The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms (original) (raw)
Abstract
The SIR genes are determinants of life span in yeast mother cells. Here we show that life span regulation by the Sir proteins is independent of their role in nonhomologous end joining. The short life span of a sir3 or sir4 mutant is due to the simultaneous expression of a and α mating-type information, which indirectly causes an increase in rDNA recombination and likely increases the production of extrachromosomal rDNA circles. The short life span of a sir2 mutant also reveals a direct failure to repress recombination generated by the Fob1p-mediated replication block in the rDNA. Sir2p is a limiting component in promoting yeast longevity, and increasing the gene dosage extends the life span in wild-type cells. A possible role of the conserved SIR2 in mammalian aging is discussed.
Keywords: Aging, yeast, SIR, rDNA, recombination, nonhomologous end-joining
In the budding yeast Saccharomyces cerevisiae, the SIR (silent information regulator) genes serve several functions. SIR1, SIR2, SIR3, and SIR4 were first identified as necessary components for the transcriptional repression of the silent mating type loci, HML and HMR (Ivy et al. 1986; Rine and Herskowitz 1987). Loss of silencing at these loci results in coexpression of a and α mating type genes and sterility in haploid strains. An additional function of SIR2, SIR3, and SIR4 was demonstrated by showing that reporter genes positioned at telomere-proximal sequences exhibit positional effect variegation (PEV) of gene expression (Gottschling et al. 1990). Sir3p and Sir4p can be visualized at telomeric locations microscopically, and sir mutations result in a loss of PEV, telomere shortening, and the constitutive expression of telomeric reporter genes (Aparicio et al. 1991; Palladino et al. 1993).
A growing body of evidence suggests that the Sir proteins are also involved in nonhomologous end joining (NHEJ), which is used to repair breaks in DNA by ligation of the free ends (for review, see Critchlow and Jackson 1998). In mammalian cells, DNA protein kinase and KU70/85 carry out NHEJ, which can occur in response to double strand breaks. In yeast, HDF1 and HDF2 encode Ku70p and Ku80p and, together with other genes including DNL4, XRS2, MRE11, and RAD50, are required for NHEJ (Boulton and Jackson 1996a,b; Moore and Haber 1996; Teo and Jackson 1997). Sir4p interacts with Hdf1p in a two-hybrid assay, and efficient NHEJ requires SIR2, SIR3, and SIR4 (Tsukamoto et al. 1997; Boulton and Jackson 1998). Furthermore, genetic and biochemical experiments imply a direct role for the Sir proteins and Hdf1p in NHEJ (Martin et al. 1999; Mills et al. 1999). Immunofluorescence and chromatin immunoprecipitation have shown that Sir3p, as well as Hdf1p, relocalizes from the telomeres to sites of double-strand breaks created by the _Eco_RI or HO endonucleases (Martin et al. 1999; Mills et al. 1999).
In addition to its roles in HM silencing, telomeric silencing, and NHEJ, Sir2p also functions at the ribosomal DNA (rDNA) locus. In yeast, the rDNA consists of a 9.1 kb unit that is tandemly repeated 100–200 times on chromosome XII (Petes and Botstein 1977; Philippsen et al. 1978; Rustshenko and Sherman 1994). Each unit contains genes encoding the 35S rRNA and the 5S rRNA, separated by a nontranscribed spacer (NTS). Transcription of these genes and ribosome assembly takes place in a subnuclear compartment called the nucleolus (Scheer and Benavente 1990, Melese and Xue 1995; Shaw and Jordan 1995). SIR2 was initially shown to play a role in the suppression of mitotic recombination in the rDNA (Gottlieb and Esposito 1989). More recently, SIR2 was shown to be required for transcriptional silencing of reporter genes integrated at the rDNA (Bryk et al. 1997; Smith and Boeke 1997). The majority of cellular Sir2p, as assayed by immunofluoresence, is found in the nucleolus (Gotta et al. 1997), and the accessibility of rDNA chromatin is responsive to Sir2p dosage (Fritze et al. 1997).
Recently, it was demonstrated that a cause of aging in yeast is the accumulation of circular species of rDNA (Sinclair and Guarente 1997). These extrachromosomal rDNA circles (ERCs) are able to replicate via an ARS sequence contained within the rDNA repeat, and are preferentially segregated to mother cells during division. Deletion of RAD52, a gene required for homologous recombination, results in the absence of ERCs, thereby implicating a recombinational repair process in their formation (Park et al. 1999).
Replication within the rDNA repeats is blocked in one direction at a site in the NTS termed the replication fork barrier or RFB (Brewer and Fangman 1988). Thus, rDNA replication is unidirectional, with the replication fork progressing in the same direction as transcription of the 35S rRNA (Brewer et al. 1992; Kobayashi et al. 1992). The replication block at the RFB requires the FOB1 gene (Kobayashi and Horiuchi 1996), and Fob1p has been shown to localize to the nucleolus (Defossez et al. 1999). Strains with fob1 mutations have a reduced rate of rDNA recombination (Kobayashi et al. 1998) and ERC formation, and have an extended life span compared with wild-type cells. (Defossez et al. 1999). The _FOB1_-dependent replication block may cause a DNA double-strand break within the rDNA. This break can be repaired by homologous recombination, which results in the formation of an ERC (Gangloff et al. 1996; Defossez et al. 1999; Johnson et al. 1999).
Interestingly, Sir3p and Sir4p both relocalize from telomeres to the nucleolus in aging yeast cells (Kennedy et al. 1997). In addition, the sir4-42 allele results in both a 30% increase in life span (Kennedy et al. 1995) and a constitutive redistribution of Sir3p and Sir4p to the nucleolus in young cells (Kennedy et al. 1997). These observations led to a model proposing that Sir3p and Sir4p move to the nucleolus to delay an event that leads to the accumulation of ERCs and ultimately, cell death. Taken together with the role of the SIR genes in DNA damage repair, the question that emerges is: Do the Sir proteins move to the nucleolus via an active mechanism to forestall or repair DNA damage and thereby slow the aging process?
In this paper, we investigate the roles of SIR2, SIR3, and SIR4 in yeast aging. Our results indicate that the sir3Δ and sir4Δ mutations cause a moderate shortening of haploid life span, which is due to derepression of the silent mating type loci and the simultaneous expression of both a and α mating-type information. In contrast, the sir2Δ mutation has a much more severe effect on life span, which is largely due to an inability to suppress the generation of ERCs. Genetic analysis suggests that the primary role of Sir2p is to prevent homologous recombination at the blocked replication fork in the rDNA. Over-expression of SIR2 increases life span beyond that of wild type, showing that Sir2p is a limiting component of the aging process in yeast.
Results
The SIR2/3/4 complex regulates life span
To examine roles of the Sir proteins in yeast aging, null alleles of either sir2, sir3, or sir4 were introduced into a W303 MATa RAD5 strain (W303R). Disruption of either sir3 or sir4 resulted in a 20% decrease in mean life span (Fig. 1A). In contrast, the sir2 mutant strain demonstrated a more severe reduction in mean life span of ∼50% (Fig. 1A). To ascertain whether the short life span of a sir mutant could be the result of a defect in NHEJ, hdf1 and hdf2 were disrupted in W303R, and the life spans of the mutants were determined. Both hdf mutants had life spans that were significantly shorter than wild type (Fig. 1B). Thus, these observations initially raised the possibility that the short life span of sir mutants was due to a defect in NHEJ.
Figure 1.

SIR2, SIR3, and SIR4, but not NHEJ, are required for longevity of haploid mother cells. (A) Mutation of either sir3 or sir4 results in a 20% reduction in mean life span. Mutation of sir2 reduces mean life span by ∼50%. Life spans were determined for W303R, and isogenic sir4Δ, sir3Δ, and sir2Δ mutants. Mean life spans and the number of mothers examined were as follows: W303R 21.4 (n = 50), sir4Δ 18.8 (n = 50), sir3Δ 18.0 (n = 50), and sir2Δ 11.6 (n = 50), (♦) W303R; (█) sir4; (▴) sir3; (x) sir2. (B) Mutation of hdf1 or hdf2 shortens life span. Life spans were determined for haploid W303R, hdf1Δ, and hdf2Δ strains. Mean life spans and the number of mothers examined were as follows: W303R 21.2 (n = 50), hdf1Δ 17.3 (n = 50), and hdf2Δ 14.3 (n = 50), (♦) W303R; (█) hdf1; (▴) hdf2. (C) Mutation of dnl4 does not affect life span. Life spans were determined for haploid W303R and dnl4Δ strains. Mean life spans and the number of mothers examined were as follows: W303R 23.7 (n = 50) and dnl4Δ 23.4 (n = 50), (♦) W303R; (█) dnl4.
Because hdf1 and hdf2 mutants are also defective for telomere maintenance and are temperature sensitive (Feldmann et al. 1996; Boulton and Jackson 1996a,b; Barnes and Rio 1997; Gravel et al. 1998), we wished to measure the life span of a mutant specifically defective for NHEJ, without the pleiotropic effects associated with hdf mutations. The yeast ligase IV gene, DNL4, is required for NHEJ (Schar et al. 1997; Teo and Jackson 1997; Wilson et al. 1997), but is not required for growth at 37°C in the W303R strain background (not shown). The life span of the dnl4 mutant was indistinguishable from wild type (Fig. 1C), demonstrating that NHEJ does not affect aging, and suggesting that the short life span of a sir mutant is not caused by a defect in NHEJ.
The short life span of sir3 and sir4 mutant haploid strains is due to the simultaneous expression of a and α mating-type genes
In addition to their role in NHEJ, the SIR genes are also required for transcriptional repression. Mutation in any of the sir genes results in a loss of silencing at the HM loci, coexpression of a and α mating-type genes, and a corresponding sterility phenotype in haploid S. cerevisiae. This raised the possibility that the short life span of sir mutants was due to the inappropriate expression of loci that are normally silenced in a _SIR_+ strain (e.g., see Astrom et al. 1999). Therefore, we wished to test whether the simultaneous expression of a and α mating-type genes affected life span.
_HML_α was deleted from each of the sir mutant strains above. As expected, deletion of _HML_α resulted in a restoration of the a mating phenotype, as demonstrated by the ability of the sir hml strains to mate with a _MAT_α strain (not shown). Strikingly, haploid sir4 hml and sir3 hml strains showed a significant extension of mean life span relative to the sir4 and sir3 strains (Fig. 2A,B), and were indistinguishable from W303R. In contrast, the sir2 hml strain had a life span that was indistinguishable from the sir2 strain (Fig. 2C). Deleting HML from the wild-type parent did not alter the life span (not shown). Similarly, the sir4 gene was disrupted in an isogenic strain of mating type α. The HMRa locus was then deleted from this strain, restoring the ability of these cells to mate. Once again, a 20% shortening of life span was observed in the sir4 mutant, and this life span defect was suppressed by deletion of hmr (not shown). Thus, it appears that the life span defect caused by a sir4 or sir3 mutation, but not a sir2 mutation, is due to derepression of the HM loci.
Figure 2.

The Sir2/3/4 complex extends life span by silencing the HM loci and preventing a/α coexpression. (A) The life span defect of a W303R MATa sir4Δ mutant is suppressed by deletion of _hml_α. Life spans were determined for wild-type W303R, and isogenic derivatives, sir4Δ and sir4Δ hmlΔ. Mean life spans and the number of mothers examined were as follows: W303R 21.6 (n = 37), sir4Δ 18.2 (n = 49), and sir4Δ hmlΔ 22.4 (n = 48). (♦) W303R; (█) sir4; (▴) sir4 hml. (B) The life span defect of a W303R MATa sir3Δ mutant is suppressed by deletion of _hml_α. Life spans were determined for W303R, and isogenic derivatives, sir3Δ and sir3Δ hmlΔ. Mean life spans and the number of mothers examined were as follows: W303R 20.3 (n = 48), sir3Δ 15.2 (n = 47), and sir3Δ hmlΔ 21.3 (n = 46). (♦) W303R; (█) sir3; (▴) sir3 hml. (C) The short life span of a W303R MATa sir2Δ mutant is not suppressed by deletion of _hml_α. Life spans were determined for wild-type W303R, and isogenic derivatives, sir2Δ, and sir2Δ hmlΔ. Mean life spans and the number of mothers examined were as follows: W303R 21.4 (n = 58), sir2Δ 11.8 (n = 48), and sir2Δ hmlΔ 11.9 (n = 43). (♦) W303R; (█) sir2; (▴) sir2 hml. (D) Mating-type heterozygosity is sufficient to shorten haploid life span to the level of a sir3Δ or sir4Δ mutant. Life spans were determined for wild-type W303R, and isogenic derivatives, sir3Δ, sir4Δ, and _W303R URA3/MAT_α. Mean life spans and the number of mothers examined were as follows: W303R 20.4 (n = 48), sir3Δ 15.2 (n = 47), sir4Δ 15.4 (n = 49), and _W303R URA3/MAT_α 15.2 (n = 48). (♦) W303R; (█) sir4; (▴) sir3; (×) W303a/α.
To determine whether mating-type heterozygosity in an otherwise wild-type background (SIR+) is sufficient to reduce haploid longevity, we directly compared the life spans of the sir4, sir3, and W303R strains with an isogenic strain with _MAT_α integrated at the URA3 locus (W303Ra/α). The mean life span of the W303Ra/α strain was significantly shorter than the wild-type strain and indistinguishable from the sir3 and sir4 strains (Fig. 2D). Thus, simultaneous expression of a and α mating-type information causes a short life span even in the presence of a functional Sir2/3/4 complex.
To further test the hypothesis that mating-type heterozygosity shortens life span, we constructed homozygous sir4/sir4 and sir2/sir2 mutant diploids. The life span of the homozygous sir4/sir4 diploid was indistinguishable from the wild-type diploid, and ∼20% shorter than isogenic a or α haploid strains (Fig. 3A). We have found previously in other strain backgrounds that the life spans of a/α diploids and isogenic haploids were not significantly different (Kennedy et al. 1995; Park et al. 1999). The effect of a/α heterozygosity in diploid cells therefore appears to be strain specific, for reasons that are presently unclear.
Figure 3.
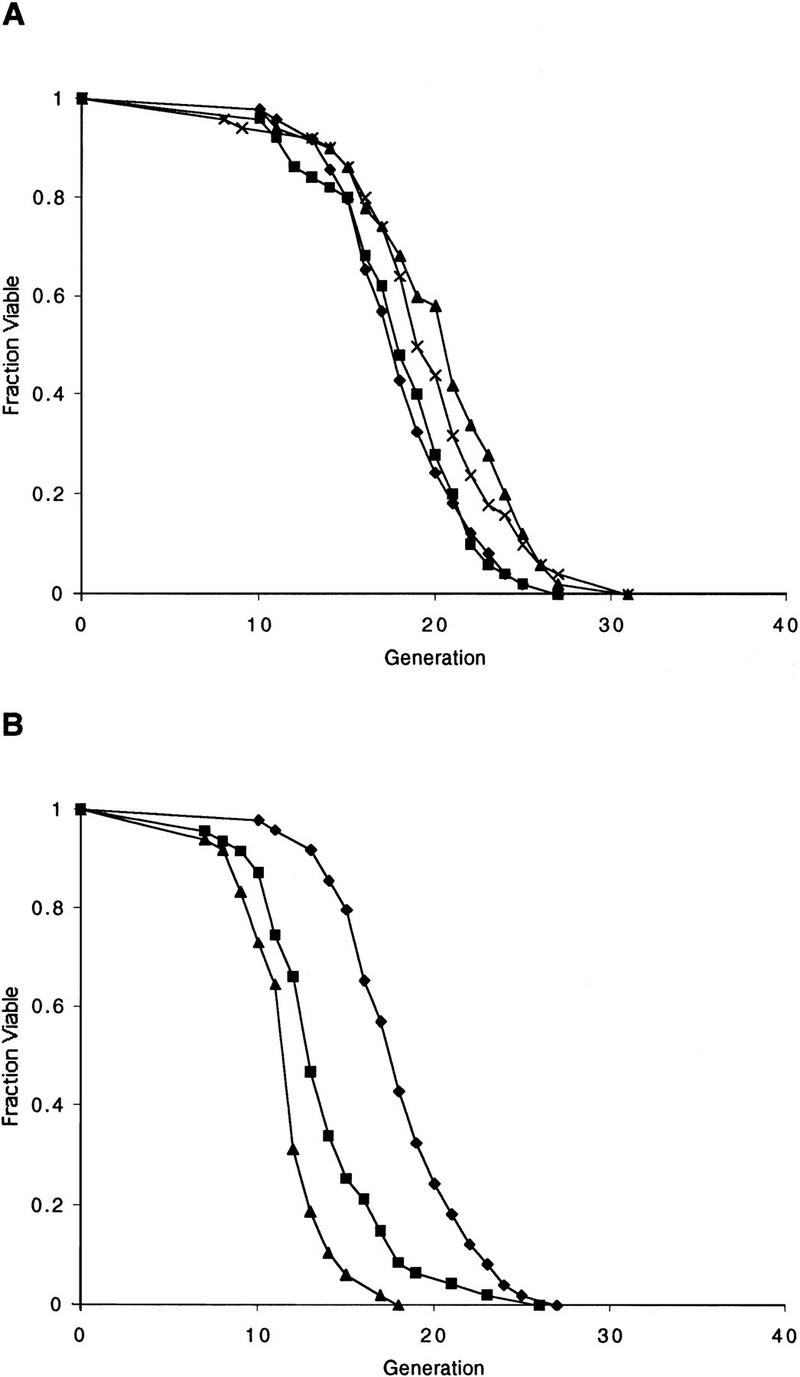
SIR2, but not SIR4, is required for longevity in diploid cells. (A) Diploid life span is not regulated by SIR4. Life spans were determined for W303R/W303R, the isogenic derivative, sir4Δ/sir4Δ, _W303R MAT_α, and W303R MATa. Mean life spans and the number of mothers examined were as follows: W303R/W303R 18.2 (n = 49), sir4Δ/sir4Δ 18.1 (n = 50), _W303R MAT_α 21.1 (n = 50), and W303R MATa 20.5 (n = 50). (♦) W303R/W303R; (█) sir4/sir4; (▴) W303R MATα; (×) W303R MATa. (B) Homozygous mutation of sir2 causes a significant reduction in diploid life span. The sir2Δ/SIR2 heterozygote has a life span that is intermediate between the wild-type diploid and the sir2Δ/sir2Δ homozygote. Life spans were determined for W303R/W303R, and isogenic derivatives, sir2Δ/SIR2, and sir2Δ/sir2Δ. Mean life spans and the number of mothers examined were as follows: W303R/W303R 18.8 (n = 50), sir2Δ/SIR2 14.0 (n = 47), and sir2Δ/sir2Δ 12.1 (n = 48). (♦) W303R/W303R; (█) sir2/SIR2; (▴) sir2/sir2.
In contrast, the homozygous sir2/sir2 diploid displayed a life span significantly shorter than the wild-type or sir4/sir4 homozygous strains (Fig. 3B), consistent with the conclusion that the short life span of the sir2 mutant is not related to cell mating type. Interestingly, the sir2/SIR2 heterozygous diploid had a life span that was much closer to the sir2/sir2 homozygote than to wild type, suggesting that the levels of Sir2p are limiting for life span, at least in diploid strains.
The short life span of sir2, sir3, or sir4 haploids is likely due to an increase in the rate of ERC formation
As deleting _HML_α extends the life span of a sir3 or a sir4 mutant, but fails to suppress the life span defect of a sir2 mutant, Sir2p must function to forestall senescence by a mechanism(s) that does not involve silencing at HM loci. Sir2p is known to localize to the rDNA independently of Sir3p and Sir4p, in which it functions to repress homologous recombination (Gottlieb and Esposito 1989). Because homologous recombination is absolutely required for ERC formation (Park et al. 1999), the short life span of a sir2 mutant is likely to be caused, at least in part, by an increased rate of ERC formation. As a direct test of this hypothesis, we quantitated ERC levels in age-matched W303R and the isogenic sir2 hml mutant cells. In both young (unsorted) and old (sorted) cells, the sir2 hml strain accumulated a greater number of high molecular weight ERCs than W303R (Fig. 4). The fact that we can detect more ERCs in an unsorted population of sir2 hml cells, relative to wild-type, indicates that a significant fraction of the sir2 hml cells form an ERC in an early mother cell division. This suggests that increased homologous recombination at the rDNA in a sir2 mutant directly results in the formation of ERCs, which shortens the life span.
Figure 4.
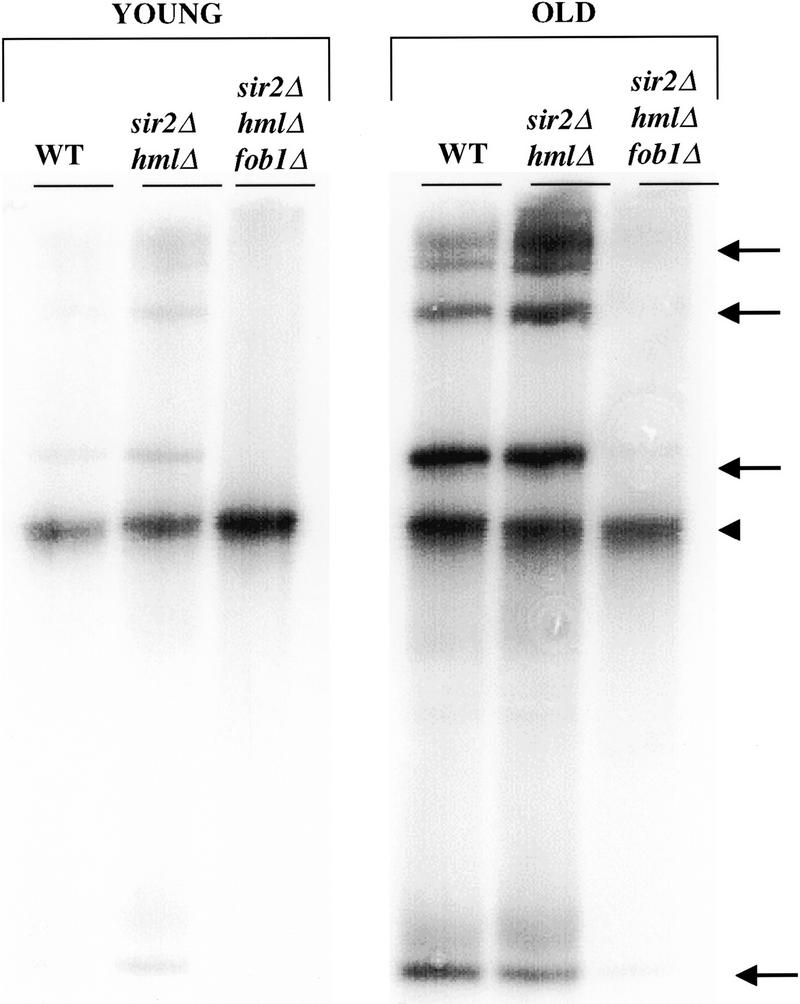
_FOB1_-dependent accumulation of ERCs occurs more rapidly in a sir2 mutant than in wild-type mother cells. DNA from young (unsorted) and old (mean age = 7 generations) mother cells was isolated and electrophoresed as described (Defossez et al. 1999). The gel was transferred and probed with rDNA. Chromosomal DNA is denoted by an arrowhead and ERC species by arrows. Both sorted and unsorted W303R sir2Δ mother cells show a greater accumulation of higher oligomeric species of ERCs than wild type. Mutation of fob1 in sir2Δ hmlΔ mother cells decreases ERC accumulation below levels observed in wild-type cells. Intensity of bands was quantitated on a PhosophorImager. The ratio of ERC DNA to genomic rDNA in young wild-type and sir2Δ hmlΔ cells is 0.2 and 0.4, respectively. In old wild-type and sir2Δ hmlΔ cells, this ratio is 2.1 and 3.1, respectively.
We were not able to observe a clear increase in the levels of ERCs in sir3 or sir4 mutants relative to wild type (not shown). This does not rule out the possibility that sir3 or sir4 mutations increase ERC formation, because an undetectable difference in levels of ERCs might be able to cause the small difference in the life spans of these sir mutants relative to wild type. As a more accurate measure of recombination rates in the rDNA, we measured the loss rate of the ADE2 marker integrated into a single rDNA repeat. This marker was lost at a two- to threefold higher frequency in a sir3 or sir4 mutant strain compared with wild type (Table 1). Furthermore, this elevated rate of rDNA recombination was suppressed by deleting _HML_α to restore the a mating type. These data suggest that the short life span in the sir3 or sir4 nonmating haploids is caused by a small increase in recombination at the rDNA due to the expression of both a and α mating-type genes.
Table 1.
Mating-type heterozygosity increases rDNA marker loss of the W303R haploid—deletion of fob1 suppresses the rDNA recombination defect of a sir2 hml mutant
| Strain | Rate of rDNA marker loss (× 103) |
|---|---|
| W303R | 1.1 (0.1) |
| sir4 | 2.3 (0.1) |
| sir4 hml | 0.9 (0.3) |
| sir2 | 18.4 (4.6) |
| sir2 hml | 9.0 (0.6) |
| sir2 fob1 hml | 0.8 (0.1) |
Also consistent with the notion that sir3 and sir4 mutants have an elevated rate of ERC formation, the release of an ERC into wild-type or sir4 mother cells caused an identical shortening of life span. For this experiment, we used the method of Sinclair and Guarente (1997) to release an ERC into the nucleus of wild-type and sir4 virgin cells. On release of an ERC, W303R+ERC cells had a mean life span that was shortened by ∼25% and did not differ significantly from the sir4+ERC strain (Fig. 5). This finding indicates that the sir4 (and presumably sir3) mutation does not affect the rate of accumulation of ERCs and strengthens the idea that it increases the rate of formation of ERCs.
Figure 5.

Sir4p does not function to extend life span after release of an ERC. ERCs were released in virgin cells as described (Sinclair and Guarente 1997). Life spans were determined for W303R, W303R induced to release an ERC (W303R + ERC), the isogenic derivative, sir4, and sir4 induced to release an ERC (sir4 + ERC). Mean life spans and the number of mothers examined were as follows: W303R 17.6 (n = 58), W303R + ERC 13.5 (n = 68), sir4 13.5 (n = 55), and sir4 + ERC 13.0 (n = 51). (♦) W303R; (█) W303R + ERC; (▴) sir4; (×) sir4 + ERC.
Deletion of FOB1 suppresses the rDNA hyperrecombination phenotype and the life span defect of a sir2 mutant
A key gene that regulates rDNA recombination and life span is FOB1. Deletion of fob1 gives rise to bidirectional rDNA replication (Kobayashi and Horiuchi 1996), decreases the rate of rDNA recombination (Kobayashi et al. 1998), dramatically reduces ERC formation, and extends life span by 30%–60% (Defossez et al. 1999). We therefore wished to examine the relationship between sir2 and fob1 with respect to life span, rDNA recombination, and ERC formation.
To eliminate any effects of the sir2 mutation on HM expression, the sir2 hml strain was used. When fob1 was deleted in this strain, the short life span of the sir2 mutant was largely suppressed, giving rise to a life span extended by ∼50% (Fig. 6). The mean and maximum life span of the sir2 fob1 hml strain was comparable with W303R, but slightly shorter than the fob1 single mutant. This suppression, along with the experiment described below (Fig. 7B), suggests that SIR2 and FOB1 act in the same pathway to determine yeast life span (see Discussion).
Figure 6.

Mutation of fob1 suppresses the life span defect of a sir2 mutant. Life spans were determined for W303R, and the isogenic derivatives, sir2Δ hmlΔ, and sir2Δ hmlΔ fob1Δ. Mean life spans and the number of mothers examined were as follows: W_303R_ 20.7 (n = 38), fob1Δ 27.0 (n = 37), sir2Δ hmlΔ 13.0 (n = 47) and sir2Δ hmlΔ fob1Δ 21.9 (n = 48). (♦) W303R; (█) fob1; (▴) sir2 hml; (×) sir2 hml fob1.
Figure 7.

Sir2p is a limiting component of yeast longevity. (A) Overexpression of SIR2 extends the life span of W303R by ∼30%. A second copy of SIR2 was integrated at the URA3 locus (denoted SIR2/URA3) of strains W303R and sir2Δ. Life spans were determined for W303R, and the isogenic derivatives sir2Δ, W303R SIR2/URA3, and sir2Δ SIR2/URA3. Mean life spans and the number of mothers examined were as follows: W303R 23.0 (n = 48), W303R SIR2/URA3 28.8 (n = 50), sir2Δ 11.0 (49), and sir2Δ SIR2/URA3 21.3 (49). (♦) W303R; (█) W303R SIR2/URA3; (▴) sir2; (×) sir2 SIR2/URA3. (B) Overexpression of SIR2 does not affect the life span of a fob1 mutant. A second copy of SIR2 was integrated at the URA3 locus of fob1Δ. Life spans were determined for W303R, and the isogenic derivatives W303R SIR2/URA3, fob1Δ, and fob1Δ SIR2/URA3. Mean life spans and the number of mothers examined were as follows: W303R 22.2 (n = 50), W303R SIR2/URA3 29.9 (n = 50), fob1Δ 29.0 (n = 50), and fob1ΔSIR2/URA3 29.4 (n = 49). (♦) W303R; (█) W303R SIR2/URA3; (▴) fob1; (×) fob1 SIR2/URA3.
Consistent with the life span data, deletion of fob1 also suppressed the rDNA hyperrecombination phenotype of the sir2 hml strain (Table 1). An ADE2 marker inserted into the rDNA array was lost at an ∼10-fold higher rate in the sir2 hml strain than in W303R. Deletion of fob1 in this background reduced this high rate of marker loss to approximately that of the SIR2 FOB1 strain.
To test whether the fob1 deletion reduced the rate of ERC accumulation in the sir2 mutant, we determined ERC levels in young and old sir2 hml fob1 cells. Deletion of fob1 reduced ERC levels in both young and old cells, even below those seen in W303R (Fig. 4). A similarly dramatic reduction in ERCs is observed when fob1 is mutated in a wild-type background (Defossez et al. 1999). The fact that the sir2 hml fobl strain does not live significantly longer than W303R, even though the levels of ERCs are lower than wild type, suggests that Sir2p may play an additional role as a regulator of longevity.
Increasing the dosage of SIR2 extends life span
The above analysis of SIR2 suggested that this gene played a central role in yeast aging. The short life span of the SIR2/sir2 heterozygous diploid further suggested that the levels of this protein may be limiting. If this were so, then increasing the levels of Sir2p might extend the life span of wild-type cells.
Thus, a second copy of SIR2 was integrated into the genome of W303R and W303R sir2 (see Materials and Methods), and the life spans of the resulting transformants were determined. The integrated SIR2 restored the wild-type life span (Fig. 7A) and mating ability (not shown) to the sir2 mutant, indicating that the cloned SIR2 was functional. Strikingly, integration of a second copy of SIR2 into the wild-type strain gave rise to an ∼30% extension of life span (Fig. 7A,B). This finding shows that the levels of Sir2p are limiting for the life span of wild-type mother cells.
The above finding afforded another possibility to test whether FOB1 and SIR2 function in the same pathway. If the role of SIR2 were to prevent recombination at blocked replication forks in the rDNA, then a second copy of SIR2 should not extend further the elongated life span in a fob1Δ strain. Thus, a second SIR2 copy was integrated into the fob1Δ strain and life spans were determined (Fig. 7B). The increased levels of Sir2p did not extend further the long life span in the fob1Δ strain, providing additional evidence that the role of SIR2 in extending life span involves preventing recombination at the Fob1p-arrested replication fork.
Discussion
The results presented here define two distinct roles of the SIR genes as negative regulators of aging in S. cerevisiae. First, the SIR2/3/4 encoded complex acts indirectly to extend haploid life span by repressing transcription at HML and HMR. In sir3 and sir4 mutants, coexpression of the a and α mating-ype genes results in an elevated rate of rDNA recombination, likely increasing the rate of formation of ERCs. The short life span of a sir3 or sir4 mutant is suppressed by deleting the relevant HM locus. Second, SIR2 acts directly to suppress ERC formation by inhibiting homologous recombination at a blocked replication fork in the rDNA (see below). Thus, the short life span of a sir2 mutant is not significantly altered by deleting the HM loci. Sir2p appears to be a limiting component in yeast aging, because increasing the dosage of this gene extends the life span well beyond that of wild type.
The role of cell type in aging
Several previous experiments led us to propose that the role of the Sir complex in yeast longevity might be to repair damage in the rDNA. First, the wild-type Sir complex redistributes from telomeres to the nucleolus in aging mother cells (Kennedy et al. 1997). Second, the sir4-42 mutation, identified previously as an allele-specific suppressor for the short life span of a strain carrying a carboxy-terminal deletion in uth4 (Kennedy et al. 1995), results in the constitutive localization of the Sir complex to the nucleolus. One specific possibility was that the Sir complex might function by repairing DSBs in the rDNA via NHEJ. Although one study indicated that the role of the Sir complex in NHEJ was an indirect consequence of the a/α cell type (Astrom et al. 1999), more recent studies showed that the Sir complex redistributes directly from telomeres to induced DSBs and likely functions directly in promoting their repair (Martin et al. 1999; Mills et al. 1999).
Mutations in hdf1 and hdf2 shorten life span significantly. However, the short life spans of these mutants do not result from a defect in NHEJ. This follows from the fact that the dnl4 mutant is also defective in NHEJ, but has a wild-type life span. Hdf mutants show an aberrant organization of telomeres (Gravel et al. 1998) and are temperature sensitive for growth (Feldmann et al. 1996; Boulton and Jackson 1996a,b; Barnes and Rio 1997). Thus, the short life spans of hdf1 and hdf2 strains are caused by defects in processes other than NHEJ, perhaps related to telomere maintenance or function.
Rather, it is the expression of both a and α mating-type genes in sir3 or sir4 mutants that appears to cause the short life span in these strains. Deletions of the HM loci that preserve haploid mating reverse the short life span of sir3 or sir4 mutants. Further, homozygous deletion of sir4 in an a/α diploid does not shorten life span.
How might a/α expression in haploids shorten life span? It has been suggested that _RAD52_-mediated recombination is increased in cells expressing both a and α mating-type genes (Heude and Faber 1993; Schild 1995; Fasullo et al. 1999), thus accounting for the increased resistance of diploid cells to certain DNA damaging agents. Consistent with these findings, we demonstrate an increase in the rate of marker loss from the rDNA due to a mutation in sir4, and this increase is suppressed by deleting _HML_α (thereby preventing a/α coexpression). In addition, RAD52 is absolutely required for the formation of ERCs (Park et al. 1999). Thus, we propose that ERCs are generated at a higher rate in sir mutants, and that this increase in ERC formation results in a shortened life span (Fig. 8).
Figure 8.

The Sir proteins regulate longevity by reducing the rate of ERC formation by two mechanisms. The Sir2/3/4p complex acts indirectly to inhibit ERC formation by preventing the simultaneous expression of a and α mating-type genes. When expressed, the a/α cell type increases recombination in the rDNA and, presumably, the formation of ERCs. Sir2p acts by a second mechanism in the rDNA to inhibit Fob1p-mediated formation of ERCs, presumably at blocked replication forks.
How then can we understand the sir4-42 mutation and the redistribution of the wild-type Sir complex in old cells? We speculate that the constitutive localization of the sir4-42 mutant complex to the rDNA might alter the recombinational properties in that locus to forestall the generation of ERCs. In this way, the mutation is really a neomorph and does not correspond to any normal activity of the Sir complex. In addition, accumulation of ERCs in old cells results in an amplification of rDNA sequence, which may titrate the Sir2/3/4p complex to the nucleolus.
A direct role of Sir2p in forestalling aging
The extremely short life span of a sir2 mutant is not a consequence of the a/α cell type. Rather, elevated levels of rDNA recombination (Gottlieb and Esposito 1989) result in an increase in the rate of formation of ERCs and thus accelerated aging. Strikingly, deletion of the replication fork block protein gene, FOB1, largely suppresses the short life span of a sir2 mutant. This epistasis of FOB1 to SIR2 implies that these genes function in the same pathway. The inability of a second SIR2 copy to extend life span in a fob1 strain strengthens this view, although we cannot exclude the possibility that fob1 mutant cells senesce for reasons unrelated to SIR2 function.
We propose that Sir2p stabilizes blocked replication forks in the rDNA to prevent DNA breaks, recombination, and the generation of ERCs (Fig. 8). In the absence of Fob1p, the replication fork is not blocked in the rDNA, and Sir2p is not required to prevent the formation of ERCs. This suppression of the short life span of sir2 mutants by mutation in fob1 contrasts with the short life span in mutants lacking the Sgs1p DNA helicase, which is not suppressed by fob1 (Defossez et al. 1999). Therefore, unlike the case in a sir2 mutant, the short life span of an sgs1 mutant is not caused by a Fob1p-blocked replication fork.
Further support for this model comes from observations that rad52 or fob1 mutations suppress the rDNA hyperrecombination phenotype of a sir2 mutant (Gottleib and Esposito 1989; Table 1). This demonstrates that in the absence of Sir2p, the predominant form of repair at the rDNA is through the RAD52 pathway of homologous recombination, most likely at the replication fork block. Correspondingly, ERCs are dramatically reduced in fob1 and sir2 fob1 hml mutant strains (Defossez et al. 1999; Fig. 4) and eliminated in a rad52 strain (Park et al. 1999).
However, because the sir2 fob1 hml strain has a shorter life span than the fob1 mutant, Sir2p must function to extend life span by an, as yet undefined, additional mechanism. Thus, FOB1 is only partially epistatic to SIR2 for life span, but is completely epistatic to SIR2 for ERC formation. Therefore, we propose that SIR2 promotes longevity by at least three distinct mechanisms; first, Sir2p functions, along with Sir3p and Sir4p, to silence HM loci; second, Sir2p directly inhibits ERC formation by antagonizing homologous recombination at the blocked replication fork in the rDNA; finally, SIR2 apparently helps extend life span by a third mechanism that is independent of ERCs or cell type.
Sir2p as the limiting component in yeast aging
It is of interest that introduction of a second copy of SIR2 into the yeast genome extends the life span 30% beyond wild type. Sir2p is, therefore, limiting in wild-type cells, and rDNA recombination at the replication fork block is not fully suppressed. In the fob1 mutant, in which the replication fork block is absent and life span is extended, a second copy of SIR2 does not extend life span further. The short life span of a sir2/SIR2 heterozygous diploid reveals a haplo insufficiency, also indicating that levels of Sir2p are limiting for life span. This is consistent with the observation that rDNA silencing is extremely sensitive to Sir2p dosage (Smith et al. 1998), and suggests that rDNA silencing, rDNA recombination, and life span are all tightly coupled to nucleolar levels of Sir2p.
Of all of the SIR genes, SIR2 is evidently the only one conserved in higher eukaryotes. Whereas the function of _SIR2_-related genes in mammals is not presently known, it is interesting to speculate that an alteration in Sir2p levels or activity may alter the pace of aging in these organisms, as well. If so, a key molecular link in the aging process of many organisms would be highlighted in the form of the function of this gene.
Materials and methods
Strains, plasmids, and media
The yeast strains used in this work are listed in Table 2. Strain W303R is described in Mills et al. (1999), in which it is referred to as W303AR5. The sir3Δ::URA3 disruption was generated as described in Sinclair and Guarente (1997). The sir4Δ::HIS3, sir2Δ::TRP1, and hmlΔ::LEU2 disruptions were generated as described in Mills et al. (1999).
Table 2.
Yeast strains used in this study
| Strain | Genotype |
|---|---|
| W303R | W303-1A MATaRDN1::ADE2 RAD5 |
| DSY1036 | W303R sir4Δ::HIS3 |
| DSY880 | W303R sir3Δ::URA3 |
| DSY1034 | W303R sir2Δ::TRP1 |
| MMY98 | W303R hdf2Δ::LEU2 |
| MMY134 | W303R dn14Δ::URA3 |
| MKY576 | W303R hdf1Δ::URA3 |
| MKY545 | W303R sir4ΔHIS3 hmlΔ::LEU2 |
| MKY543 | W303R sir3ΔURA3 hmlΔ::LEU2 |
| MKY541 | W303R sir2ΔTRP1 hmlΔ::LEU2 |
| MKY549 | W303R URA3/MATα |
| MKY1033 | W303-1A MATα RDN1::ADE2 RAD5 sir4Δ::HIS3 |
| MKY1046 | MKY1033 hmrΔ::URA3 |
| MKY547 | MKY545 fob1Δ::URA3 |
| MKY501 | W303R fob1Δ::URA3 |
| MKD104 | W303R/W303R MATa/MATα sir4Δ::HIS3/sir4Δ::HIS3 hmlΔ::LEU2/HML hmrΔ::URA3/HMR |
| MKD105 | W303R/W303R MATa/MATα hmlΔ::LEU2/HML hmrΔ::URA3/HMR |
| MKD106 | MKD105 sir2Δ::TRP1/SIR2 |
| MKD107 | MKD105 sir2Δ::TRP1/sir2Δ::TRP 1 |
| MKY546 | MKY541 fob1Δ::URA3 |
| MMY094 | W303R URA3/SIR2 |
| MMY095 | W303R sir2Δ::TRP1 URA3/SIR2 |
| MMY112 | W303R fob1Δ::URA3 |
| MMY113 | W303R fob1Δ::URA3 URA3/SIR2 |
All other gene disruptions were generated by targeted PCR disruption. Marker genes were amplified from the appropriate pRS integrating vector (Sikorski and Heiter 1989) with primers designed to incorporate 40 nucleotides of homology to genomic DNA at each end of the disruption cassette. In each case, the entire coding region of the disrupted gene and up to 100 nucleotides of flanking sequence were removed. All disruptants were verified by PCR prior to subsequent analysis. Additionally, hdf1 and hdf2 mutations were verified by the inability of these strains to grow at 37°C. Lack of NHEJ in the dnl4 and hdf1 mutants was verified by a plasmid end-joining assay as described (Mills et al. 1999).
Integration of SIR2 at URA3 was accomplished by transforming cells with plasmid p306SIR2 digested with _Xcm_I. In addition to the entire coding region of SIR2, ∼1000 nucleotides of upstream sequence and 200 nucleotides of downstream sequence are present. Plasmid p306SIR2 was constructed by ligating the PCR-amplified SIR2 gene into the _Xba_I and _Xho_I sites of pRS306.
Unless otherwise stated, all strains were cultured at 30°C and grown on YEPD containing 2% glucose.
Life span analysis
Micromanipulation and life span analysis were performed as described previously (Kennedy et al. 1994), with the following modifications. Prior to analysis, strains were patched onto solid YPD for at least two consecutive days. Cells were then transferred at low density to fresh YPD and allowed to grow for ∼3 hr. Virgin daughter cells were then isolated as buds from mother cells and subjected to life span analysis. Each experiment consisted of 37–50 mother cells and was carried out at least twice, independently. Statistical significance was determined by a Wilcoxon rank sum test. Average life span is stated to be different for P < 0.05. Each figure represents data derived from a single experiment.
Release of ERCs into virgin daughter cells
The method of Sinclair and Guarente (1997) was used to release an ERC into virgin cells, with the following modifications. Life spans were performed on plates lacking adenine and containing 1% galactose and 2% raffinose. Mother cells whose daughters formed large colonies on this medium failed to induce the Cre recombinase and were therefore not included in the data set.
Determination of rDNA recombination rate
Strains carrying an ADE2 marker integrated into the rDNA array were grown overnight and then plated onto solid YPD. Colonies were allowed to grow for 48 hr at 30°C then placed at 4°C for 48 hr. The number of half-red/half-white colonies was determined; each was assumed to represent a marker loss event during the first cell division after plating. The number of half-sectored colonies divided by the total number of colonies (not including entirely red colonies) was reported as the rate of marker loss.
Purification of old cell DNA and ERC analysis
Old cells were obtained by the biotin-labeling procedure (Smeal et al. 1996) and age was determined by counting bud scars (Sinclair et al. 1997). ERC levels were detemined as described (Defossez et al. 1999), with the following modifications. ERC species were separated on a 0.6% agarose gel (without ethidium bromide) run at 1 Volt/cm for 44 hr.
Acknowledgments
We thank T. Isaacson, K. Mills, D. Sinclair, and H. Tissenbaum for reading the manuscript, and we thank other members of the Guarente laboratory for discussion and insight. This work was supported by an National Institutes of Health (NIH) grant to LG. M.K. and M.M. are supported by an NIH predoctoral training grant (T32 GM07287).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked 'advertisement' in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL leng@mit.edu; FAX (617) 253–8699.
References
- Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- Astrom SU, Okamura SM, Rine J. Yeast cell-type regulation of DNA repair. Nature. 1999;397:310. doi: 10.1038/16833. [DOI] [PubMed] [Google Scholar]
- Barnes G, Rio D. DNA double-strand-break sensitivity, DNA replication, and cell cycle arrest phenotypes of Ku-deficient Saccharomyces cerevisiae. Proc Natl Acad Sci. 1997;94:867–872. doi: 10.1073/pnas.94.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: Roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996a;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double- strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996b;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- ————— Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell. 1988;55:637–643. doi: 10.1016/0092-8674(88)90222-x. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Lockshon D, Fangman WL. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell. 1992;71:267–276. doi: 10.1016/0092-8674(92)90355-g. [DOI] [PubMed] [Google Scholar]
- Bryk M, Banerjee M, Murphy M, Knudsen KE, Garfinkel DJ, Curcio MJ. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes & Dev. 1997;11:255–269. doi: 10.1101/gad.11.2.255. [DOI] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP. DNA end-joining: From yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol Cell. 1999;3:447–455. doi: 10.1016/s1097-2765(00)80472-4. [DOI] [PubMed] [Google Scholar]
- Fasullo M, Bennett T, Dave P. Expression of Saccharomyces cerevisiae MATa and MAT alpha enhances the HO endonuclease-stimulation of chromosomal rearrangements directed by his3 recombinational substrates. Mutat Res. 1999;433:33–44. doi: 10.1016/s0921-8777(98)00059-7. [DOI] [PubMed] [Google Scholar]
- Feldmann H, Driller L, Meier B, Mages G, Kellermann J, Winnacker EL. HDF2, the second subunit of the Ku homologue from Saccharomyces cerevisiae. J Biol Chem. 1996;271:27765–27769. doi: 10.1074/jbc.271.44.27765. [DOI] [PubMed] [Google Scholar]
- Fritze CE, Verschueren K, Strich R, Easton Esposito R. Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 1997;16:6495–6509. doi: 10.1093/emboj/16.21.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Zou H, Rothstein R. Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 1996;15:1715–1725. [PMC free article] [PubMed] [Google Scholar]
- Gotta M, Strahl-Bolsinger S, Renauld H, Laroche T, Kennedy BK, Grunstein M, Gasser SM. Localization of Sir2p: The nucleolus as a compartment for silent information regulators. EMBO J. 1997;16:3243–3255. doi: 10.1093/emboj/16.11.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- Gravel S, Larrivee M, Labrecque P, Wellinger RJ. Yeast Ku as a regulator of chromosomal DNA end structure. Science. 1998;280:741–744. doi: 10.1126/science.280.5364.741. [DOI] [PubMed] [Google Scholar]
- Heude M, Fabre F. a/alpha-control of DNA repair in the yeast Saccharomyces cerevisiae: Genetic and physiological aspects. Genetics. 1993;133:489–498. doi: 10.1093/genetics/133.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy JM, Klar AJ, Hicks JB. Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol. 1986;6:688–702. doi: 10.1128/mcb.6.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NR, Jr, Guarente L. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol. 1994;127:1985–1993. doi: 10.1083/jcb.127.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell. 1997;89:381–391. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996;1:465–474. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Hidaka M, Nishizawa M, Horiuchi T. Identification of a site required for DNA replication fork blocking activity in the rRNA gene cluster in Saccharomyces cerevisiae. Mol & Gen Genet. 1992;233:355–362. doi: 10.1007/BF00265431. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Heck DJ, Nomura M, Horiuchi T. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes & Dev. 1998;12:3821–3830. doi: 10.1101/gad.12.24.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA Strand Breaks in Yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- Melese T, Xue Z. The nucleolus: An organelle formed by the act of building a ribosome. Curr Opin Cell Biol. 1995;7:319–324. doi: 10.1016/0955-0674(95)80085-9. [DOI] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-srand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino F, Laroche T, Gilson E, Axelrod A, Pillus L, Gasser SM. SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell. 1993;75:543–555. doi: 10.1016/0092-8674(93)90388-7. [DOI] [PubMed] [Google Scholar]
- Park PU, Defossez PA, Guarente L. Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in saccharomyces cerevisiae. Mol Cell Biol. 1999;19:3848–3856. doi: 10.1128/mcb.19.5.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD, Botstein D. Simple Mendelian inheritance of the reiterated ribosomal DNA of yeast. Proc Natl Acad Sci. 1977;74:5091–5095. doi: 10.1073/pnas.74.11.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippsen P, Thomas M, Kramer RA, Davis RW. Unique arrangement of coding sequences for 5 S, 5.8 S, 18 S and 25 S ribosomal RNA in Saccharomyces cerevisiae as determined by R-loop and hybridization analysis. J Mol Biol. 1978;123:387–404. doi: 10.1016/0022-2836(78)90086-4. [DOI] [PubMed] [Google Scholar]
- Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustchenko EP, Sherman F. Physical constitution of ribosomal genes in common strains of Saccharomyces cerevisiae. Yeast. 1994;10:1157–1171. doi: 10.1002/yea.320100904. [DOI] [PubMed] [Google Scholar]
- Schar P, Herrmann G, Daly G, Lindahl T. A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks. Genes & Dev. 1997;11:1912–1924. doi: 10.1101/gad.11.15.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer U, Benavente R. Functional and dynamic aspects of the mammalian nucleolus. BioEssays. 1990;12:14–21. doi: 10.1002/bies.950120104. [DOI] [PubMed] [Google Scholar]
- Schild D. Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics. 1995;140:115–127. doi: 10.1093/genetics/140.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Jordan EG. The nucleolus. Annu Rev Cell Dev Biol. 1995;11:93–121. doi: 10.1146/annurev.cb.11.110195.000521. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Smeal T, Claus J, Kennedy B, Cole F, Guarente L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell. 1996;84:633–642. doi: 10.1016/s0092-8674(00)81038-7. [DOI] [PubMed] [Google Scholar]
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes & Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Pillus L, Boeke JD. Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics. 1998;149:1205–1219. doi: 10.1093/genetics/149.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo SH, Jackson SP. Identification of Saccharomyces cerevisiae DNA ligase IV: Involvement in DNA double-strand break repair. EMBO J. 1997;16:4788–4795. doi: 10.1093/emboj/16.15.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Kato J, Ikeda H. Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature. 1997;388:900–903. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates nonhomologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]