3583, Chr3_185.822, Chr3_185.860, Chr3_185.870, D3S3592, Chr3_186.199, D3S1530, D3S1262, D3S2436, D3S3686, D3S3651, and D3S1580 (Table S2). Eighteen of these STR markers are novel (prefixed with Chr3_ as “D segment” numbers are no longer available) and were designed by searching for repeat polymorphisms in 6.77 Mb of genomic sequence spanning the locus. One primer of each pair was labeled with a fluorescent tag; amplified sequences are available on Table S1. PCR reactions were performed under standard conditions and products were run on an ABI 3730 genetic analyzer. Results were analyzed with GeneMapper 4.0 software (Applied Biosystems). Five SNPs located in EIF4G1, rs4912537, rs2178403, rs2293605, rs1879244 and rs2230571, were included. Marker-allele frequencies for the European population were obtained from the CEPH genotype database, the Human HapMap database, or estimated by genotyping 100 unrelated healthy subjects from North America of European descent. NCBI build 36.1 is referenced throughout.
Testing for EIF4G1 Copy-Number Variation
To determine whole-gene copy number, primer pairs, and probes were designed against exon 10, 18, and 24 of EIF4G1. An endogenous control assay was designed against exon 5 of the presenilin 2 gene. All primers and probes were purchased from Applied Biosystems and sequences are available on Table S1. Quantitative PCR was carried out with TaqMan expression chemistry protocol; 25 ng genomic DNA was amplified with 0.25 μl primer probe and 2.5 μl TaqMan 2X Universal PCR Master Mix (Applied Biosystems). The thermal cycle conditions were performed at 50°C for 2 min and at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s for denaturation and at 60°C for 1 min for annealing and extension. All assays were performed in triplicate on the ABI 7900HT Fast Real-Time PCR System and analyzed with ABI SDS 2.2.2 Software (Applied Biosystems).
EIF4G1 Cloning and Coimmunoprecipitation Studies of Protein-Complex Binding Partners
Full-length EIF4G1 wild-type (WT) and a dominant negative mutant were amplified from plasmids as previously described14 and subcloned into pcDNA6.2-V5 and pcDNA6.2/C-EmGFP-Dest expression vectors. The eIF4G1 p.Ala502Val and p.Arg1205His mutations were subsequently introduced with site-specific mutagenesis (primers available in Table S1). HEK293T cells, maintained in Opti-mem 1+GlutaMAX1 with 10% of fetal calf serum and penicillin/streptomycin (all GIBCO) at 37°C and 5% CO2, were transfected with cDNA encoding the respective WT or mutant eIF4G1-V5 proteins with Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Cells were harvested 48 hr after transfection and lysed in 50 mM Tris/HCL, 150 mM NaCl, and 0.1% Triton X-100. To avoid unspecific binding during immunoprecipitation, we precleared crude lysates by rotating them at 4°C for 1 hr with Protein A/G ultralink resin (Thermo Scientific) followed by centrifugation at 13,000 × g for 10 min. After performing a BCA Protein assay (Pierce), equal amounts of supernatant were combined with protein G Dyna beads (Invitrogen) preconjugated with monoclonal mouse anti-V5 antibodies (Invitrogen) and rotated on a spinning wheel for 4 hr at 4°C. The resulting immunocomplex was stringently washed with intraperitoneal (IP) buffer/PBS and eluted in SDS-Sample buffer (Invitrogen). Immunoprecipitated and coprecipitated proteins were analyzed with SDS PAGE/immunoblot technologies. For protein detection, monoclonal anti-V5 (Invitrogen) and polyclonal rabbit eIF4E, eIF3e, and eIF4A (Abcam) were applied. ImageJ was used to quantify the relative amounts of copurified interactors eIF4A1, eIF4E, and eIF3e, taking into account the amounts in the full lysates and the amount of immunoprecipitated eIF4G1 proteins. One-way ANOVA was applied for statistical analysis.
Assessment of Mitochondrial Function by Flow Cytometry
HEK293T cells were transfected with plasmids encoding GFP-tagged eIF4G1 proteins (WT, p.Ala502Val, and p.Arg1205His) and subsequently subjected to viability studies with fluorescence activated cell sorting (FACS) analysis. Incorporation of the mitochondrial-permeable dye tetramethyl rhodamine ethyl ester (TMRE; Invitrogen) has been shown to reflect mitochondrial activity and was used as an indicator for cellular viability. Cells, maintained at 37°C in Opti-mem 1+GlutaMAX1 with 10% fetal calf serum medium (both GIBCO), were seeded at a density of 150,000 cells/6-well and transfected 24 hr prior to FACS analysis. Duplicates of each condition were treated with either 0 or 0.5 mM H2O2 for 6 hr. Labeling with 100 nM TMRE was performed by directly adding TMRE from a DMSO stock solution (0.2 mM) to the growth medium and incubating for 30 min at 37°C. After two washing steps with warm PBS containing 1% FBS, cells were trypsinized, resuspended, and immediately subjected to FACS analysis. We established specific settings for the separation into transfected (GFP positive) and untransfected (GFP negative) as well as viable (TMRE positive) and TMRE negative cells by using appropriate controls including (1) untransfected, no TMRE; (2) transfected, no TMRE; (3) untransfected, TMRE stained; and (4) transfected, TMRE stained samples. For each sample, 100,000 events were acquired and subsequently analyzed. After removal of background counts and the exclusion of untransfected (GFP negative) cells, average TMRE loading values were based on at least 25,000 cells. The number of TMRE positive cells were calculated as the percentage of the total number of GFP positive cells in the respective sample.
Results
Genetic and Physical Mapping
Genome-wide analysis of the P30 pedigree led us to identify two regions with suggestive two-point LOD scores > 2 (θ = 0), a 31 cM interval between D3S1763 and D3S1580, and a 15 cM interval between D5S2055 and D5S393. The two loci on chromosomes 3 and 5 were saturated with STRs at <2.0 cM resolution to maximize genetic information, and genotype segregation with disease was reanalyzed with additional DNA samples from family P30. Significant linkage was obtained only for chromosomal region 3q26-q28 (mLOD = 3.01) with a haplotype shared by all affected relatives.
Identification of an EIF4G1 Mutation in Family P30
In silico analysis revealed that the critical interval contained 159 genes (1432 exons). We sequenced each of these along with their donor and acceptor splice sites by using genomic DNA samples from three affected first cousins of P30 (Figure 1A; III-2, III-3, III-5). We found 236 known SNPs and 33 novel variants with only five that were present in all three affected family members sequenced; three intronic variants were located more than 20 bases from exon-intron boundaries. Of those novel variants, only EIF4G1 mutation c.3614G>A (p.Arg1205His) was found to segregate with disease in all ten blood-related, affected family members (family P30 two-point LOD = 3.55, θ = 0). The mutation was absent in 146 unrelated control subjects from the same ethnical and geographical origin and in 370 unrelated North American control subjects of European descent. Because chromosome 3q26-q28 is a fragile site in cancer and genomic multiplications are associated with malignancy,10 probes in EIF4G1 were used to grossly assess copy number variants but none were found (n = 225 affected subjects).
eIF4G1 c.3614G>A is located in exon 24 of a 33 exon gene. It predicts the substitution of a conserved arginine to histidine at residue 1205 (p.Arg1205His) of the protein. A cross-species alignment of the eIF4G1 demonstrated the p.Arg1205His is highly conserved across species (Figure S1). In silico analysis predicts the mutation might be damaging to protein function (Polyphen-2 PSIC score = 0.99).
Screening of EIF4G1 c.3614G>A (p.Arg1205His) in Individuals with PD
To further investigate the pathogenicity of the EIF4G1 c.3614G>A (p.Arg1205His), we genotyped 4050 control subjects and 4708 individuals with idiopathic PD. We identified nine affected heterozygotes from seven families originating from the US (385-1493, 2498), Canada (2499), Ireland (32150- 50), Italy (GE0925 and GE1843), and Tunisia (Tun83) (Figure 1A). A family history of parkinsonism was not evident in the Irish (32150-50), one Italian (GE0925), or the Tunisian (Tun83) subjects. In contrast, eIF4G1 p.Arg1205His was absent in control subjects.
To determine whether the EIF4G1 c.3614G>A (p.Arg1205His) mutation segregates with disease in these seven cases, we genotyped 42 STRs distributed across the 3q26-q28 locus flanking EIF4G1 c.3614G>A (Figure 2A; Table S2A). Haplotype analysis shows that each affected member of all seven families shares a minimal interval of 45,676bp (chr3:185,521,663-185,567,338) in common with family P30. With the exception of EIF4G1 c.3614G>A (p.Arg1205His) no other novel coding variant segregates with the disease in this shared haplotype, in which linkage disequilibrium between neighboring variants is marginal (Figure 2B). The results suggest the deleterious EIF4G1 c.3614G>A (p.Arg1205His) mutation originates from an ancestral founder and segregates with disease in seemingly unrelated families.
Figure 2.
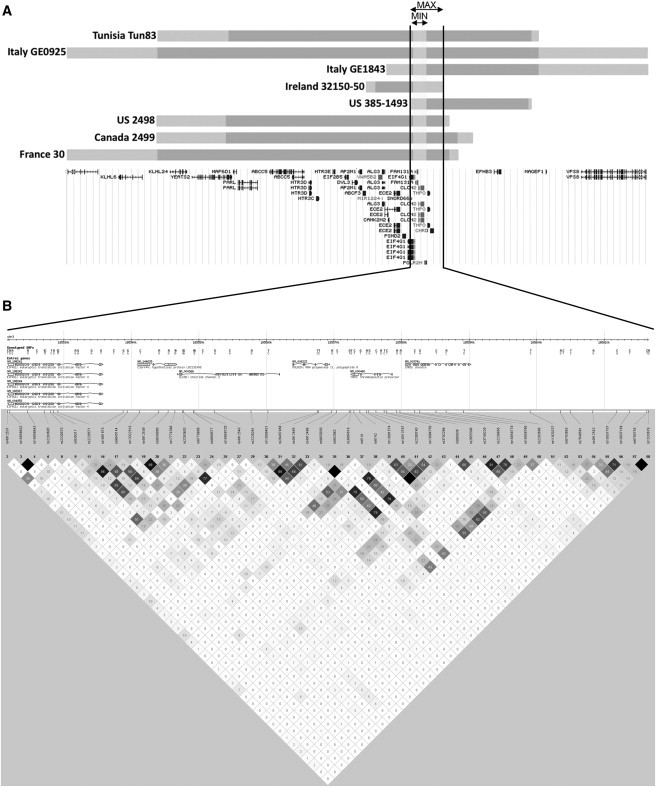
Disease-Segregating EIF4G1 c.3614G>A Haplotypes among Families
(A) Comparison of haplotype data among all families with EIF4G1 mutations shows alleles between rs4912537 and D3S3578 are shared (Table S2). The maximum shared interval is 99,330 bp (MAX; chr3:185,519,706–185,619,035; gray and blue bars) and contains six gene transcripts, EIF4G1, FAM131A, CLCN2, POLR2H, THPO, and CHRD. The minimal shared interval is 45,676 bp (MIN; chr3:185,521,663-185,567,338; gray) and harbors four genes (EIF4G1, FAM131A, CLCN2, and POLR2H). EIF4G1 c.3614G>A (p.Arg1205His) is the only novel coding variant that segregates with disease.
(B) The CEU haplotype map suggests negligible linkage disequilibrium across the rs4912537–D3S3578 region.
Screening for Novel EIF4G1 Variants in Other Families
To further evaluate the EIF4G1 mutations in PD, the 31 coding exons were sequenced in 95 randomly selected affected probands with autosomal-dominant parkinsonism, 130 pathologically-defined cases with Lewy body disease, and 185 ethnically matched controls. Sequence analysis identified eight novel coding variants in affected subjects (4 missense, 4 silent), three novel changes in controls (two missense, one silent), and three novel coding variants in affected and control subjects (Table S3). The four missense mutations not observed in controls were c.1505C>T (p.Ala502Val), c.2056G>T (p.Gly686Cys), c.3490A>C (p.Ser1164Arg), and c.3589C>T (p.Arg1197Trp) (Table S3). They were detected in two affected probands with parkinsonism (US 623, US 385-1391) and two autopsied cases with Lewy body disease (US 331-5, US 331-95) (Figure 1B).
These four variants were subsequently genotyped in a case-control series consisting of 4483 individuals with idiopathic PD and 3865 age, gender, and ethnically matched control subjects. Screening identified only affected heterozygotes p.Ala502Val (US 385-284 and Poland 1736-155) and p.Gly686Cys (2 with PD in the family French P141), whereas the p.Ser1164Arg and p.Arg1197Trp mutations were not observed again in affected or control subjects.
Consistent with eIF4G1 p.Arg1205His, the p.Gly686Cys, p.Ser1164Arg, and p.Arg1197Trp mutations are also evolutionarily conserved in mammals (Figure S1). Similarly, eIF4G1 p.Ala502Val is conserved in most mammals with the exception of the rabbit that has a valine at this position. Limited cross-species conservation typically argues against pathogenicity, but the first alpha-synuclein p.Ala53Thr mutation linked to PD is a notable precedent.15 Assessment of segregation with disease was not possible for p.Ala502Val, p.Gly686Cys, p.Ser1164Arg, and p.Arg1197Trp mutations, and as a consequence genetic evidence for pathogenicity is limited. STR marker analysis of chromosomal region 3q26-q28 in all heterozygotes with p.Ala502Val suggested an ancestral founder, although the phase was only available for one pedigree (Table S2A).
Clinical Findings and Neuropathological Analyses
Within family P30, the clinical phenotype among EIF4G1 c.3614G>A (p.Arg1205His) heterozygotes is consistent with late-onset PD, with a mean onset of 64 ± 10.3 standard deviation (SD) years (n = 10), but spans a broad range (50–80 years) (Figure 1A). Symptoms start insidiously with asymmetric resting tremor or akinetic rigidity and become progressively mixed. The parkinsonism has a relatively long, mild course, and cognition seems preserved. Patients' symptoms respond well to L-DOPA, but many remain untreated or use only dopamine agonists. In clinically symptomatic individuals' dopaminergic imaging with I-123 Ioflupan, DaTSCAN is abnormal and asymmetric.
The clinical phenotype among unrelated subjects with a heterozygous EIF4G1 c.3614G>A (p.Arg1205His) mutation was comparable to family P30 with a mean symptom onset of 59 ± 13.0 SD years (n = 9), range 32–77 years (Figure 1A). Overall, affected subjects with an EIF4G1 c.1505C>T (p.Ala502Val) mutation have a mean age of symptom onset of 52 ± 8.1 SD years (range 41–58 years, n = 4) (Figure 1B).
The ages at onset of subjects who have PD with other EIF4G1 substitutions (c.2056G>T [p.Gly686Cys] and c.3490A>C [p.Ser1164Arg]) were 55, 62, and 64, respectively, in the France P141 and US 385-1391 families. EIF4G1 mutations were also observed in two autopsy-confirmed cases with Lewy body disease: an EIF4G1 c.1505C>T (p.Ala502Val) mutation in family US 331-5 and the double EIF4G1 mutant c.2056G>T (p.Gly686Cys) and c.3589C>T (p.Arg1197Trp) in family US 331-95 (Figures 1B and 1D). The Lewy body pathology in these cases is presented in Figure S2.
Functional Analyses of eIF4G1 p.Arg1205His and p.Ala502Val Substitutions
eIF4G1 p.Arg1205His is just distal to the putative eIF3e binding domain (amino acids 1015–1118; Figure S3), a key partner of the translation initiation complex that recruits the 40 S ribosomal subunit.16–18 Thus, we evaluated WT and mutant eIF4G1 binding to eIF3e. Plasmids containing eIF4G1 WT or eIF4G1 p.Arg1205His or eIF4G1 LacZ were transfected into HEK293 cells. Coimmunoprecipitation studies of protein lysates from mutant eIF4G1-transfected cells show that p.Arg1205His perturbs eIF3e binding, but the WT protein does not (Figure 3).
Figure 3.
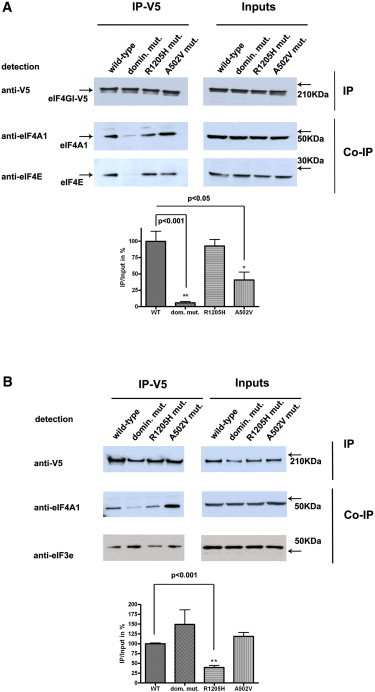
Coimmunoprecipitation Studies of eIF4G1 p.Ala502Val and p.Arg1205His with eIF4A and eIF3e
Coimmunoprecipitation (CO-IP) of endogenous eIF4A, eIF4E, and eIF3e with transfected full-length eIF4G1-V5 (WT, a dominant negative mutant,13 or the p.Ala502Val and p.Arg1205His mutants) in HEK293T cells.
(A) The p.Ala502Val mutant perturbs the interaction with eIF4E but not eIF4A (IP-V5). The inputs on the upper right panel represent the amounts of transfected eIF4G1-V5 (WT and mutants) and the endogenous proteins (eIF4A1 or eIF4E) introduced in the immunoprecipitation assay.
(B) Coimmunoprecipitation of eIF3e with eIF4G1-V5 (IP left; inputs right). In the presence of the p.Arg1205His mutation significantly reduced amounts of eIF3e protein are coprecipitated with eIF4G1-V5. Experiments were performed on three separate occasions as depicted in the graphs. Representative immunoblots are shown.
eIF4G1 p.Ala502Val is the most frequent substitution encountered in other familial cases, although genetic evidence for segregation with disease and thus pathogenicity is limited. Its physical location predicts that the substitution might perturb eIF4E binding (amino acids 612-618). Coimmunoprecipitation studies of protein lysates from mutant eIF4G1 transfected cells show p.Ala502Val perturbs eIF4E binding compared to WT protein (Figure 3). Both p.Ala502Val and p.Arg1205His are located within or proximal to the eIF4A-interacting domains (amino acids 761-988 and 1240-1450) but do not appear to affect its binding, although a nonsignificant trend toward increased binding was observed for p.Ala502Val.
When TMRE, a marker for mitochondrial membrane polarization, was applied to either WT or mutant (p.Ala502Val; p.Arg1205His) eIF4G1 transfected cells, no differences could be detected under basal conditions.10 However, with hydroperoxide treatment, a profound loss in mitochondrial membrane potential was observed for both mutations but not for cells overexpressing WT protein (Figure 4). Complementary results were obtained with patient-derived lymphoblastoid cell lines (data not shown). When starvation was used as an alternate stressor, the induction of autophagy was comparable in WT and mutant (p.Ala502Val; p.Arg1205His) overexpressing cells. The lack of response might reflect the presence of endogenous eIF4G1 (Figure S4).9
Figure 4.

Fluorescence-Activated Cell-Sorting Analysis of Mutant and Control Cell Lines HEK293T Cells Transiently Overexpressing WT or Mutant EIF4G1 (p.Ala502Val, p.Arg1205His)
eIF4G1-GFP proteins were subjected to FACS analysis, with TMRE labeling applied as a marker of mitochondrial polarization. Untreated eIF4G1-GFP-positive cells are comparable in terms of TMRE loading. In contrast, with hydrogen peroxide treatment p.Ala502Val- and p.Arg1205His eIF4G1-GFP-positive cells show a 10% decrease in cell survival compared to WT cells. For each sample, 100,000 events were acquired and subsequently analyzed. After the removal of background counts and the exclusion of untransfected (GFP negative) cells, average TMRE loading values were based on at least 25,000 cells. The number of TMRE-positive cells was calculated as a percentage of the total number of GFP-positive cells in the respective sample. Experiments with two duplicates per condition were performed on three separate occasions.
Discussion
eIF4G1 is the core scaffold of a multisubunit translation initiation complex that regulates the translation initiation of mRNAs encoding mitochondrial, cell survival and growth genes in response to different stresses.9,10 The pathogenicity of EIF4G1 mutations is supported by (1) the segregation of eIF4G1 p.Arg1205His with disease in a family with multi-incident autosomal-dominant, parkinsonism; (2) the absence of eIF4G1 p.Ala502Val and p.Arg1205His in control subjects (n = 4050), although observed in several multi-incident families with autosomal-dominant late-onset PD; (3) an eIF4G1 p.Arg1205His haplotype suggestive of one ancestral founder, and de-limiting the candidate gene interval; (4) the sequence conservation of eIF4G1p.Ala502Val and p.Arg1205His substitutions suggesting these mutations might be functionally deleterious; and (5) impaired binding of eIF4G1 p.Ala502Val and p.Arg1205His to eIF4E or eIF3e, interactions that are normally required for translation initiation.17,18 Both p.Ala502Val and p.Arg1205His directly impair formation of the larger eIF4 complex and support the genetic argument for a dominant-negative loss-of-function compatible with an age-dependent neurodegenerative disorder; (6) the loss of mitochondrial membrane potential and viability in transfected cells subjected to oxidative stress. The functional data presented are cursory and further analyses of mitochondrial biogenesis, autophagy, and transcription and translation, in additional models is warranted. A caveat of linkage within a multi-incident family is that there remains a remote possibility of a missing segregating variant, most likely within the disease-segregating haplotype. Although rare, additional EIF4G1 mutations in other families will be important to discover.
EIF4G1 mutations directly implicate mRNA translation initiation in parkinsonism and might help unify other monogenic forms, toxin, and perhaps virally-induced disease within a convergent pathway.19,20 Availability of eIF4E is generally the rate-limiting step of translation initiation and is largely determined by phosphorylation of eIF4E-binding proteins (4E-BP) through the mammalian target of the rapamycin (mTOR) pathway.9,21 Constitutive activity of mTOR signaling normally leads to phosphorylation of 4E-BPs; this dislodges eIF4E and enables assembly of the eIF4F complex to promote cap-dependent translation (Figure S3).18 Notably, 4E-BP is a substrate of human Lrrk2 and the Drosophila ortholog (Lrrk).22 Lrrk2 pathogenic mutations cause hyperphosphorylation of 4E-BP leading to reduced oxidative stress resistance and dopaminergic neurodegeneration. Conversely, inhibition of mTOR signaling during development or overexpression of 4E-BP in Drosophila mutants with PINK1 or Parkin loss-of-function suppresses the flies' pathologic phenotypes.23
The variability in clinical and pathologic presentation of affected members of EIF4G1 families is similar to genetically-defined parkinsonism but most consistent with idiopathic, late-onset Lewy body PD. Although EIF4G1 mutations represent a molecular mechanism for PD, genetic defects in translation have been implicated in many neurological diseases.24 Most are private mutations in orphan families. Nevertheless, the three-fold increase in rare EIF4G1 coding variants in PD compared to control subjects warrants further study. Common polymorphic variability in the EIF4G1 locus was not associated with PD in case-control samples.
These data suggest that eIF4G1 p.Ala502Val and p.Arg1205His substitutions affect scaffold function and consequently impair the ability of cells to rapidly and dynamically respond to stress, presumably through changes in the translation of existing mRNAs essential to cell survival.9,10 Perturbations in mTOR/4E-BP signaling and eIF4G1-mediated changes in mRNA translation will be important to elucidate in neurons in vivo. The pathway highlighted might represent a central theme in PD for which targeted interventions already have therapeutic potential.25
Acknowledgments
We thank subjects who generously participated in this study. We also thank the many clinicians who have contributed to the follow-up of family P30 over the past 15 years, including Estelle Becquet, Serge Brique, Delphine Prince, Xavier Douay, and Nawal Waucquier. We acknowledge the laboratory support of Clotilde Levecque, Vincent Mouroux, Pierre Semaille, and Trevor Tyson. Studies in France would not have been possible without financial support from CHR de Lille, Université Lille 2, Inserm, French Ministry Hospital Clinical Research Programs (PHRCs) (1994/, 2002/1918, 2005/1914), Association France Parkinson (2005), Fondation de France 2004-013306, 2011-00016815, Fondation de la Recherche Médicale (2006), PPF (synucléothèque 2005-2009), the two Centres de Resources Biologiques (IPL-Lille, CHRU-Lille) and their scientific committee (A.D., M.-C.C.-H., Philippe Amouyel, Florence Pasquier, Régis Bordet). Studies in Ireland were supported by the Program for Research in Third Level Institutions (PRTLI). Studies in Italy were supported by the Italian Ministry of Health (Ricerca Corrente 2011, Ricerca Finalizzata, Giovani Ricercatori). Studies in the PROGENI sample were supported by R01NS37167 and the Parkinson Study Group. Clinicogenetic studies in Tunisia were funded in part by the Institute of Neurology, Tunis, and the Michael J. Fox Foundation. GlaxoSmithKline (GSK) funded and provided a subset of the Tunisian case:control samples and associated phenotypic data, and we gratefully acknowledge the PD Programme Team, most notably Tina Stapleton and Carole Stapleton for sample management. We thank James Weber and NIH National Heart, Lung, and Blood Institute/Marshfield Medical Research Foundation for genome-wide genotyping (genotyping award to M.J.F.). Studies performed at Mayo Clinic were financed by NIH National Institute of Neurological Disorders and Stroke (Morris K. Udall Parkinson's Disease Research Center of Excellence; P50 #NS40256 #NS072187; M.J.F., Z.K.W., D.W.D.), 2R01 ES10751 (D.M.M.), the Michael J. Fox Foundation (M.J.F.) and a Herb Geist gift for Lewy body research (M.J.F.). We thank Alice McKinney for supplementary Figure S3. This research was undertaken, in part, thanks to funding from the Canada Excellence Research Chairs program (M.J.F.; C.V.-G.). In addition, Leading Edge Endowment Funds provided by the Province of British Columbia, Life Labs and Genome BC support the Dr. Donald Rix BC Leadership Chair (M.J.F.). The authors (J.O.A., D.W.D., M.J.F., D.M.M.) declare provisional patents relevant to Parkinson's disease, but not to EIF4G1. Royalties have been obtained from licensing related to alpha-synuclein (Alnylam Pharmaceuticals [M.J.F., D.M.M.], Isis Pharmaceuticals [M.J.F.]) and leucine-rich repeat kinase 2 (J.O.A., D.W.D., M.J.F., Z.K.W.) but no conflict of interest is declared.
Supplemental Data
Document S1. Four Figures and Three Tables
Web Resources
The URLs for data presented herein are as follows:
- Human Polymorphism Study Center (CEPH) database, http://www.cephb.fr
- dbSNP homepage, http://www.ncbi.nlm.nih.gov/SNP/
- Genethon, http://www.genethon.fr
- Haploview, http://www.broad.mit.edu/mpg/haploview/
- International HapMap Project, http://www.hapmap.org
- MAP-O-MAT genotype database, http://compgen.rutgers.edu/mapomat/
- National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
- Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
- PolyPhen, http://genetics.bwh.harvard.edu/pph2/
- RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/
- UCSC human genome browser, http://genome.cse.ucsc.edu/cgi-bin/hgGateway
Accession Numbers
NCBI accessions NM_198241.2 and NP_937884.1 were used to number all variants within the EIF4G1 gene and eIF4G1protein.
References
- 1.Gelb D.J., Oliver E., Gilman S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 2.Dickson D.W., Braak H., Duda J.E., Duyckaerts C., Gasser T., Halliday G.M., Hardy J., Leverenz J.B., Del Tredici K., Wszolek Z.K., Litvan I. Neuropathological assessment of Parkinson's disease: Refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 3.de Lau L.M., Breteler M.M. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilariño-Güell C., Wider C., Ross O.A., Dachsel J.C., Kachergus J.M., Lincoln S.J., Soto-Ortolaza A.I., Cobb S.A., Wilhoite G.J., Bacon J.A. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 7.Büeler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp. Neurol. 2009;218:235–246. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Farrer M., Destée T., Becquet E., Wavrant-De Vrièze F., Mouroux V., Richard F., Defebvre L., Lincoln S., Hardy J., Amouyel P., Chartier-Harlin M.C. Linkage exclusion in French families with probable Parkinson' s disease. Mov. Disord. 2000;15:1075–1083. doi: 10.1002/1531-8257(200011)15:6<1075::aid-mds1004>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Ramírez-Valle F., Braunstein S., Zavadil J., Formenti S.C., Schneider R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008;181:293–307. doi: 10.1083/jcb.200710215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silvera D., Arju R., Darvishian F., Levine P.H., Zolfaghari L., Goldberg J., Hochman T., Formenti S.C., Schneider R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009;11:903–908. doi: 10.1038/ncb1900. [DOI] [PubMed] [Google Scholar]
- 11.Ott J. Computer-simulation methods in human linkage analysis. Proc. Natl. Acad. Sci. USA. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 13.Kong X., Matise T.C. MAP-O-MAT: Internet-based linkage mapping. Bioinformatics. 2005;21:557–559. doi: 10.1093/bioinformatics/bti024. [DOI] [PubMed] [Google Scholar]
- 14.Yanagiya A., Svitkin Y.V., Shibata S., Mikami S., Imataka H., Sonenberg N. Requirement of RNA binding of mammalian eukaryotic translation initiation factor 4GI (eIF4GI) for efficient interaction of eIF4E with the mRNA cap. Mol. Cell. Biol. 2009;29:1661–1669. doi: 10.1128/MCB.01187-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 16.Prévôt D., Darlix J.L., Ohlmann T. Conducting the initiation of protein synthesis: The role of eIF4G. Biol. Cell. 2003;95:141–156. doi: 10.1016/s0248-4900(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 17.LeFebvre A.K., Korneeva N.L., Trutschl M., Cvek U., Duzan R.D., Bradley C.A., Hershey J.W., Rhoads R.E. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3e subunit. J. Biol. Chem. 2006;281:22917–22932. doi: 10.1074/jbc.M605418200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkes C.H., Del Tredici K., Braak H. Parkinson's disease: The dual hit theory revisited. Ann. N Y Acad. Sci. 2009;1170:615–622. doi: 10.1111/j.1749-6632.2009.04365.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhou C., Huang Y., Przedborski S. Oxidative stress in Parkinson's disease: A mechanism of pathogenic and therapeutic significance. Ann. N Y Acad. Sci. 2008;1147:93–104. doi: 10.1196/annals.1427.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma X.M., Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 22.Gehrke S., Imai Y., Sokol N., Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tain L.S., Mortiboys H., Tao R.N., Ziviani E., Bandmann O., Whitworth A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheper G.C., van der Knaap M.S., Proud C.G. Translation matters: Protein synthesis defects in inherited disease. Nat. Rev. Genet. 2007;8:711–723. doi: 10.1038/nrg2142. [DOI] [PubMed] [Google Scholar]
- 25.Santini E., Heiman M., Greengard P., Valjent E., Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009;2:ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Four Figures and Three Tables