Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies (original) (raw)
Abstract
Bcl-2 family proteins are key regulators of apoptosis and function as cell death antagonists (e.g., Bcl-2, Bcl-XL, and Mcl-1) or agonists (e.g., Bax, Bad, and Bak). Here we report that among the Bcl-2 family of proteins tested (Bcl-2, Bcl-XL, Mcl-1, Bax, Bad, and Bak), Bcl-XL was unique in that its protein levels were tightly regulated by hemopoietins in both immortal and primary myeloid progenitors. Investigating signaling pathways utilized by cytokine receptors established that the regulation of Bcl-XL protein levels is mediated by the Jak kinase pathway and is independent of other signaling effectors including STATs, PI-3′ kinase, and Ras. Moreover, we provide the first direct evidence that Bcl-X is altered in cancer, because bcl-X expression was activated selectively by retroviral insertions in murine myeloid and T-cell hemopoietic malignancies. Tumors harboring bcl-X insertions had altered bcl-X RNAs, expressed elevated levels of Bcl-XL protein, and lacked the requirements for cytokines normally essential for cell survival. Finally, overexpression of Bcl-XL effectively protected IL-3-dependent myeloid cells from apoptosis following removal of trophic factors. Therefore, Bcl-XL functions as a key cytokine regulated anti-apoptotic protein in myelopoiesis and contributes to leukemia cell survival.
Keywords: Bcl-XL, Jak kinase, apoptosis, leukemia
Committed hematopoietic progenitors continuously require hemopoietins for both growth and survival. Depletion of these trophic factors leads to apoptosis, or programmed cell death (PCD), and this endogenous cell suicide mechanism ensures strict control of hematopoietic cell numbers (Williams et al. 1990). In leukemia, cytokine-dependent signals required for survival are circumvented by oncoproteins that provide signals that suppress apoptosis. Thus, a major emphasis of leukemia research has been focused on identifying cytokine-dependent signaling pathways that govern cell survival and defining the effectors targeted by these pathways that regulate apoptosis.
There are many initiators of apoptosis, but the ultimate downstream effectors of the suicide program are a family of conserved cysteine-dependent, aspartate-specific proteases, termed caspases, that degrade key targets required for cell and nuclear integrity, including lamin A, hnRNPs, poly(ADP-ribose) polymerase, and DNA-dependent protein kinase (for review, see Villa et al. 1997). Genetic and biochemical evidence has demonstrated that the activation of caspases is influenced by the Bcl-2 family of proteins, which either suppress or induce apoptosis (for review, see Yang and Korsmeyer 1996). For example, the family members Bcl-2, Bcl-XL, Mcl-1, and A-1 function as cell death antagonists against a variety of apoptotic stimuli, whereas the pro-apoptotic members Bax, Bak, and Bad augment the apoptotic program. Thus, some Bcl-2 family members would be predicted to be key targets for activation, whereas others would be targets for inactivation in cancer. Indeed, BCL-2 expression is activated by the t(14:18) reciprocal translocation of BCL-2 into the immunoglobulin heavy-chain locus in human follicular lymphoma (Bakshi et al. 1985; Tsujimoto et al. 1985; Cleary and Sklar 1985), and high levels of BCL-2 have been reported in other cancers without obvious chromosomal alterations (Pezella et al. 1990). Elevated levels of BCL-2 in these scenarios are thought to suppress apoptosis by sequestering Bax, which dimerizes with many Bcl-2 family members, yet is a potent inducer of apoptosis as a homodimer (Oltvai et al. 1993; Sedlak et al. 1995). In support of this concept, Bax functions as a tumor suppressor in murine model systems (Yin et al. 1997) and is down-regulated or inactivated by mutations in some human malignancies including leukemia and colon and breast carcinomas (Krajewski et al. 1995; Rampino et al. 1997; Brimmell et al. 1998). However, to date, other Bcl-2 family members have not been shown to be mutated directly in cancer.
Because Bcl-2 family members are key gatekeepers of the cell-death pathway, a reasonable expectation would be that their activity and/or expression is regulated by signaling pathways that suppress or induce apoptosis, and there is mounting evidence that this is the case. First, in some cell contexts Bcl-2 family members are regulated transcriptionally by mitogens and by other signals that regulate apoptosis. In these cases, the mRNA levels of the anti-apoptotic members Bcl-2, Bcl-X, A-1, and Mcl-1 are generally mitogen dependent (Reed et al. 1987; Boise et al. 1993; Lin et al. 1993; Broome et al. 1995; Kozopas et al. 1993; Yang and Korsmeyer 1996; Lomo et al. 1997), whereas the expression of the pro-apoptotic family members Bax and Bak is induced under some scenarios that lead to apoptosis (Zhan et al. 1994; Miyashita and Reed 1995; Ossina et al. 1997). Second, the activity of certain family members is regulated by post-translational modifications. For example, phosphorylation of Bcl-2 following treatment of some cells with paclitaxel has been associated with apoptosis (Haldar et al. 1994). More compelling is the recent demonstration that phosphorylation of the pro-apoptotic family member, Bad, ablates its activity as an apoptotic agonist in response to survival factors by inhibiting its association with Bcl-XL (Zha et al. 1996). Moreover, phosphorylation of Bad occurs via the Akt serine/threonine kinase (Datta et al. 1997; Delpeso et al. 1997), a key signaling effector of the PI-3′ kinase pathway (Franke et al. 1995) essential for the survival of at least some cell types (Yao and Cooper 1995; Dudek et al. 1997). Finally, it has been also reported that Bcl-2 and Bcl-XL are direct substrates for caspases, and that they are cleaved into pro-apoptotic forms during apoptosis induced by specific stimuli, suggesting a mechanism that ensures apoptosis goes to completion (Cheng et al. 1997; Clem et al. 1998).
To resolve which Bcl-2 family members might be involved in the regulation of programmed cell death that occurs following the withdrawal of trophic factors, we assessed the regulation of steady-state levels of these proteins in both immortal and primary myeloid cells. Here we report that of all the Bcl-2 family members tested, Bcl-XL protein was unique in its tight regulation by hemopoietins, and that this regulation depends upon a Jak kinase-dependent signaling pathway. Moreover, in screens of murine hematologic malignancies we demonstrate bcl-X is selectively activated by retroviral insertions in myeloid and T-cell leukemia. This is the first direct evidence that Bcl-X is activated in cancer and underscores the important role of this protein in regulating apoptosis during hematopoiesis and in leukemia.
Results
Bcl-XL proteins levels are selectively regulated by hemopoietins in immortal and primary myeloid cells
Hemopoietins are required to suppress the PCD of committed hematopoietic progenitors (Williams et al. 1990). In T- and B-lymphoid cells, cytokines have been variably reported to regulate the expression (Reed et al. 1987; Boise et al. 1993; Lin et al. 1993; Broome et al. 1995; Leverrier et al. 1997; Lomo et al. 1997; Yang et al. 1997) and/or cleavage (Cheng et al. 1997; Clem et al. 1998) of the anti-apoptotic family members Bcl-2, Bcl-XL, Bcl-Xγ, A-1, or Mcl-1, or the activity of pro-apoptotic family members such as Bad (Zha et al. 1996). To determine the family members that play roles in the physiologic PCD observed when myeloid progenitors are deprived of trophic factors, we assessed initially whether regulation of their mRNA or protein levels was dependent upon hemopoietins in immortal and primary myeloid cells. For analyses of immortal myeloid progenitors we used murine 32D.3 and FDC-P1 myeloid cells which, like primary myeloid cells, are diploid, able to differentiate along different lineages in response to specific hemopoietins, and require IL-3 for growth and survival (Dean et al. 1987; Askew et al. 1991). When examined for regulation of Bcl-2 family protein levels by immunoblots following IL-3 withdrawal, only the down-regulation of Bcl-XL protein correlated with the PCD of either 32D.3 or FDC-P1.2 cells (Fig. 1A,C). Marked down-regulation of Bcl-XL was coincident with the commitment point for the PCD of these cells (16–20 hr for 32D.3 cells and 24 hr for FDC-P1.2 cells; Fig. 1C). By contrast, when examined for regulation at the RNA level, transcripts for the cell-death antagonists bcl-2, bcl-X, A-1, and _mcl_-1 were all dependent on IL-3, whereas RNA levels of the cell-death agonists bax, bad, and bak were independent of ligand (Fig. 2A; data not shown). Thus, although IL-3 signaling apparently regulates the transcription of all family members that function as cell-death antagonists, only the down-regulation of Bcl-XL protein was consistent kinetically with a key role in regulating the PCD of myeloid progenitors.
Figure 1.

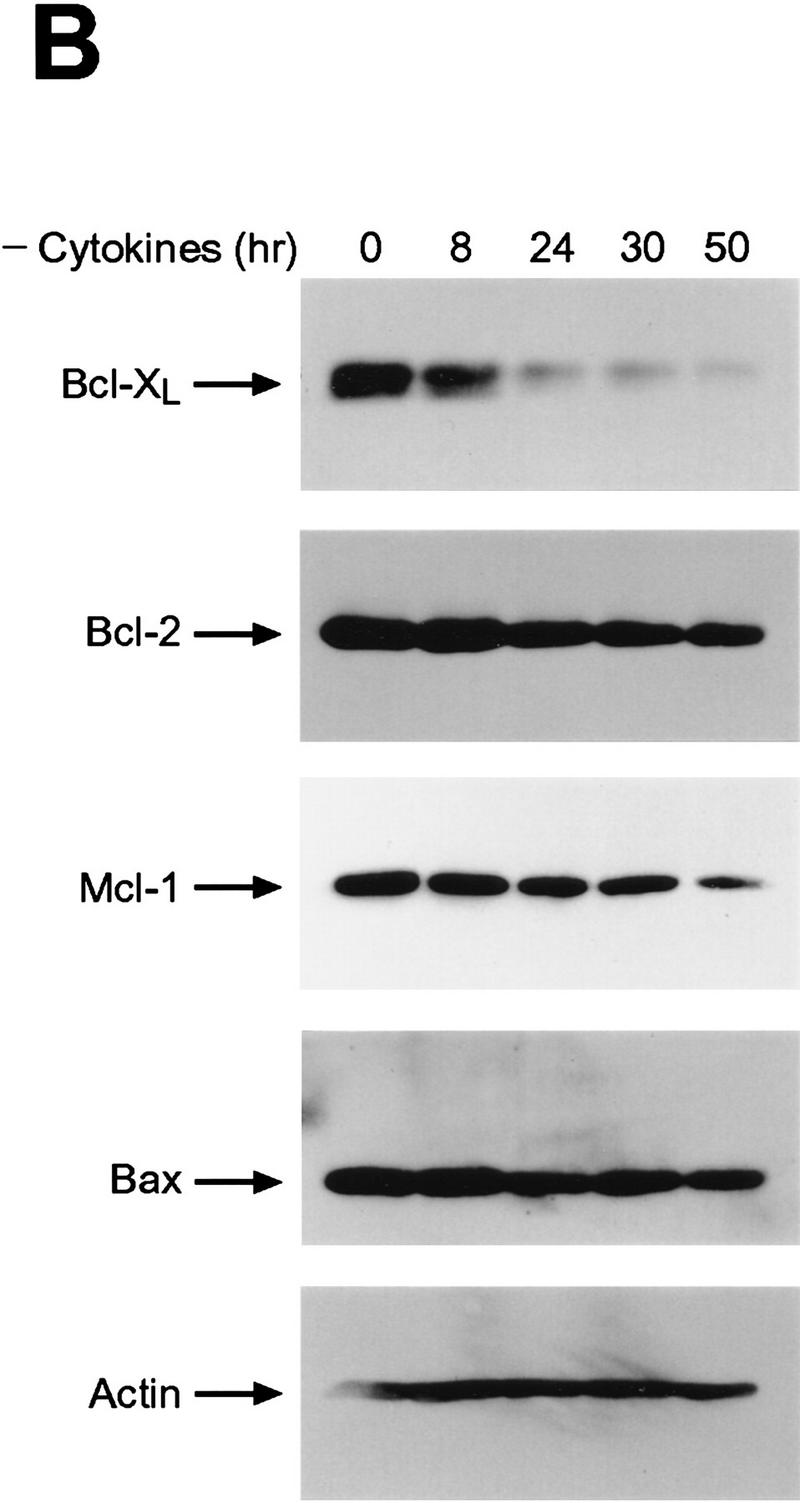
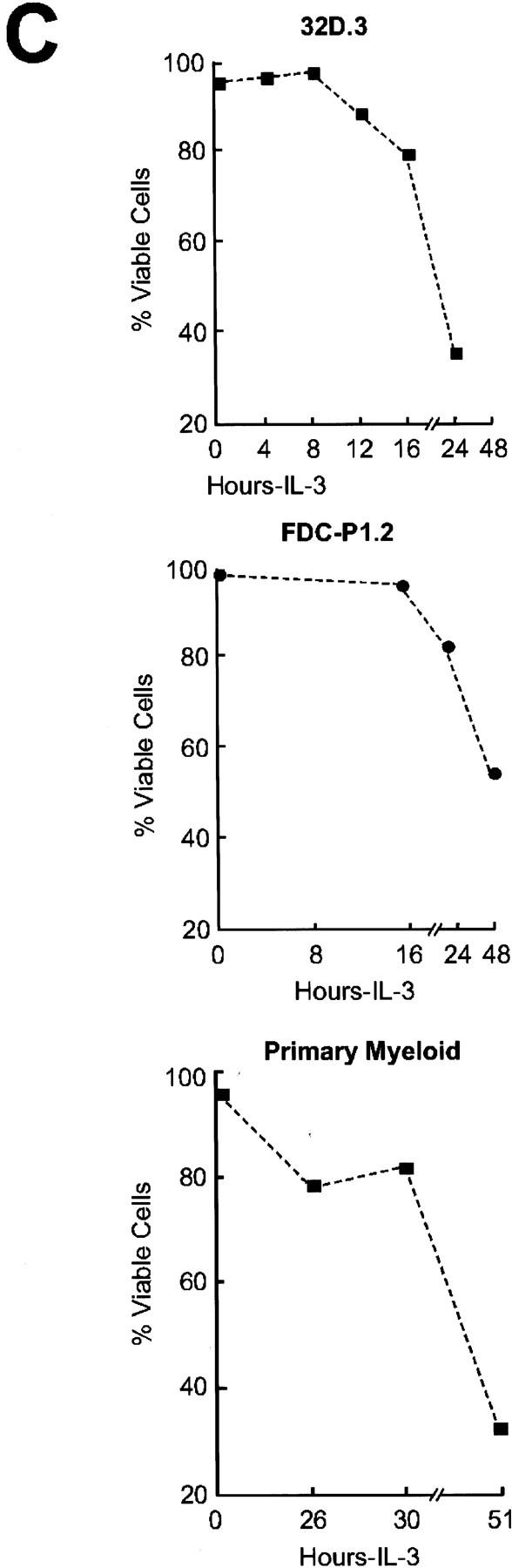
Bcl-XL protein levels are selectively regulated by hemopoietins. (A) Levels of Bcl-2 family members were assessed by immunoblot analyses with antibodies that detect murine Bcl-2, Bcl-XL, Mcl-1, Bax, Bad, and Bak proteins. Protein extracts (50 μg each) were prepared from 32D.3 (left) and FDC-P1.2 (right) myeloid cells growing in IL-3 or deprived of IL-3 for the indicated intervals and analyzed by immunoblots. Results shown are representative of six independent experiments. (B) Fetal liver-derived myeloid cells were grown in IL-3 (20 U/ml), IL-6 (10 ng/ml), and SCF (10 ng/ml) and then were deprived of all hemopoietins. Protein extracts (40 μg each) were prepared from cells after the indicated times after removal of hemopoietins and analyzed by immunoblot. Results shown are representative of three independent experiments. (C) The rates of death of 32D.3, FDC-P1.2, and primary myeloid cells were assessed following withdrawal of cytokines by trypan blue dye exclusion. Cell death was always apoptotic based on morphological criteria, and AnnexinV and TUNEL assays (data not shown). Results shown are representative of 10 independent experiments for 32D.3 and FDC-P1 cells and three for primary myeloid cells.
Figure 2.

Induction of Bcl-XL protein parallels the regulation of bcl-X gene expression. (A) bcl-X is a delayed-early class gene. Asynchronously growing 32D.3 cells were deprived of IL-3 for 14 hr and then treated with 100 U/ml of IL-3, 25 μg/ml cycloheximide (CHX), or pretreated with 25 μg/ml CHX before adding IL-3. This dose of CHX blocked ⩾95% of protein synthesis within 15 min (data not shown). Total RNA was prepared from cells at the indicated intervals and 20 μg analyzed for bcl-X RNA levels using a coding-region probe. Control hybridizations showed equal loading (data not shown). (B) Kinetics of Bcl-XL induction by IL-3. Levels of Bcl-2 and Bcl-XL were assessed in 32D.3 myeloid cells deprived of IL-3 for 15 hr or following stimulation of ligand-starved cells with IL-3 (20 U/ml) for the indicated intervals. Extracts were prepared and 50 μg of protein was analyzed by immunoblot for changes in Bcl-2 and Bcl-XL levels.
To confirm that the observed regulation of Bcl-XL in these immortal myeloid cell lines was representative of the response of primary progenitors, we also analyzed the regulation of Bcl-2 family members in primary, fetal-liver-derived, myeloid cell cultures (Pierce et al. 1985) grown in IL-3, IL-6, and stem cell factor. These cells had hallmarks of myeloid stem cells, being CD34+, c-Kit+, Sca-1+, and lin− (data not shown). As observed in the immortal cells, Bcl-XL was the only cell-death antagonist whose levels were significantly down-regulated following the withdrawal of cytokines from these primary cells (Fig. 1B), and the down-regulation of Bcl-XL levels was coincident with the commitment point for the PCD of primary myeloid cells (Fig. 1C).
The observations that Bcl-XL protein was down-regulated in myeloid progenitors following withdrawal of hemopoietins suggested that readdition of cytokines to ligand-starved cells would up-regulate Bcl-XL protein levels, and parallel regulation of bcl-X RNA levels. In 32D.3 myeloid cells deprived of ligand, IL-3 induced robust increases of bcl-X transcripts, yet the kinetics of bcl-X induction were delayed relative to that of immediate early genes like c-myc (Dean et al. 1987; Askew et al. 1991), suggesting that induction of bcl-X might require de novo protein synthesis. Indeed, pretreatment of ligand-starved cells with cycloheximide blocked the ability of IL-3 to induce bcl-X (Fig. 2A), indicating that bcl-X is a delayed-early class gene whose induction requires the synthesis of a cytokine-regulated factor. Examination of Bcl-XL protein levels by immunoblots demonstrated that the kinetics of induction of Bcl-XL followed temporally the induction of bcl-X transcripts. Bcl-XL levels were increased markedly after 3 hr of addition of IL-3 and continued to accumulate (Fig. 2B). By contrast, the steady-state protein levels of other Bcl-2 family members were not regulated by IL-3 stimulation of ligand-starved cells (Fig. 2B, and data not shown). Thus, unlike other Bcl-2 family members, Bcl-XL levels are regulated tightly by IL-3, and this regulation parallels that observed at the RNA level.
In contrast to observations in pro-B cells, we failed to detect smaller forms of Bcl-2 or Bcl-XL in immortal or primary myeloid progenitors following IL-3 withdrawal that were comparable in size to those suggested to be caspase-cleaved forms of Bcl-2 and Bcl-XL (Cheng et al. 1997; Clem et al. 1998), even when cells were <40% viable as measured by trypan blue dye exclusion (Fig. 1C). Thus, the down-regulation of bcl-X mRNA expression and parallel decreases in the steady-state levels of Bcl-XL protein, rather than cleavage to a pro-apoptotic form (or cleavage of Bcl-2), correlates with the PCDs of these myeloid progenitors. Although the murine bcl-X gene expresses three Bcl-X isoforms, Bcl-XL, Bcl-Xβ, and Bcl-Xγ (González-García et al. 1994; Yang et al. 1997), only the expression of Bcl-XL protein was detected in these cells.
To confirm that the selective down-regulation of Bcl-XL levels was relevant to PCD following IL-3 withdrawal, we generated 32D.3-derived cells that constitutively overexpressed (SFFV–LTR-driven; Boise et al. 1993) murine Bcl-XL (Fig. 3A). As expected (Boise et al. 1993; Vaux et al. 1988) pools and clones of 32D.3 myeloid cells overexpressing Bcl-XL displayed remarkably extended survival when shifted to medium lacking IL-3 relative to vector-only pools and clones (Fig. 3B). Thus, the selective down-regulation of Bcl-XL is relevant for PCD observed following the withdrawal of hemopoietins.
Figure 3.
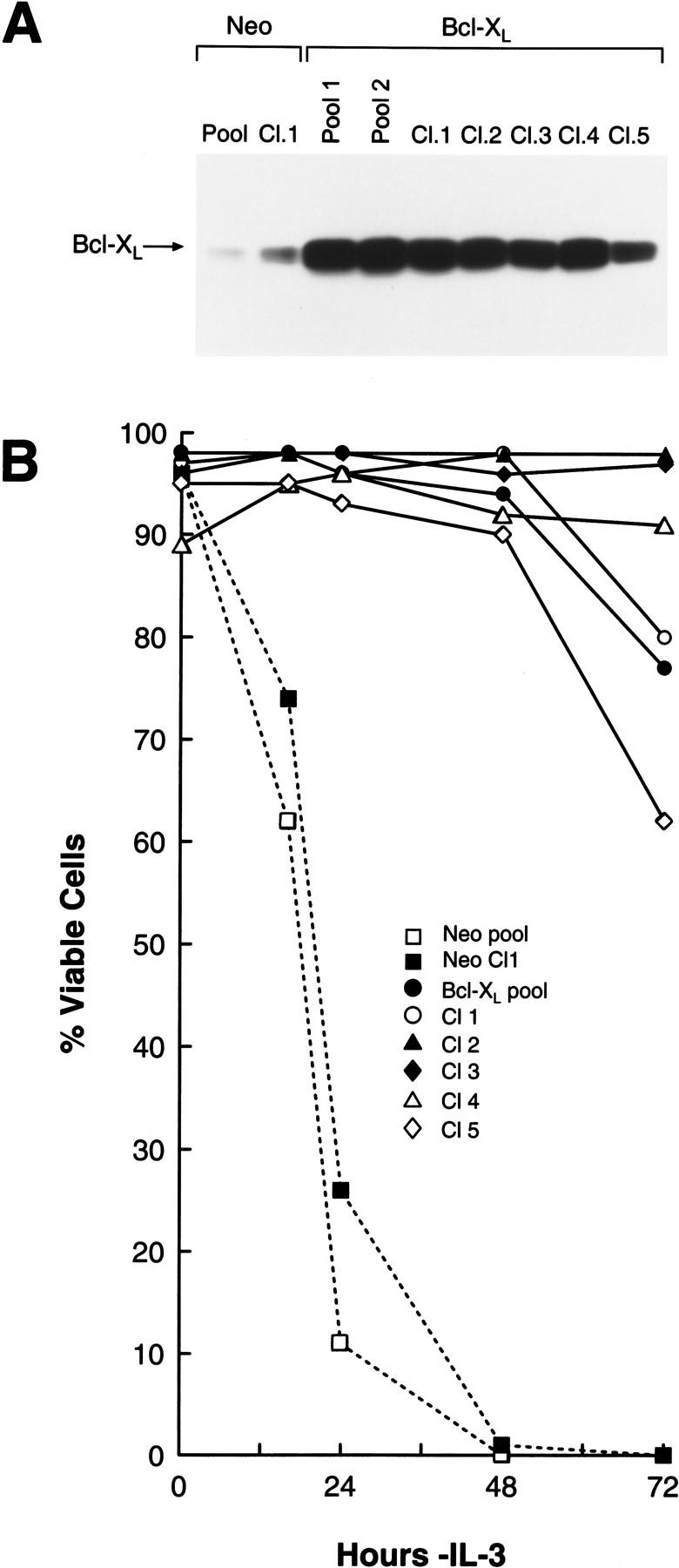
Enforced expression of Bcl-XL suppresses PCD of myeloid cells in the absence of survival factors. (A) Immunoblot analyses of pools and clones of 32D.3-derived, murine Bcl-XL-overexpressing cells exponentially growing in IL-3 medium. Whole cell extract (50 μg) was analyzed for levels of Bcl-XL and Bax. (B) Overexpression of Bcl-XL suppresses apoptosis. Control and Bcl-XL overexpressing pools and clones were deprived of IL-3 and at the indicated interval the percentage of viable cells was assessed by trypan blue dye exclusion. Cells overexpressing Bcl-XL constitutively expressed Bcl-XL in the absence of IL-3 and arrested in G0/G1 (data not shown). Data shown are representative of four separate experiments.
The Jak kinase pathway is necessary and sufficient for the regulation of Bcl-XL protein levels by hemopoietins
Mutational analyses have demonstrated that cytokine receptors utilize multiple signaling pathways following ligand binding, and many of these, including the PI-3′ kinase/Akt kinase, Ras/Raf, and Stat pathways, have been implicated as regulators of cell survival (Cleveland et al. 1994; Kinoshita et al. 1995; Fukada et al. 1996; Canman et al. 1995; Delpeso et al. 1997; Songyang et al. 1997). For cytokines such as IL-3 and Epo, activation of these pathways requires the activity of Jak kinases that associate with and phosphorylate ligated cytokine receptors and their signaling effectors (Witthuhn et al. 1993; Miura et al. 1994a; Damen et al. 1995; Ihle 1995; Quelle et al. 1996).
To identify which hemopoietin-signaling pathways regulate Bcl-XL protein levels, we took advantage of a series of well-characterized 32D.3-derived transfectants engineered to express different versions of the erythropoietin receptor (EpoR). The EpoR has been very well characterized in terms of its signaling pathways and mutants of the EpoR have been created that abolish selectively the PI-3′ kinase/Akt, Ras/MAPK, and STAT pathways (Miura et al. 1993, 1994a,b; Quelle et al. 1996). The EpoR carboxy-terminal truncation mutants EpoR-H and EpoR-S cannot activate the PI-3′ kinase and Ras/MAPK pathways, and EpoR-S is defective additionally in activation of the STAT pathway (Ihle 1995; Quelle et al. 1996). In contrast, the internal deletion mutant in homology Box1 of the Epo receptor, EpoR–PB, is defective in activation of Jak2, the only Jak kinase activated by Epo stimulation (Witthuhn et al. 1993). To address which signal was required for maintenance of Bcl-XL levels, cells expressing these mutated EpoRs were shifted from IL-3 to Epo and expression of Bcl-2 family members was assessed by immunoblots. As expected, EpoR-H and EpoR-S transfectants maintained viability and continued to proliferate when shifted to medium containing Epo, whereas EpoR–PB transfectants began to lose viability at 16 hr following transfer to Epo (Miura et al. 1993 and data not shown). Bcl-XL levels were maintained in Epo-stimulated cells bearing EpoR-H and EpoR-S receptors, but were down-regulated in cells bearing EpoR-PB at a time coincident with the PCD of these cells (Fig. 4A). By contrast, and as expected, levels of Bcl-2 did not change appreciably in any of the transfectants when they were shifted to Epo. Thus, the selective regulation of Bcl-XL protein levels is dependent upon a Jak kinase-regulated survival pathway and is independent of activation of PI-3′ kinase/Akt, Ras, and STAT pathways.
Figure 4.


Activation of Jak2 kinase is necessary and sufficient for the induction of Bcl-XL by hemopoietins. (A) Maintenance of Bcl-XL levels requires activation of the Jak kinase pathway. Exponentially growing cultures of the indicated cell lines in IL-3 (lane 0) were deprived of IL-3 by washing cells in medium lacking IL-3 and then cultured in medium supplemented with 3 units Epo/ml. At the indicated intervals extracts were prepared and 50 μg of protein was analyzed by immunoblots for changes in Bcl-2 and Bcl-XL levels. Results shown are representative of three independent experiments. 32D.3 cells engineered to overexpress the wild-type EpoR (Miura et al. 1993) behaved like EpoR-H and EpoR-S cells in their regulation of Bcl-XL (data not shown). (B) Jak2 is sufficient for the induction of Bcl-XL. 32D.3 cells engineered to overexpress the indicated chimeric EGFR-Jak2 kinase domain receptors have been previously described (Nakamura et al. 1996) and express comparable numbers of chimeric receptors (Quelle et al. 1998). Exponentially growing cultures of the indicated cells in IL-3 were deprived of IL-3 for 12 hr by washing cells in medium lacking IL-3 and then stimulated with 50 ng/ml purified EGF or 20 U/ml IL-3. At the indicated intervals extracts were prepared and 50 μg of protein was analyzed by immunoblots for changes in Bcl-2 and Bcl-XL levels. Results shown are representative of three independent experiments.
Because activation of a Jak kinase pathway was necessary for regulation of Bcl-XL, we also tested whether activation of Jak kinase was sufficient for the induction of Bcl-XL. To address this issue we took advantage of 32D.3 transfectants harboring epidermal growth factor receptor (EGFR)–Jak2 kinase domain chimeric receptors that contain the extracellular ligand binding and transmembrane domains of the EGFR fused to the kinase domain of Jak2 (Nakamura et al. 1996). Stimulation of these cells with EGF activates this chimeric kinase and supports survival of these cells in the absence of IL-3. By contrast, stimulation of cells harboring a chimeric receptor containing multiple mutations in the ATP binding site of the Jak2 kinase domain (kinase dead) fails to support cell survival (Quelle et al. 1998). EGF treatment of ligand-starved cells harboring the wild-type, but not the kinase-dead EGFR-Jak2 chimeric receptor induced Bcl-XL levels (Fig. 4B), and up-regulation in wild-type EGFR–Jak2 transfectants correlated with the extended survival of these cells in EGF (data not shown). By contrast, treatment with IL-3 resulted in a similar up-regulation of Bcl-XL in both chimeric derivatives. Levels of Bcl-XL induced by EGF in wild-type EGFR-Jak2 chimeric-receptor-expressing cells were equivalent to those induced by IL-3, albeit with slightly delayed kinetics. Again induction of Bcl-XL was selective, as Bcl-2 levels remained relatively unchanged after treatment with EGF or IL-3 (Fig. 4B). Thus, activation of Jak kinase pathways is sufficient for up-regulation of Bcl-XL and is required for cell survival.
Bcl-X is selectively activated by retroviral insertions in murine myeloid and T-cell malignancies
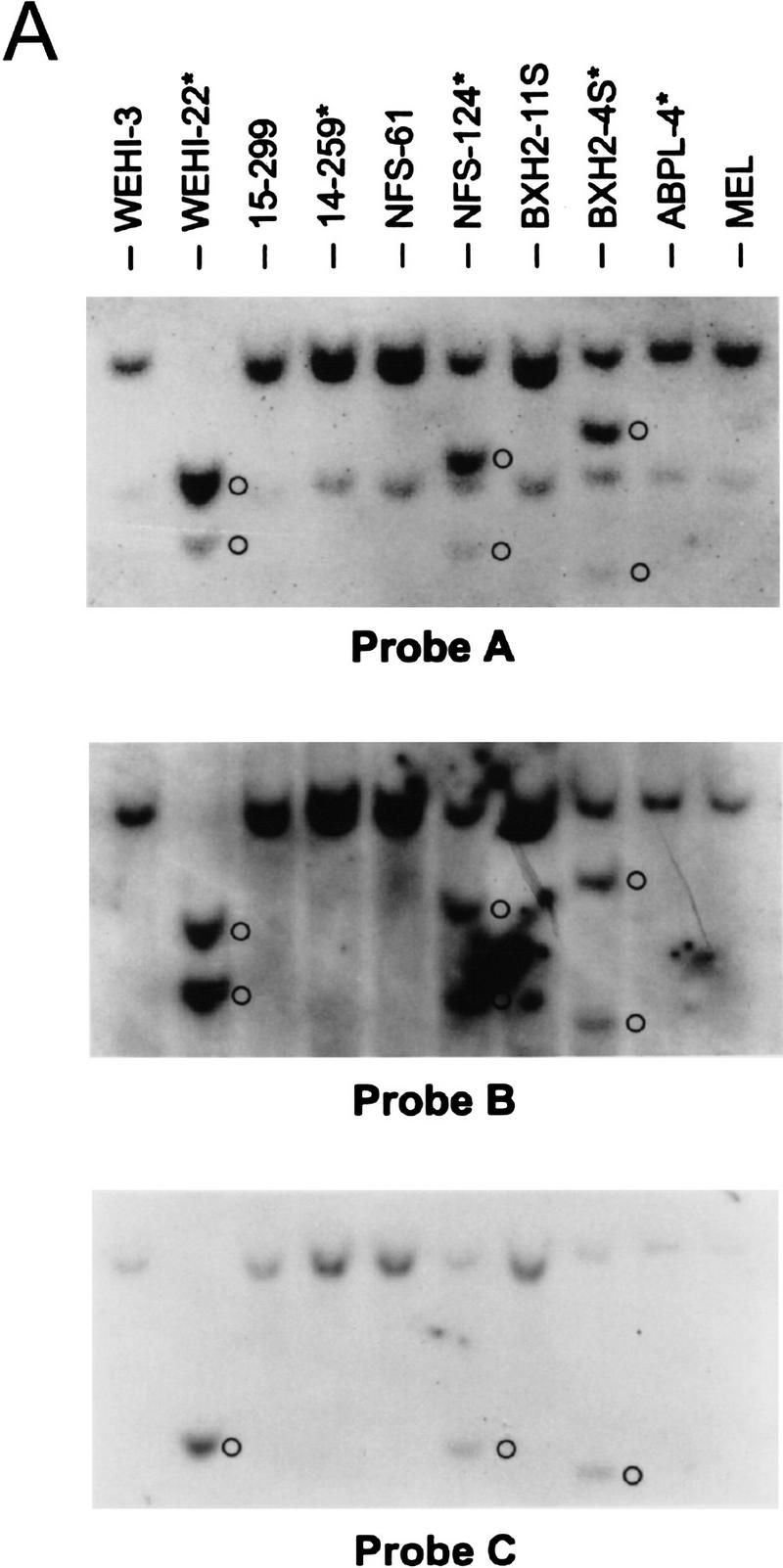
Activation of BCL-2 and mutational inactivation of BAX are observed in only a fraction of hematopoietic malignancies. Given the selective cytokine-dependent nature of Bcl-XL regulation, and its ability to protect hematopoietic progenitors from apoptosis, we addressed whether Bcl-X might contribute to hematopoietic cell transformation. We screened a large panel of spontaneous, retrovirus- or radiation-induced murine leukemia and lymphoma cell lines [_n_ = 51, of myeloid, T-, and B-lymphoid cell lineage (Table 1; Askew et al. 1993)] for changes in the expression of bcl-X RNA by Northern blot analyses with a bcl-X coding-region probe. Although most tumor lines expressed the expected 2.7-kb bcl-X RNA, we detected elevated levels of altered, high-molecular-weight bcl-X RNAs in 5 (9.8%) of the tumor-derived lines (14–259, NFS-124, BXH2-4S, WEHI-22, and ABPL-4 cells; Fig. 5A; Table 1). WEHI-22 cells also expressed a smaller bcl-X RNA and failed to express the normal 2.7-kb RNA, whereas the other tumor lines also expressed low levels of the normal 2.7-kb transcript (Fig. 5A). Altered bcl-X RNA expression was restricted to leukemic cells of myeloid or T-cell lineage that lacked requirements for specific hemopoietins normally essential for cell survival. Altered bcl-X transcripts were not evident in any of the B-cell-derived malignancies tested (n = 11). Overall, aberrant bcl-X RNAs were detected in 4/18 (22%) myeloid and 1/6 (16.6%) T-cell growth-factor-independent leukemias. Although levels of transcripts of some of the other Bcl-2 family members varied, we failed to detect any tumors having visibly altered RNAs (Table 1; data not shown), demonstrating that the bcl-X alterations were selective. Leukemic cells having altered bcl-X transcripts constitutively expressed bcl-X RNA independent of cytokines or serum (data not shown). Elevated levels of Bcl-XL protein were observed in four of the tumor lines relative to controls matched for mouse strain and tumor-cell lineage (Fig. 5B). We did not detect Bcl-X proteins with altered mobilities in these leukemias, suggesting that the bcl-X open reading frame (ORF) had been preserved.
Table 1.
Expression of Bcl-2 family members in murine myeloid and T-cell hematologic malignancies
| Tumor line | Cytokine dependencea | Lineageb | bcl-2 | bcl-x | mcl-1 | A-1 | bax | bad | bak |
|---|---|---|---|---|---|---|---|---|---|
| DA-1 | IL-3 | myeloid | − | + | + | + | + | − | +c |
| DA-3 | IL-3 | myeloid | + | + | + | N.T.d | + | N.T. | N.T. |
| DA-7 | IL-3 | myeloid | + | + | + | + | + | ++ | − |
| DA-13 | IL-3 | myeloid | + | + | + | +++ | + | + | +e |
| DA-22 | IL-3 | myeloid | + | + | + | N.T. | + | N.T. | N.T. |
| DA-24 | IL-3 | myeloid | + | + | + | − | + | + | +++e |
| DA-28 | IL-3 | myeloid | + | + | + | − | + | + | +c |
| DA-29 | IL-3 | myeloid | − | + | + | N.T. | + | N.T. | N.T. |
| DA-31 | IL-3 | myeloid | − | + | + | N.T. | − | N.T. | N.T. |
| DA-34 | IL-3 | myeloid | + | + | + | N.T. | + | N.T. | N.T. |
| NFS-36 | IL-3 | myeloid | + | + | + | − | + | ++ | +c |
| NFS-58 | IL-3 | myeloid | ++ | + | + | N.T. | + | N.T. | N.T. |
| NFS-60 | IL-3 | myeloid | + | + | + | N.T. | + | N.T. | N.T. |
| NFS-78 | IL-3 | myeloid | + | + | ++ | N.T. | + | N.T. | N.T. |
| NFS-107 | IL-3 | myeloid | ++ | + | + | + | + | + | +e |
| DA-33 | − | myeloid | + | + | + | + | + | + | − |
| NFS-56 | − | myeloid | + | + | + | − | + | + | + |
| NFS-61 | − | myeloid | + | ++ | + | − | + | + | +c |
| NFS-124f | − | myeloid | + | +++ | + | +++ | + | + | +c |
| C6 | − | myeloid | ++ | + | + | − | + | + | +c |
| C10 | − | myeloid | + | + | + | N.T. | + | N.T. | N.T. |
| 14-122 | − | myeloid | + | + | + | − | + | + | +c |
| 14-166 | − | myeloid | + | + | + | − | + | + | +c |
| 14-259f | − | myeloid | + | +++ | + | +++ | + | + | +c |
| 15-299 | − | myeloid | + | + | + | − | + | + | +c |
| 7M12 | − | myeloid | + | + | + | − | + | + | +c |
| M1 | − | myeloid | ++ | + | + | +++ | + | + | +c |
| ABPL-4f | − | myeloid | + | +++ | + | +++ | + | + | +c |
| BXH2-4Sf | − | myeloid | + | ++ | + | +++ | + | + | +c |
| BXH2-11S | − | myeloid | − | + | + | +++ | + | + | +c |
| WEHI-3 | − | myeloid | + | + | + | ++ | + | + | +c |
| AFSTL-1 | − | mast | + | + | + | + | + | + | +c |
| AFSTL-2 | − | mast | + | + | + | − | + | + | +c |
| WEHI-22f | − | T-cell | + | +++ | + | − | + | + | +c |
| WEHI-164 | − | T-cell | + | + | + | − | + | + | +c |
| WEHI-279 | − | T-cell | − | + | ++ | +++ | + | + | +c |
| RL-12 | − | T-cell | + | + | + | − | + | + | +c |
| EL-4 | − | T-cell | + | + | ++ | +++ | + | ++ | ++c |
| DA-2 | − | T-cell | + | ++ | ++ | − | + | + | +c |
| CTLL | IL-2 | T-cell | + | + | + | − | + | + | +c |
Figure 5.
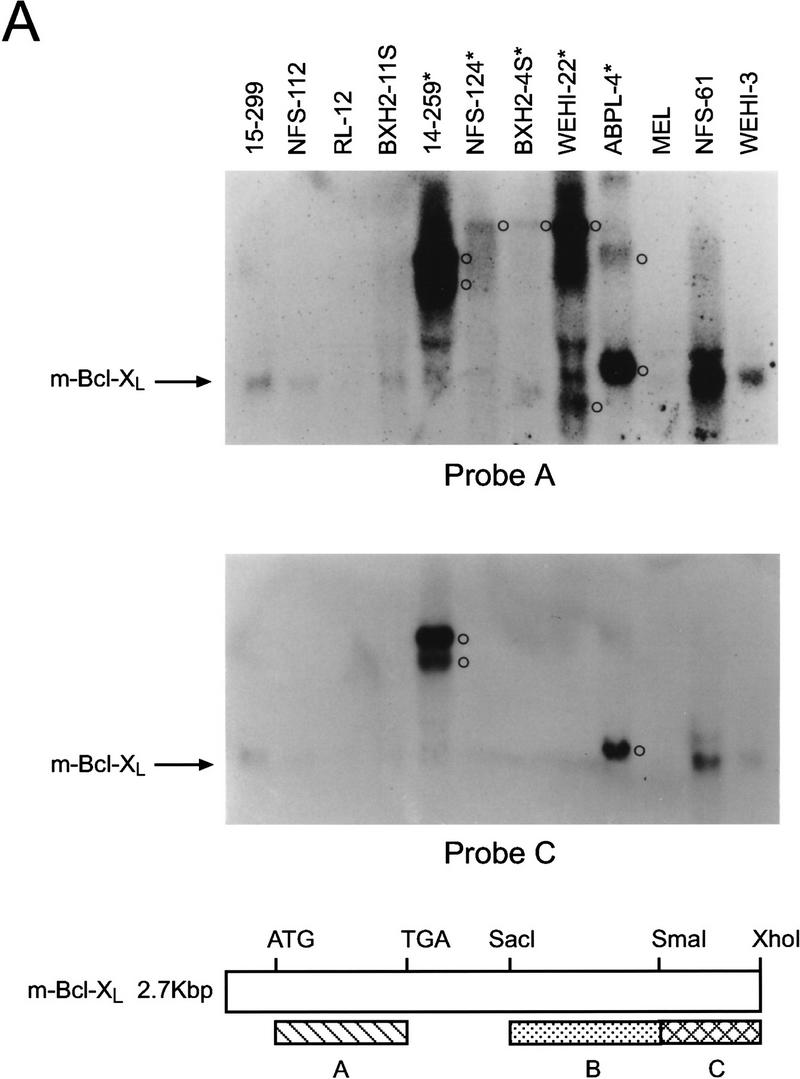

Bcl-X is activated in hematopoietic malignancies. (A) Total RNA was prepared from the indicated tumor cells and analyzed by Northern blot with bcl-X probes spanning the coding portion of the cDNA (Probe A, top) or the 3′ UTR (Probe C, a _Sma_I–_Xho_I fragment, see schematic). (*) Tumor lines having altered bcl-X transcripts; (○) altered transcripts. (B) Bcl-X proteins were evaluated by immunoblot in cells having altered bcl-X transcripts compared to control tumors lines matched for lineage and strain. (*) Tumor lines having altered bcl-X transcripts. To control for loading the membrane was blotted with antibody specific for murine Bag-1 (Packham et al. 1997).
Many tumor lines with altered bcl-X RNAs were induced by retroviral infection (Askew et al. 1993), suggesting that the altered RNAs were caused by fusion with retroviral transcripts generated from insertions in the bcl-X gene. Consistent with this interpretation, altered bcl-X transcripts also hybridized with an LTR-specific probe (data not shown). The RNA encoding Bcl-XL contains three exons (Grillot et al. 1997) and to assess the structure of altered bcl-X RNAs, we initially used a probe derived from the 3′ untranslated region (UTR) of bcl-X exon 3 in Northern blots. This probe failed to detect the altered bcl-X RNAs in NFS-124, BXH2-4S, and WEHI-22 cells (Fig. 5A), indicating that the putative insertion site in bcl-X in these cells is likely 5′ of the _Sma_I site in the 3′ UTR of exon 3. In contrast, the 3′ bcl-X UTR probe detected altered bcl-X RNAs in APBL-4 and 14–259 cells. Thus, the putative retroviral insertions in these lines lie either 5′ of the bcl-X ORF or within the large intron 2 of bcl-X (Grillot et al. 1997).
By Southern blot analysis, bcl-X gene rearrangements were evident in WEHI-22, NFS-124, and BXH2-4S cells and mapped within a 793-bp region between the _Sac_I and Sma_I sites present in the noncoding portion of bcl-X exon 3 (Fig. 6A). In WEHI-22 cells, which lacked expression of the normal 2.7-kb bcl-X RNA (Fig. 5A), no bands were shared with matched controls (from tumors with normal bcl-X transcripts) indicating that, in addition to gene rearrangement, they had lost the normal bcl-X allele. Duplication of the altered bcl-X allele was also evident in WEHI-22 cells (Fig. 6A). By contrast, NFS-124 and BXH2-4S cells retained an apparently normal bcl-X allele in addition to the rearranged allele, consistent with the expression of both normal and altered bcl-X RNAs (Fig. 5A). There was no evidence for bcl-X gene rearrangements in APBL-4 and 14–259 cells by conventional Southern blot analysis (Fig. 6A), yet alterations of larger DNA fragments were detected by pulse-field gel electrophoresis (Fig. 6B). Therefore, these tumor lines also have retroviral insertions that alter bcl-X expression, but the sites of insertion are relatively distant or are within intron 2. In these leukemias retroviral–_bcl-X fusion transcripts (Fig. 5A) must include splicing into bcl-X exon 2, which contains the Bcl-XL initiator codon.
Figure 6.

bcl-X is rearranged in tumor lines having altered bcl-X transcripts. (A) Southern blot analyses of _Kpn_I + _Eco_RI-restricted genomic DNA of tumor lines having altered bcl-X transcripts (*) relative to tumors matched for lineage and mouse strain. No polymorphisms were detected with five distinct restriction digests of these matched tumor line DNAs (data not shown). Blots were hybridized sequentially to probes A, B, and C (see Fig. 5A). (B) Blot analyses of pulsed-field inversion gels of genomic DNA from bcl-X rearranged tumors (*) vs. matched controls (15–299 and ABPL-2) using the full-length bcl-X cDNA as region probe. (○) Altered bands. Genomic DNA was digested with _Sfi_I (odd numbered lanes) or _Mlu_I (even-numbered lanes).
To pinpoint the retroviral insertion sites in WEHI-22, BXH2-4S, and NFS-124 cells, we performed 3′ rapid amplification of cDNA ends (RACE) and the PCR products were cloned and sequenced. Three unique retrovirus integration sites were identified (Fig. 7A) and, in agreement with Northern and Western blot analyses (Fig. 5), the integrations were outside of the Bcl-XL ORF in the 3′ UTR of the gene (Fig. 7A). Sequence analysis of the viral portion of the _bcl-X_–retrovirus fusion RNAs demonstrated they were identical to those of the virus used to derive the tumors (data not shown). The integration sites were further characterized by performing RT–PCR using _bcl-X_- and LTR-specific primers (Fig. 7B) and sequencing these products (data not shown). Overall these data demonstrate that activation of bcl-X in hematopoietic malignancies preserves the ORF and results in enhanced and constitutive expression of the gene, underscoring its important role as a regulator of PCD of hematopoietic progenitors.
Figure 7.


bcl-X is activated by retroviral insertions. (A) 3′ RACE extended bcl-X cDNAs were generated from WEHI-22, NFS-124, and BXH2-4S RNAs and the products amplified by PCR. The PCR products were then cloned and sequenced. (Top) Diagrammatic of bcl-X exon 3 (Grillot et al. 1997) and the sites of retroviral insertions; (bottom) the sequence of these junctions. The shaded region indicates bcl-X sequences and number denotes nucleotide number of bcl-X exon 3. Identical results were obtained in two additional 3′ RACE analyses. Retroviral integrations occurred at the following position of bcl-X exon 3: WEHI-22, nucleotide 576; and NFS-124, nucleotide 652; BXH2-4S, nucleotide 823. (B) RT–PCR analyses of _bcl-X_–LTR fusion transcripts. Primers spanning the identified _bcl-X_-retroviral junctions were used to amplify cDNA made from the indicated RNA samples, and the resultant PCR products were analyzed by agarose-gel electrophoresis. Products of the expected lengths were obtained (see Materials and Methods) and identity was confirmed by sequencing (data not shown). Control lane is the positive RT–PCR reaction provided with the kit.
Discussion
We demonstrate here that Bcl-XL protein levels are regulated selectively by hemopoietins in primary and immortal myeloid progenitors, and that the murine bcl-X gene is activated in myeloid and T-cell malignancies that lack requirements for hemopoietins. These facts, coupled with observations linking the regulation of Bcl-XL to Jak kinase-dependent pathways that are required for cytokine-receptor signaling (Ihle 1995), support the hypothesis that Bcl-XL plays a key role as a cytokine-regulated cell-death antagonist. In agreement with this concept, deletion of bcl-X in mice leads to embryonic lethality that may in part be caused by the rampant apoptosis of hematopoietic progenitors (Motoyama et al. 1995). Thus, it appears that one function of Bcl-XL is as an important and limiting mediator of hemopoietin-dependent survival pathways that regulate progenitor cell numbers in vivo. The bcl-X activations that we have detected in murine malignancies therefore likely contribute to tumorigenesis by providing constitutive signals for cell survival.
The selective regulation of Bcl-XL protein levels by hemopoietins, relative to other Bcl-2 family members, was surprising given that the RNA levels of all cell-death antagonists tested were dependent upon cytokines. On face value this suggests that the half-life of Bcl-XL protein is either shorter than that of other Bcl-2 family members, or that Bcl-XL is targeted selectively for degradation. The rate of decay of Bcl-XL protein following the withdrawal of survival factors roughly parallels the drop in bcl-X transcripts (data not shown). However, the decreases in steady-state levels of Bcl-XL were not associated with the appearance of smaller molecular weight forms that would be comparable to those detected in IL-3 deprived pro-B-lymphoid cells and implicated as caspase-generated pro-apoptotic forms of Bcl-XL and Bcl-2 (Cheng et al. 1997; Clem et al. 1998). This fact may in part explain the apparent increased potential of exogenous Bcl-XL to protect 32D.3 myeloid cells, relative to that seen in BaF-3 pro-B cells, from PCD following removal of IL-3 (cf. Fig. 3B and Clem et al. 1998). Thus, at least in myeloid cells, the PCD that occurs following the withdrawal of survival factors is associated with the down-regulation of steady-state levels of Bcl-XL that parallels silencing of bcl-X gene expression, rather than caspase cleavage. The concept that this form of regulation of Bcl-XL is also operational in other survival pathways is supported by the observations that Bcl-XL protein levels are also regulated by CD40 ligation, IL-3, and IL-2 in lymphocytes (Broome et al. 1995; Tuscano et al. 1996; Leverrier et al. 1997).
Post-translational modification has been proposed to link cytokine signaling and regulation of apoptosis by Bcl-2 family proteins. Phosphorylation of the cell-death agonist Bad in response to IL-3 in pro-B cells prevents the association of Bad with Bcl-XL, thereby releasing Bcl-XL to function, and this is thought to be one signaling pathway through which IL-3 suppresses cell death (Zha et al. 1996; Delpeso et al. 1997). Moreover, the Akt serine/threonine kinase, a downstream effector of the PI-3′ kinase pathway (Franke et al. 1995), has been shown to mediate Bad phosphorylation induced by both NGF and IL-3 (Delpeso et al. 1997; Datta et al. 1997), and myeloid cells engineered to overexpress constitutively activated versions of Akt, have delayed rates of death following the withdrawal of survival factors (Songyang et al. 1997). Along with the data demonstrating that treatment of neuronal cells with the PI-3′ kinase inhibitor wortmannin induces apoptosis (Yao and Cooper 1995; Dudek et al. 1997), these data suggest that the PI-3′/Akt kinase pathway plays a key role in regulating cell survival. Although we cannot strictly rule out a contribution of the PI-3′/Akt kinase pathway to myeloid cell survival, several observations are inconsistent with this pathway having an essential role. First, myeloid-cell survival is supported by versions of the EpoR that are defective in activation of PI-3′ kinase (Miura et al. 1993; 1994a; Damen et al. 1995), yet are competent to signal through Jak kinase (Witthuhn et al. 1993) and maintain Bcl-XL expression (Fig. 4A). Second, myeloid cells are remarkably resistant to the effects of wortmannin. At concentrations of wortmannin that inhibit Akt activity in myeloid cells (20 nm, Songyang et al. 1997), or at concentrations as high as 1 μm, there are no deleterious effects on the survival of primary or immortal myeloid cells (data not shown). Third, we have failed to detect any changes in the phosphorylation status of Bad potein following cytokine treatment of myeloid cells (data not shown). Therefore, at a minimum our data now suggest that regulation of Bcl-XL levels is also a critical contributor to hemopoietin-dependent survival pathways of myeloid cells.
In addition to the PI-3′/Akt kinase pathway, STAT and Ras/Raf pathways have also been implicated as mediators of cytokine survival pathways. Dominant-negative versions of STAT-3 or activated Ras or Raf-1 have been shown to modulate hematopoietic cell survival (Cleveland et al. 1994; Canman et al. 1995; Fukada et al. 1996). Moreover, forms of the GM-CSF receptor that fail to activate the Ras/Raf pathway are unable to support cell survival under serum-free conditions (Kinoshita et al. 1995). By contrast, the activation of Ras/Raf or STAT pathways in myeloid cells is dispensable for Epo-mediated cell survival (Miura et al. 1993; Quelle et al. 1996) and regulating Bcl-XL (Fig. 4A). Furthermore, the rather modest effects of Raf-1 on promoting 32D.3 cell survival (Cleveland et al. 1994), relative to Bcl-2 or Bcl-XL overexpression (Nip et al. 1997, Fig. 3B), are independent of any effects upon Bcl-XL regulation (G. Packham and J.L. Cleveland, unpubl.). Rather, our results suggest that a Jak-kinase dependent signaling pathway emanating from the membrane proximal domain of cytokine receptors is necessary and sufficient as an upstream signal that regulates cell survival and Bcl-XL. Interestingly, this same pathway has also been linked to the control of hematopoietic cell survival in response to DNA damage (Quelle et al. 1998). Up-regulation of the Jak kinase pathway has been implicated in leukemia (Lacronique et al. 1997), whereas deletion of Jak2 or Jak3 in mice compromises the development of the myeloid/erythroid or lymphoid compartments, respectively (Nosaka et al. 1995; Parganas et al. 1998). Our findings that Bcl-XL is downstream of this pathway, and is regulated selectively by hemopoietins and activated in hematopoietic malignancies, underscores the importance of this pathway in the control of PCD and in cancer.
Finally, to our knowledge this is the first direct evidence that bcl-X is activated in cancer. Of particular note are the facts that for each tumor bearing alterations of bcl-X mRNA, the rearrangements preserved the ORF and led to the constitutive, growth-factor-independent, expression of the gene, which normally is dependent upon cytokines (Fig. 2A). Furthermore, these observations suggest that components of the Jak kinase survival pathway leading to Bcl-XL expression may also be targets for activation in human cancer. Finally, these data predict that BCL-X contributes directly or indirectly to human malignancies. Although translocations involving BCL-X at 20q11 (NCBI STS Data Base, accession no. U72398) have yet to be observed, this region is associated with interstitial deletions common in myeloid malignancies (Kurtin et al. 1996), and it is possible these deletions augment expression of BCL-XL. Moreover, BCL-XL is expressed highly in some human cancers without obvious genetic alterations (Foreman et al. 1996; Pallis et al. 1997; Silva et al. 1998; Tu et al. 1998), suggesting that other mechanisms may lead to BCL-XL overexpression.
Materials and methods
Cell culture
The tumor cells used in this study were maintained in RPMI-1640/10% FCS + l-glutamine and, where necessary, 20 units murine IL-3/ml (Dean et al. 1987; Askew et al. 1991). Radiation-induced leukemias were provided by David Askew (University of Cincinnati, OH). Others have been described previously (Askew et al. 1993). 32D.3-cell-derived pools and clones that overexpress murine Bcl-XL were generated using the SFFV–Bcl-XL expression construct (Boise et al. 1993) as described previously (Packham and Cleveland 1994). Analyses of the affects of IL-3 withdrawal on apoptosis and/or expression of Bcl-2 family members in 32D.3 and FDC-P1.2 cells and stable transfectants were performed as described previously (Packham and Cleveland 1994). Exponentially growing cells were seeded on consecutive days at 5 × 105 cells per ml and on day 3 were deprived of IL-3 by washing cells in RPMI-1640 medium lacking IL-3. Cells were reset at 0.5 × 106 cells per ml in RPMI-1640/10% FCS medium and cell viability determined by trypan blue dye exclusion (Askew et al. 1991). 32D.3-derived cells overexpressing wild type and mutants of the EpoR have been described previously and express equivalent amounts of EpoR on the cell surface (Miura et al. 1993). Exponentially growing cells in IL-3 media were deprived of IL-3 by washing cells twice in RPMI-1640 and then reset at 0.3 × 106 cells/ml in RPMI-1640/10%FCS medium supplemented with 3 units Epo/ml. 32D.3 myeloid cells engineered to overexpress wild-type and mutant EGF receptor–Jak2 kinase domain chimeric receptors have been described previously and express comparable numbers of chimeric receptors on the cell surface (Nakamura et al. 1996; Quelle et al. 1998). Cells growing in IL-3 medium were deprived of IL-3 by washing in RPMI-1640 media/1% FCS and after 12 hr were stimulated with 50 ng/ml purified EGF or 20 units of IL-3/ml.
Fetal liver-derived myeloid cultures were prepared from day 17 mouse embryos by culture of suspension cells in RPMI-1640 media supplemented with IL-3 (20 U/ml), IL-6 (10 ng/ml), and SCF (10 ng/ml), 10% FCS, and l-glutamine (Pierce et al. 1985). After removal of adherent cells by consecutive passages, proliferating populations were CD34+, Sca1+, and c-Kit+, but were negative for all other more differentiated markers of mature myeloid and lymphoid cells. Antibodies for FACS analyses were purchased from Becton Dickinson. To assess rates of cell death, cells were seeded as above and cultured in RPMI-1640/10% FCS medium lacking cytokines. Cell viability was again determined by trypan blue dye exclusion.
Northern and Southern blotting
Total RNA and genomic DNA isolation and Northern and Southern blotting of agarose and pulsed-field inversion gels were performed using conventional techniques. For Northern blots the coding portions of human mcl-1 and murine bcl-2, bax, A-1, and bad cDNAs were used [provided by R. Craig (Dartmouth Medical School, Hanover, NH), D. Askew, J. Reed (The Burnham Institute, La Jolla, CA), C. Kürschner (St. Jude Children’s Research Hospital), and S. Korsmeyer (Washington University, St. Louis, MO), respectively]. The bcl-X probes were the full-length cDNA probe, a coding region probe, or 3′ UTR-specific probes (Fig. 5A). The human bak cDNA was cloned by conventional RT–PCR.
Immunoblotting
Immunoblots were performed using 40 or 50 μg of whole-cell extracts as described previously (Cleveland et al. 1994). To assure that proteins were not subject to artificial cleavage by caspases following cell lysis, cell were collected, washed once in PBS, and the dry cell pellets snap frozen in liquid nitrogen. Frozen pellets were lysed rapidly on ice in RIPA buffer containing a cocktail of protease inhibitors [AEBSF, E-64, pepstatin, aprotinin, leupeptin, and PMSF (Sigma)], clarified by centrifugation, an aliquot removed for protein assays (Bio-Rad) and immediately boiled for 5 min in 2% SDS sample buffer. In the absence of protease inhibitors, Bcl-XL protein present in lysates from cells undergoing cell death was sometimes processed to forms comparable to putative pro-apoptotic forms observed in pro-B cells (Clem et al. 1998), but was never observed when protease inhibitors were included in the lysis buffer. Antibodies used were as follows: mouse Bcl-2 (15021, PharMingen, 1:250); mouse Bcl-X (B2260, Transduction Labs, 1:250); mouse Bax (13686E, PharMingen, 1:500); Mcl-1 (B54020, Transduction Labs, 1:500); Bak (G-23, Santa Cruz, 1:100); Bad (B36420, Transduction Labs, 1:250) and Bag-1 (Packham et al. 1997). Using Bcl-2, Bcl-XL, Bak, and Bax-specific antibodies from other sources gave comparable results, including those antisera used to detect proteolytically clipped forms of Bcl-2 (Cheng et al. 1997) or Bcl-XL (Clem et al. 1998). Bound immunocomplexes were detected by enhanced chemiluminescence (Amersham) or Supersignal (Pierce).
Rapid amplification of cDNA ends and RT–PCR
3′ RACE for amplification of cDNA ends was performed according to the manufacturer’s instructions (Life Technologies). 3′ RACE products were generated by converting 1 μg RNA template into first-strand cDNA in a reaction volume of 11 μl containing 1× buffer (20 mm Tris-HCl at pH 8.4, 50 mm KCl, 2.5 mm MgCl2, 10 mm DTT, 500 nm adapter primer, 500 μm dNTPs) and 200 units of Superscript II reverse transcriptase. Two microliters of cDNA from first-strand cDNA were amplified using 200 nm gene-specific primer (bcl-X, CCACATCTCAGTTCTCTTGG) and 200 nm abridged universal amplification primer in total reaction volume of 50 μl containing 1× buffer (20 mm Tris-HCl at pH 8.4, 50 mm KCl, 1.5 mm MgCl2, 200 μm dNTPs) and 1.25 units Taq polymerase. Samples were cycled 34 times using an annealing temperature of 47°C. 20 microliters of sample were gel purified and a second amplification was done with primer bcl-X2 (CGCGTCGACTAGTCTAAACCAGCTCCTTGGAG) with the same reaction conditions. 20 microliters of sample was gel purified and subcloned into pGEM-T vector. Clones were cut with _Apa_I to check for inserts and sequenced subsequently on both strands using Taq FS dye terminator chemistry. Retroviral sequences were identified in GenBank by BLAST.
RT–PCR on tumor-derived RNAs having altered bcl-X transcripts was performed according to the manufacturers instructions (Perkin Elmer) using primers specific for the retroviral sequences for reverse transcription of RNA to cDNA, followed by amplification using primers for bcl-X. Viral and bcl-X primers were as follows. For NFS-124: bcl-X, CAGGCTGCTTGGGATAATGAG; retrovirus, CAAACAGAAGCGAGAAGCG (346 bp). For WEHI-22: bcl-X, GAGCCATTGAGTTGAAAGAC; retrovirus, GCACTGCAAGAGGTTTATTG (446 bp). For BXH2-4S: bcl-X, CCACTTGTGGTCTGAATG; retrovirus, GGGAACTTGAGACAATTCTG (412 bp).
cDNA was prepared using 1× buffer [50 mm as above but with 5 mm MgCl2, 50 pmol downstream (retroviral) primer, 1 mm dNTPs, 1 μg RNA template] and 2.5 units of MuLV reverse transcriptase and 1 unit RNase inhibitor in a total reaction volume of 20 μl. Samples were then incubated at 42°C for 15 min, 99°C for 5 min, and 5°C for 5 min. cDNA was then amplified using 1× buffer as above with 50 pmol upstream (bcl-X) primer and 1.25 units of Taq DNA polymerase in a reaction volume of 100 μl. Samples were cycled 39 times in a MJ Research PTC-100 PCR machine using an annealing temperature of 47°C. PCR products (20 μl) were run on an agarose gel and remaining product was purified using Centricon 100 columns according to manufacturer’s recommendations and sequenced by TaqFS dye terminator chemistry.
Acknowledgments
We are grateful to S.J. Korsmeyer, J.C. Reed, R.W. Craig, D. Askew, and C. Kürschner, who provided plasmids of Bcl-2 family members; Carlos Rodriguez-Galindo, Haiqing Dai, and Jinling Wang for their help with some of the experiments; and James Downing and A. Thomas Look for critical reading of the manuscript. We also thank David Askew for providing stocks of the radiation-induced leukemias. This work was supported by the Ludwig Institute for Cancer Research (G.P.), Public Health Service grants CA76379 (J.L.C.), DK42937 (J.N.I.), CA64556 (G.N.), and CA63230 (G.Z.), Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL john.cleveland@stjude.org; FAX (901) 525-8025.
References
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3 dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Askew DS, Bartholomew C, Ihle JN. Insertional mutagenesis and the transformation of hematopoietic stem cells. Hematol Pathol. 1993;7:1–22. [PubMed] [Google Scholar]
- Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:889–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson C B. Bcl-X, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–607. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Brimmell M, Mendiola R, Mangion J, Packham G. Bax frameshift mutations in cell lines derived from hemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene. 1998;16:1803–1812. doi: 10.1038/sj.onc.1201704. [DOI] [PubMed] [Google Scholar]
- Broome HE, Dargan CM, Krajewski S, Reed JC. Expression of Bcl-2, Bcl-X and Bax after T cell activation and IL-2 withdrawal. J Immunol. 1995;155:2311–2317. [PubMed] [Google Scholar]
- Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes & Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- Cheng EHY, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Cleary M, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci. 1985;82:7439–7443. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Cheng EH-Y, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffen DE, Earnshaw WC, Veliuno ME, Hardwick JM. Modulation of cell death by Bcl-XL through caspase interaction. Proc Natl Acad Sci. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland JL, Troppmair J, Packham G, Askew DS, Lloyd P, González-García M, Nuñez G, Ihle JN, Rapp UR. v-raf suppresses apoptosis and promotes growth of Interleukin-3-dependent myeloid cells. Oncogene. 1994;9:2217–2226. [PubMed] [Google Scholar]
- Damen JE, Cutler RL, Jiao J, Yi T, Krystal G. Phosphorylation of tyrosine 503 in the erythropoietin receptor (EpR) is essential for binding P85 subunit of phosphatidylinositol (PI) 3′-kinase and for EpR-associated PI 3-kinase activity. J Biol Chem. 1995;270:23402–23408. doi: 10.1074/jbc.270.40.23402. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu HA, Gotoh Y, Grennberg ME. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Dean MD, Cleveland JL, Rapp UR, Ihle JN. Role of myc in the abrogation of IL-3 dependence of myeloid FDC-P1 cells. Oncogene Res. 1987;1:279–296. [PubMed] [Google Scholar]
- Delpeso L, González-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of Bad through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Dudek H, Data SR, Franke TF, Birnbaum ML, Yao RJ, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Foreman KE, Wrone-Smith T, Boise LH, Thompson CB, Polverini PJ, Simonian PL, Nuñez G, Nickoloff BJ. Kaposis sarcoma tumor cells preferentially express Bcl-XL. Am J Path. 1996;149:795–803. [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tschlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: Involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- González-García M, Perez-Ballestro R, Ding L, Duan L, Boise LH, Thompson CB, Nuñez G. bcl-XL is the major bcl-X mRNA form expressed during mouse development and its product localizes to mitochondria. Development. 1994;120:3033–3040. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- Grillot DAM, González-García M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Nuñez G. Genomic organization, promoter region analysis and chromosomal localization of the mouse bcl-X gene. J Immunol. 1997;158:4750–4757. [PubMed] [Google Scholar]
- Haldar S, Jena N, Croce CM. Anti-apoptosis potential of bcl-2 oncogene by dephosphorylation. Biochem Cell Biol. 1994;72:455–462. doi: 10.1139/o94-061. [DOI] [PubMed] [Google Scholar]
- Ihle JN. Cytokine receptor signaling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Yokota T, Arai K, Miyajima A. Suppression of apoptotic death in hematopoietic cells by signaling through the IL-3/GM-CSF receptors. EMBO J. 1995;14:266–275. doi: 10.1002/j.1460-2075.1995.tb07000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozopas KM, Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL-2. Proc Natl Acad Sci. 1993;90:3516–3519. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Blomqvist C, Franssila K, Krajewska M, Wasenius VM, Niskanen E, Nordling S, Reed JC. Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995;55:4471–4478. [PubMed] [Google Scholar]
- Kurtin PJ, Dewald GW, Shields DJ, Hanson CA. Haematologic disorders associated with deletions of chromosome 20q—a clinicopathologic study of 107 patients. Am J Clin Path. 1996;106:680–688. doi: 10.1093/ajcp/106.5.680. [DOI] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Dellavalle V, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A Tel-Jak2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- Leverrier Y, Thomas J, Perkins GR, Mangeney M, Collins MK, Marvel J. In bone marrow derived Baf-3 cells, inhibition of apoptosis by IL-3 is mediated by two independent pathways. Oncogene. 1997;14:425–430. doi: 10.1038/sj.onc.1200845. [DOI] [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early response gene with sequence similarity to bcl-2. J Immunol. 1993;151:1979–1989. [PubMed] [Google Scholar]
- Lomo J, Blomhoff HK, Jacobsen SE, Krajewski S, Reed JC, Smeland EB. Interleukin-13 in combination with CD40 ligand potently inhibit apoptosis in human B lymphocytes: Up-regulation of Bcl-XL and Mcl-1. Blood. 1997;89:4415–4424. [PubMed] [Google Scholar]
- Miura O, Cleveland JL, Ihle JN. Inactivation of the erythropoietin receptor by point mutations in a region showing homology with other cytokine receptors. Mol Cell Biol. 1993;13:1788–1795. doi: 10.1128/mcb.13.3.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura O, Nakamura N, Ihle JN, Aoki N. Erythropoietin-dependent association of phophatidylinositol 3-kinase with tyrosine-phosphorylated erythropoietin receptor. J Biol Chem. 1994a;269:614–620. [PubMed] [Google Scholar]
- Miura Y, Miura O, Ihle JN, Aoki N. Activation of the mitogen-activated protein kinase pathway by the erythropoietin receptor. J Biol Chem. 1994b;269:29962–29969. [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negshi I, Senju S, Zhang Q, Fujii S, Loh D. Massive cell death of immature hematopoietic cells and neurons in bcl-X-deficient mice. Science. 1995;267:1506–1509. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Chin H, Miyasaka N, Miura O. An epidermal growth factor receptor/Jak2 tyrosine kinase domain chimera induces tyrosine phosphorylation of Stat5 and transduces a growth signal in hematopoietic cells. J Biol Chem. 1996;271:19483–19488. doi: 10.1074/jbc.271.32.19483. [DOI] [PubMed] [Google Scholar]
- Nip J, Strom DK, Fee BE, Zambetti G, Cleveland JL, Hiebert SW. E2F-1 cooperates with topoisomerase II inhibition and DNA damage to selectively augment p53-independent apoptosis. Mol Cell Biol. 1997;17:1049–1056. doi: 10.1128/mcb.17.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosaka T, Van Deursen JMA, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Ossina NK, Cannas A, Powers VC, Fitzpatrick PA, Knight JD, Gilbert JR, Shektman EM, Tomei LD, Umansky SR, Kiefer MC. Interferon-γ modulates p53-independent apoptotic pathway and apoptosis related gene expression. J Biol Chem. 1997;272:16351–16357. doi: 10.1074/jbc.272.26.16351. [DOI] [PubMed] [Google Scholar]
- Packham G, Cleveland JL. Ornithine decarboxylase is a mediator of c-Myc-induced apoptosis. Mol Cell Biol. 1994;9:5741–5747. doi: 10.1128/mcb.14.9.5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packham G, Brimmell M, Cleveland JL. Two isoforms of Bag-1 are generated by alternative translation initiation codons and contain Bcl-2 and activated steroid hormone receptor binding domains. Biochem J. 1997;328:807–813. doi: 10.1042/bj3280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallis M, Zhu YM, Russell NH. Bcl-XL is heterogenously expressed by acute myeloblastic leukaemia cells and is associated with autonomous growth in vitro and with P-glycoprotein expression. Leukemia. 1997;11:945–949. doi: 10.1038/sj.leu.2400705. [DOI] [PubMed] [Google Scholar]
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine J-C, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- Pezzella F, Tse AG, Cordell JL, Pulford KA, Gatter KC, Mason DY. Expression of the bcl-2 oncogene protein is not specific for the 14:18 translocation. Am J Pathol. 1990;157:225–232. [PMC free article] [PubMed] [Google Scholar]
- Pierce JH, DiFiore PP, Aaronson SA, Potter M, Pumphrey J, Scott A, Ihle JN. Neoplastic transformation of mast cells by Abelson-MuLV: Abrogation of IL-3 dependence by a non-autocrine mechanism. Cell. 1985;41:685–693. doi: 10.1016/s0092-8674(85)80049-0. [DOI] [PubMed] [Google Scholar]
- Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y, Ihle JN. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol Cell Biol. 1996;16:1622–1631. doi: 10.1128/mcb.16.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle FW, Wang J, Wang D, Feng J, Cleveland JL, Ihle JN, Zambetti GP. Cytokine rescue of p53-dependent apoptosis and cell cycle arrest is mediated by distinct Jak kinase signaling pathways. Genes & Dev. 1998;12:1099–1107. doi: 10.1101/gad.12.8.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Reed JC, Tsujimoto Y, Alpers JD, Croce CM, Nowell PC. Regulation of bcl-2 proto-oncogene expression during normal human lymphocyte proliferation. Science. 1987;235:1295–1299. doi: 10.1126/science.3495884. [DOI] [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. Multiple Bcl-2 family member demonstrate selective dimerization with Bax. Proc Natl Acad Sci. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M, Richard C, Benito A, Sanz C, Olalla I, Fernandez-Luna JL. Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N Engl J Med. 1998;338:564–571. doi: 10.1056/NEJM199802263380902. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Interleukin-3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- Tu Y, Renner S, Xu F, Fleishman A, Taylor J, Weisz J, Vescio R, Rettig M, Berenson J, Krajewski S, Reed JC, Lichtenstein A. BCL-X expression in multiple myeloma: Possible indicator of chemoresistance. Cancer Res. 1998;58:256–262. [PubMed] [Google Scholar]
- Tuscano JM, Druey KM, Riva A, Pena J, Thompson CB, Kehrl JH. Bcl-X rather than Bcl-2 mediates CD40-dependent centrocyte survival in the germinal center. Blood. 1996;88:1359–1364. [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- Williams GT, Smith CA, Spooncer E, Dexter TM, Taylor DR. Hemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature. 1990;324:76–79. doi: 10.1038/343076a0. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- Yang E, Korsmeyer SJ. Molecular thanatopsis: A discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- Yang X-F, Weber GF, Cantor H. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity. 1997;7:629–639. doi: 10.1016/s1074-7613(00)80384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhan Q, Fan S, Bae I, Guillouf C, Liebermann DA, O’Connor PM, Fornace AJ., Jr Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene. 1994;9:3743–3751. [PubMed] [Google Scholar]