SNAP19 mediates the assembly of a functional core promoter complex (SNAPc) shared by RNA polymerases II and III (original) (raw)
Abstract
The basal transcription factor SNAPc binds to the PSE, a core element in the RNA polymerase II and III human snRNA promoters. SNAPc contains at least four subunits, but it has not been possible to assemble a fully defined recombinant SNAPc. Here we reconstitute SNAPc from five recombinant subunits, SNAP43, SNAP45, SNAP50, SNAP190, and a newly identified subunit, SNAP19. This recombinant complex binds specifically to the PSE and directs both RNA polymerase II and III snRNA gene transcription. Thus, the same core SNAPc nucleates the assembly of two classes of initiation complexes.
Keywords: SNAPc, human snRNA promoters, RNA polymerases, transcription
Although considerable effort has focused on understanding RNA polymerase II transcription from mRNA-type promoters and RNA polymerase III transcription from 5S- and tRNA-type promoters, a significant portion of RNA polymerase II and III transcriptional initiation events is directed by snRNA-type promoters (Dahlberg and Lund 1988). Unraveling the mechanisms that govern snRNA gene transcription is, therefore, integral to an understanding of the global network of RNA polymerase II and III transcription.
For most promoters, RNA polymerase specificity is determined by different promoter elements that recruit distinct DNA-binding basal transcription complexes. Thus, the RNA polymerase II mRNA promoters recruit the TATA box-binding protein (TBP)-containing complex TFIID, and this results in the subsequent recruitment of TFIIB (as well as other general transcription factors) and RNA polymerase II (Orphanides et al. 1996). The 5S- and tRNA-type RNA polymerase III promoters recruit TFIIIC, either directly or through TFIIIA, and this results in the recruitment of the TBP-containing complex TFIIIB and RNA polymerase III (Geiduschek and Kassavetis 1992). In contrast, the RNA polymerase II and III human snRNA promoters contain a similar proximal sequence element (PSE), which is involved in directing transcription by both RNA polymerases. RNA polymerase III transcription is specified by the presence (and RNA polymerase II transcription by the absence) of an adjacent TATA box (Mattaj et al. 1988; Lobo and Hernandez 1989). The PSE and the TATA box are the only known core elements involved in both RNA polymerase II and III transcription. These observations raise the intriguing possibility that the PSE in the RNA polymerase II and III snRNA promoters recruits the very same basal transcription factor.
The PSE is recognized by the snRNA activating protein complex (SNAPc) (Sadowski et al. 1993), a basal transcription factor also known as PTF (Murphy et al. 1992). Previous purifications of SNAPc (Henry et al. 1995) and PTF (Yoon et al. 1995), as well as isolation of cDNA clones (Henry et al. 1995, 1996; Bai et al. 1996; Sadowski et al. 1996; Yoon and Roeder 1996; Wong et al. 1998) identified four subunits with apparent molecular masses of 43, 45, 50, and ∼200 kD. In addition, significant but substoichiometric amounts of TBP copurified extensively with PSE-binding activity (Henry et al. 1995). However, the four recombinant subunits, with or without TBP, are insufficient to reconstitute a SNAP complex with DNA-binding and transcriptional activities similar to endogenous SNAPc (Wong et al. 1998). Thus, it has not been possible to determine whether the PSE in the RNA polymerase II and III snRNA promoters is recognized by the same complex or by SNAPc variants that are responsible for nucleating different transcription initiation complexes.
Here we demonstrate that in addition to the previously characterized subunits, a small 19-kD protein, SNAP19, is also a member of the endogenous SNAP complex. SNAP19 is a novel protein of 98 amino acids characterized by an amino-terminal leucine zipper motif and a carboxy-terminal glutamic acid-rich region. Like the other members of SNAPc, SNAP19 is required for snRNA gene transcription by both RNA polymerases II and III. In coimmunoprecipitation assays, SNAP19 interacts with SNAP190. Importantly, this interaction allows the association of both SNAP43 and SNAP50, which does not occur in the absence of SNAP19. Therefore, SNAP19 is essential for mediating the assembly of a core SNAP complex containing SNAP19, SNAP43, SNAP45, SNAP50, and SNAP190. Recombinant complexes assembled with all five SNAPc proteins bind effectively to DNA in a PSE-specific manner and function to restore RNA polymerase II and III transcriptional activity in nuclear extracts that have been depleted of endogenous SNAPc. These results identify the same core SNAPc as a basal transcription factor recruited by two classes of promoters, the human RNA polymerase II and III snRNA promoters.
Results
p19 is a member of SNAPc
Because we could not assemble a recombinant SNAP complex from SNAP43, SNAP45, SNAP50, and SNAP190, we suspected the existence of an unidentified, essential subunit. During biochemical fractionation of SNAPc, a small polypeptide with an apparent molecular mass of 19 kD copurified extensively with PSE binding and U1 transcriptional activity (R.W. Henry, unpubl.). Microsequence of this material, previously thought to be a proteolytic degradation product, yielded three peptide sequences not present in the known SNAPc subunits. These new sequences were used to search expressed sequence tag (EST) databases for corresponding cDNA clones. Candidate sequences were identified and used to assemble a complete ORF, which was then confirmed by the sequencing of PCR products generated with appropriate primers from cDNA synthesized from total RNA. As shown in Figure 1, the resulting open reading frame (ORF) contains the three peptides, which encompass amino acids 11–19, 20–30, and 31–41 (shaded underlines), and encodes a novel protein of 98 amino acids with a predicted molecular mass of 11.4 kD. We refer to this protein as SNAP19. SNAP19 contains a potential leucine zipper motif at its amino terminus (amino acids 1–36) and a notable acidic region of 10 glutamic acid residues at its carboxyl terminus (amino acids 86–95). The initiation codon is preceded by a stop codon (data not shown), suggesting that this ORF encodes the full-length protein.
Figure 1.

Nucleotide sequence and predicted amino acid sequence for SNAP19. The shaded underlines indicate the peptides microsequenced from biochemically isolated SNAPc; (open circles) potential leucine zipper motif; (solid underlines) synthetic peptides used to raise rabbit polyclonal antibodies. The shaded amino acids are absent in some cDNAs, probably as a result of alternative splicing, but the observation that part of this amino acid sequence is present in one of the three microsequenced peptides suggests that the larger form of the protein is present in biochemically purified SNAPc.
Synthetic peptides corresponding to the amino (amino acids 1–13, p19Npep) and carboxyl (amino acids 82–98, p19Cpep) termini of SNAP19 (Fig. 1, solid underlines) were used to raise rabbit polyclonal antisera. These antisera were tested for their ability to recognize biochemically purified HeLa cell SNAPc in an electromobility shift assay (EMSA), and the results are shown in Figure 2. SNAPc bound specifically to the PSE (cf. lane 2 to lane 1). Addition of anti-SNAP19 (carboxy-terminal) antibodies (lane 3) but not preimmune serum (lane 10) retarded the SNAPc–PSE complex, and this effect could be inhibited by preincubation of the antibodies with increasing amounts of the peptide used to raise the antibody (lanes 4–6) but not with similar amounts of a nonspecific peptide (lanes 7–9). Thus, the 19-kD protein is indeed a member of SNAPc; hence, its name.
Figure 2.
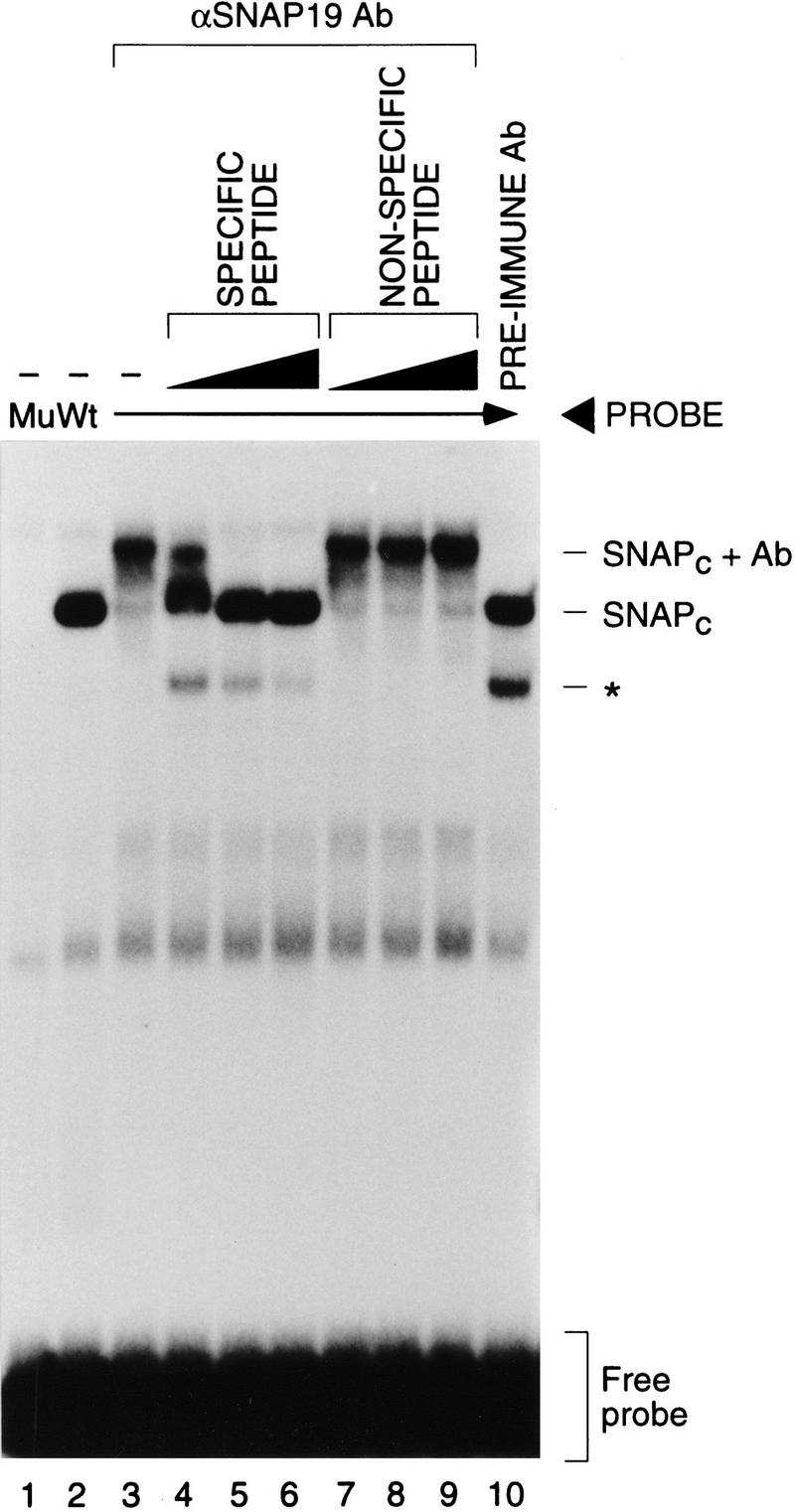
SNAP19 is a component of SNAPc. Anti-SNAP19 antibodies supershift SNAPc bound to DNA. EMSAs were performed with a biochemically purified SNAPc-containing fraction [3 μl, 0.25 μg/ml, mono Q fraction (Henry et al. 1995)] and mutant PSE (lane 1) or wild-type PSE (lanes 2–10) probes as described previously (Sadowski et al. 1993). Lanes 3–9 contained 0.3 μl of anti-SNAP19 serum (anti-p19Cpep); lanes 4–6 contained 0.1, 0.3, and 1.0 μl, respectively, of specific peptide (p19Cpep: 10 μg/μl); and lanes 7–9 contained similar amounts of a nonspecific peptide (p19Npep). Lane 10 contained 0.3 μl of rabbit preimmune serum. The band labeled with a an asterisk represents a proteolytic degradation product of SNAPc.
SNAP19 is required for snRNA gene transcription by RNA polymerases II and III
To determine whether SNAP19 is required for transcription of snRNA genes by both RNA polymerases II and III or whether it might be part of a complex dedicated to transcription by only one of these two polymerases, we used rabbit preimmune or anti-SNAP19 antibodies to deplete whole cell extracts. First, the effects of the depletions on levels of SNAP43 were determined by immunoblotting. As shown in Figure 3A, there was little difference in the amount of the SNAP43 component of SNAPc present in untreated whole cell extract (lane 1), and extracts treated with preimmune antibody beads (lane 2) or with anti-SNAP19 antibody beads preincubated with the peptide used to raise the antibody (lane 7). However, SNAP43 was efficiently immunodepleted when extracts were treated with anti-SNAP19 antibody beads (lanes 3–5) or anti-SNAP19 antibody beads preincubated with the nonspecific peptide (lane 6). Thus, immunodepletion with anti-SNAP19 antibodies resulted in efficient depletion of another member of the SNAP complex, SNAP43. These results suggest that SNAP43 is not associated with any other non-SNAP19-containing complexes in the cell and that SNAPc is removed effectively by SNAP19 immunodepletion.
Figure 3.
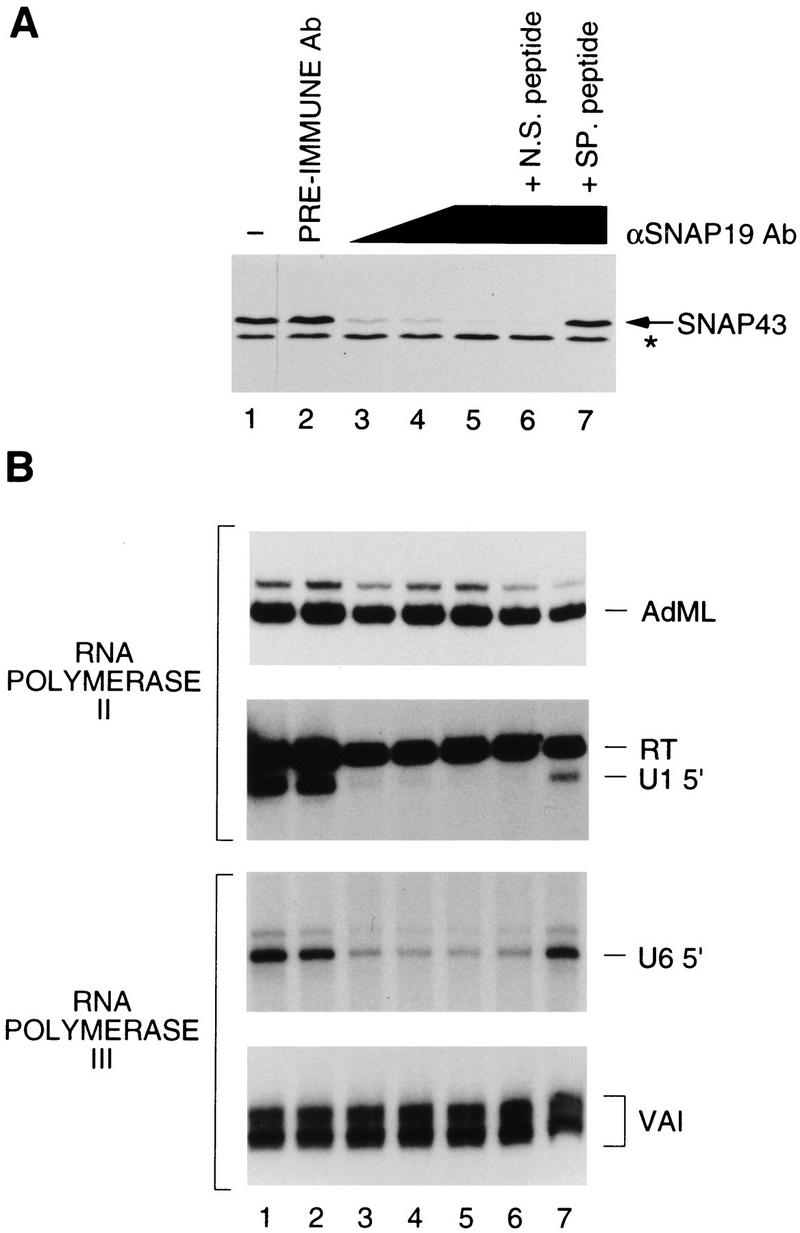
SNAP19 is required for human snRNA transcription by both RNA polymerases II and III. (A) Anti-SNAP43 immunoblot analysis of depleted extracts. The samples in the various lanes correspond to 7 μl of HeLa whole cell extract (lane 1), 10-μl aliquot of extract depleted with a constant amount of beads consisting of only preimmune antibody beads (lane 2), preimmune antibody beads plus anti-SNAP19 antibody beads at 3:1 (lane 3) and 1:1 (lane 4) ratios, or only anti-SNAP19 antibody beads (lanes 5–7). In lanes 6 and 7, the immunodepletions were performed in the presence of 5 μg of nonspecific (p19Npep) or specific (p19Cpep) peptide, respectively. The band labeled with an asterisk corresponds to an unidentified cross-reacting protein and was used as a loading control. (B) The same extracts analyzed in A were tested for their ability to support in vitro transcription from the RNA polymerase II AdML (top panel) and U1 snRNA (second panel) promoters, and the RNA polymerase III U6 snRNA (third panel), and Ad2 VAI (bottom panel) promoters. The correctly initiated transcripts are labeled AdML, U1 5′, U6 5′, and VAI, respectively. Readthrough transcripts from cryptic mRNA-type promoters in the U1 transcription experiment (Sadowski et al. 1993) are labeled RT.
The depleted extracts were tested for their ability to support in vitro transcription of the U1 snRNA gene by RNA polymerase II and the U6 snRNA gene by RNA polymerase III. We also tested the depleted extracts for transcription directed by the adenovirus 2 (Ad2) major late promoter (AdML), a typical RNA polymerase II mRNA-type promoter, and by the Ad2 VAI promoter, a typical gene-internal RNA polymerase III promoter. As shown in Figure 3B, all four promoters directed efficient transcription in untreated extract or extract treated with preimmune antibody beads (lanes 1,2). In addition, neither RNA polymerase II transcription from the AdML promoter (top panel) nor RNA polymerase III transcription from the Ad2 VAI promoter (bottom panel) was significantly affected by SNAPc depletion with anti-SNAP19 antibody beads or any of the conditions tested in this assay. Therefore, SNAP19 apparently is not involved in transcription of these genes. In contrast, RNA polymerase II transcription from the U1 snRNA promoter was reduced strongly in extracts treated with increasing levels of anti-SNAP19 antibody beads (second panel, lanes 3–5), whereas in the same lanes, transcription of a readthrough transcript (labeled RT), which probably results from cryptic mRNA-type promoters located within vector sequences (Sadowski et al. 1993), was not affected. Similarly, RNA polymerase III transcription from the U6 snRNA promoter was decreased in extracts treated with anti-SNAP19 antibody beads (third panel). The inhibitory effect of the anti-SNAP19 antibody beads was specific because preincubation of the antibodies with the peptide used to raise the antiserum (p19Cpep, lane 7) but not a nonspecific peptide (p19Npep, lane 6) reduced or prevented the inhibition of transcription. These results suggest that SNAP19 itself or SNAP19-associated subunits are required for both RNA polymerase II and III snRNA gene transcription.
SNAP19 mediates assembly of a core SNAPc
The precise architecture of SNAPc is unknown. We showed previously that SNAP43 and SNAP50 associate (Henry et al. 1996) and that SNAP190 and SNAP45 associate (Wong et al. 1998), to form SNAP43/50 and SNAP190/45 protein pairs. However, we were unable to demonstrate any strong interactions between proteins in the different pairs. The identification of SNAP19 prompted us to test whether SNAP19, together with the four previously identified SNAPc components, could form a complete complex. Pair-wise studies of SNAP19 association with the other four SNAP components showed that SNAP19 can associate on its own with SNAP190 but not with SNAP43, SNAP45, or SNAP50 (data not shown). We then performed an in vitro cotranslation reaction containing all five subunits and used this material in a coimmunoprecipitation experiment with anti-SNAP43 antibodies. The results are shown in Figure 4A. Strikingly, in contrast to our previous experiments in which SNAP19 was missing (Wong et al. 1998), when all five subunits were cotranslated (lane 2), each subunit was efficiently immunoprecipitated by the anti-SNAP43 antibodies (lane 8). This suggests that in the presence of SNAP19, a complex containing all five subunits can be assembled, as shown schematically in Figure 4B (top panel).
Figure 4.

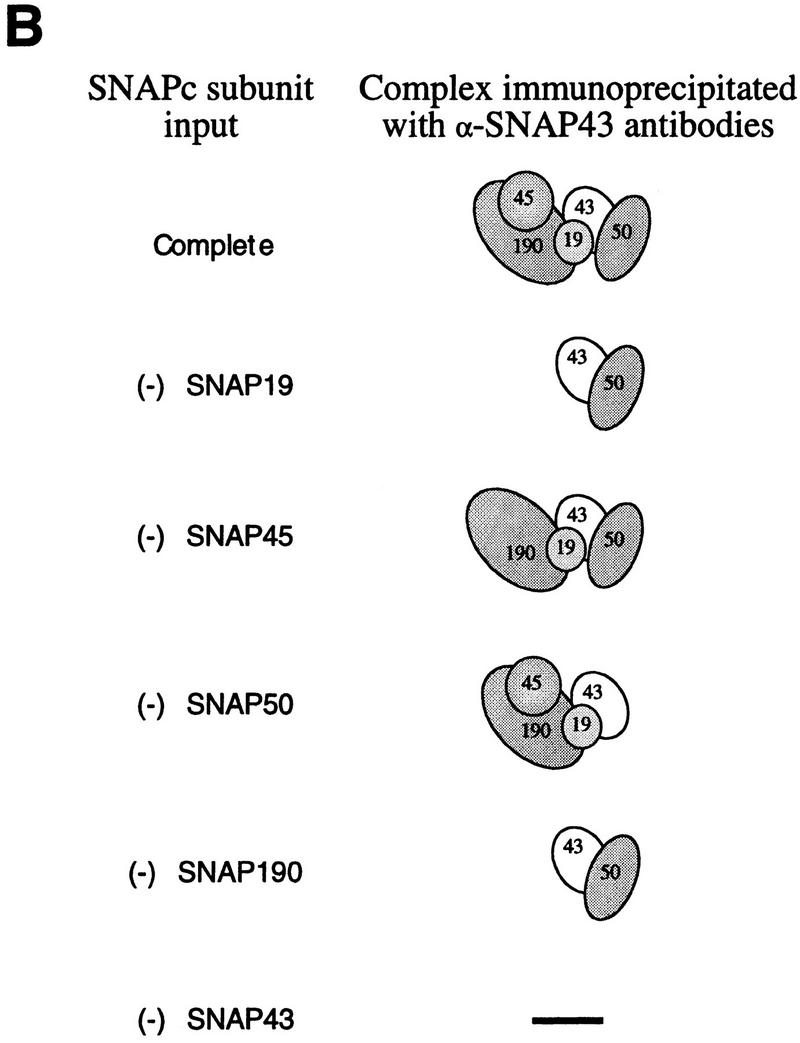
SNAP19 is essential for assembly of SNAPc. [35S]Methionine-labeled SNAPc subunits were cotranslated in rabbit reticulocyte lysates. The cotranslated products were used for immunoprecipitations with anti-SNAP43 antibodies, and the immunoprecipitates were fractionated by 15% SDS-PAGE. Radiolabeled proteins were detected by autoradiography. (Lane 1 ) Protein size markers; (lanes 2–7) 10% (1 μl) of the input materials used for the coimmunoprecipitations. In lane 2, all five proteins were cotranslated; in lanes 3–7, one SNAPc subunit was omitted in turn as indicated above the lanes. (Lanes 8–13) Immunoprecipitated proteins. The identity of each full-length protein is indicated. Note that the weak band migrating very slightly ahead of SNAP50 in lane 11 was not reproducibly observed but is probably a proteolytic breakdown product of SNAP190. (B) Schematic representation of the anti-SNAP43 immunoprecipitation results. The stoichiometry of the various subunits is not known.
To gain a better understanding of the architecture of SNAP complex, we performed cotranslations in which only SNAP19 (Fig. 4A, lane 3), SNAP45 (lane 4), SNAP50 (lane 5), or SNAP190 (lane 6) had been omitted out of the five SNAPc subunits. As a control for anti-SNAP43 antibody specificity, SNAP43 was also omitted from one reaction (lane 7). Strikingly, when SNAP19 was omitted, neither SNAP190 nor SNAP45 was recovered in the anti-SNAP43 immunoprecipitation (lane 9; see also Fig. 4B, panel labeled −SNAP19). Thus, in the absence of SNAP19, SNAP43 could only interact with SNAP50 in this assay, consistent with our previous results (Henry et al. 1996). When SNAP45 (Fig. 4A, lane 10) or SNAP50 (lane 11) was omitted, all of the remaining subunits were coimmunoprecipitated with SNAP43, suggesting that, as observed before and as illustrated in Figure 4B, SNAP45 interacts strongly only with SNAP190 and SNAP50 interacts strongly only with SNAP43. When SNAP190 was omitted, neither SNAP19 nor SNAP45 was immunoprecipitated with the SNAP43/SNAP50 protein pair (lane 12). And when SNAP43 was omitted, no proteins were immunoprecipitated with the anti-SNAP43 antibody, as expected, except for weak background bands in the 45- to 50-kD range (lane 13). Thus, the coimmunoprecipitation results obtained with just pairs of in vitro-translated SNAPc subunits (Henry et al. 1996; Wong et al. 1998; data not shown) are entirely consistent with those obtained when four of the five SNAPc subunits are present in the starting material, suggesting that individual protein–protein interactions detected in this assay accurately reflect protein–protein interactions occurring within the SNAP complex.
Together, these observations provide us with a detailed view of the architecture of the SNAP complex (Fig. 4B, top panel). Although the stoichiometry of the various subunits is not known, the results suggest that SNAP190 forms a backbone on which SNAP45 and SNAP19 can assemble directly and independently. SNAP19 is essential for the subsequent assembly of SNAP43 with SNAP190, perhaps because SNAP19 bridges the two proteins. The observation that SNAP43 does not interact with SNAP19 in a pairwise combination suggests, however, that SNAP43 is involved in weak protein interactions with both SNAP19 and SNAP190, only the sum of which is sufficient to promote coimmunoprecipitation in this assay. SNAP50 assembles with the complex via interactions with SNAP43. Consistent with this view of the complex, we can assemble a trimeric complex containing SNAP190, SNAP19, and SNAP43 (data not shown).
Recombinant SNAPc mediates transcription by both RNA polymerases II and III
The observation that in the presence of SNAP19, all SNAPc subunits could be coimmunoprecipitated with SNAP43 suggested that we might be able to assemble a functional SNAP complex from recombinant subunits (rSNAPc). We therefore coexpressed the five SNAPc subunits in a baculovirus expression system. As a control, we also coexpressed all SNAPc subunits except SNAP190. The resulting complexes were then purified by immunoaffinity chromatography with anti-SNAP43 antibodies followed by peptide elution. As shown in Fig. 5A, the complete recombinant SNAPc bound efficiently to the wild-type (lanes 4) but not to a mutant (lane 5) PSE. In contrast, proteins present in the control fraction did not bind to either the wild-type or mutant PSE (data not shown). The rSNAPc–PSE complex comigrated with the HeLa cell SNAPc–PSE complex (lane 2), suggesting that recombinant and biochemically purified SNAP complexes are similar. Addition of antibodies directed against each SNAPc component—SNAP190, SNAP45, SNAP43, SNAP19, or SNAP50—retarded the migration of the rSNAPc–PSE complex (lanes 6–10), indicating that each of the five subunits is present in the recombinant SNAP complex.
Figure 5.


Recombinant core SNAPc binds specifically to the PSE and reconstitutes transcription of the human snRNA genes by RNA polymerase II and III. (A) rSNAPc or partial SNAP complexes missing SNAP190 were assembled by coexpression of the appropriate subunits in a baculovirus expression system and purified by immunoaffinity. The EMSA was performed as described previously (Sadowski et al. 1993) with either wild type (Wt) or mutant (Mu) PSE probe, as indicated above the lanes, and no added fraction (lane 1) or 1 μl of biochemically purified SNAPc (mono-Q) (lanes 2,3) or 10 μl of rSNAPc (lanes 4–10). In lanes 6–10, 0.5–1 μl of the antibodies indicated above the lanes was added to the binding reactions. The retarded complexes are labeled SNAPc + Ab. Preimmune antibodies had no effect on the complex (data not shown). (B) Untreated HeLa nuclear extracts (lane 1) or extracts immunodepleted with rabbit preimmune (lane 2) or anti-SNAP43 antibody beads (lanes 3–11) were tested for their ability to support in vitro transcription of U1 snRNA by RNA polymerase II (top panel) and U6 snRNA by RNA polymerase III (bottom panel) as described (Sadowski et al. 1993). In lanes 4 and 5, 5 and 10 μl of tagged (top panel) or untagged (bottom panel) rSNAPc was added. In lanes 6 and 7, 5 and 10 μl of control fractions were added. In lanes 8–11, 1, 2, 4, and 8 μl (top panel) or 2, 4, 8, or 16 μl (bottom panel) of biochemically purified SNAPc (mono-Q) were added.
We have shown previously that independent depletion with anti-SNAP43 (Henry et al. 1995), anti-SNAP45 (Sadowski et al. 1996), anti-SNAP50 (Henry et al. 1996), and anti-SNAP190 (Wong et al. 1998) antibodies inhibits both RNA polymerase II and III snRNA gene transcription, and we have shown above that the same is true for depletions with anti-SNAP19 antibodies. Together, these data suggest that each of these subunits is involved in both RNA polymerase II and III snRNA gene transcription, but they do not address whether the same SNAP complex performs both functions. Therefore, rSNAPc was tested for its ability to reconstitute transcription in vitro, and the results are shown in Figure 5B. Nuclear extracts were treated with anti-SNAP43 or preimmune antibody beads and then tested for their ability to support transcription of U1 snRNA by RNA polymerase II (top panel) and U6 snRNA by RNA polymerase III (bottom panel). Efficient U1 and U6 snRNA transcription was observed in untreated (lane 1) and mock-depleted (lane 2) nuclear extracts. As expected, treatment with anti-SNAP43 antibody beads greatly diminished both U1 and U6 transcriptional activity (lane 3). Strikingly, upon addition of increasing amounts of the rSNAPc (lanes 4,5), but not control fractions (lanes 6,7) to the SNAPc-depleted extract, both U1 and U6 transcription were reconstituted efficiently. The levels of transcriptional activity restored by rSNAPc were similar to those observed with biochemically purified SNAPc for equivalent ‘PSE-binding units’ as estimated by EMSA (data not shown). Thus, rSNAPc assembled from the five core subunits is functional as a basal transcription factor for both the RNA polymerase II and III transcriptional pathways.
rSNAPc exhibits a similar protein profile as endogenous SNAPc
The core SNAP complex assembled entirely from recombinant proteins is functional both for DNA binding and transcription of snRNA genes by two different RNA polymerases. To determine the protein profile of rSNAPc, a double-tagged rSNAP complex containing His-tagged SNAP190 and HA-tagged SNAP50 was assembled in the baculovirus expression system. As a negative control, an untagged recombinant complex was also assembled. The recombinant complexes were then chromatographed over nickel columns in 350 mm KCl. Bound proteins were then eluted with imidazole and used directly as the starting material for immunopurification with anti-HA antibody beads followed by peptide elution with the HA peptide. As a comparison, endogenous SNAPc was partially purified (mono S step) from HeLa cell S-100 extracts as described previously (Henry et al. 1995). Proteins present in the negative control, the endogenous HeLa SNAPc, and the rSNAPc preparations were then size fractionated by SDS-PAGE and visualized by staining with silver.
The negative control fraction did not contain any detectable proteins (data not shown). As shown in Figure 6, the HeLa cell SNAPc preparation, which corresponds to the penultimate step in the purification of SNAPc (Henry et al. 1995), still contained a number of proteins, but the individual SNAPc subunits (identified by comigration with in vitro-translated, [35S]methionine-labeled individual SNAPc subunits fractionated on the same gel) were visible. In the purified rSNAPc preparation, five major bands were visible, four of which either comigrated or migrated close to the HeLa SNAPc subunits (lane 4). However, as expected, the mobility of HA-tagged rSNAP50 was reduced as compared to that of untagged HeLa SNAP50. In addition, both rSNAP19 and rSNAP43 migrated slightly differently from the corresponding HeLa SNAPc subunits. These differences may be due to differential post-translational modification of SNAP19 and SNAP43 in the two SNAPc preparations. Both of these proteins can be phosphorylated to various extents and, at least in the case of SNAP43, this affects its mobility on SDS–polyacrylamide gels (R.W. Henry, unpubl.). The functional significance of these modifications is, however, not known. These results indicate that although the natural SNAPc and the rSNAPc are highly similar, they are not identical. However, the subtle differences in migration do not result in functional differences for DNA binding and transcription detectable in our in vitro assays. These results also show that SNAP19 can be readily identified in both HeLa SNAPc and near homogenous rSNAPc, consistent with the contention that this protein is a bona fide member of the SNAP complex.
Figure 6.
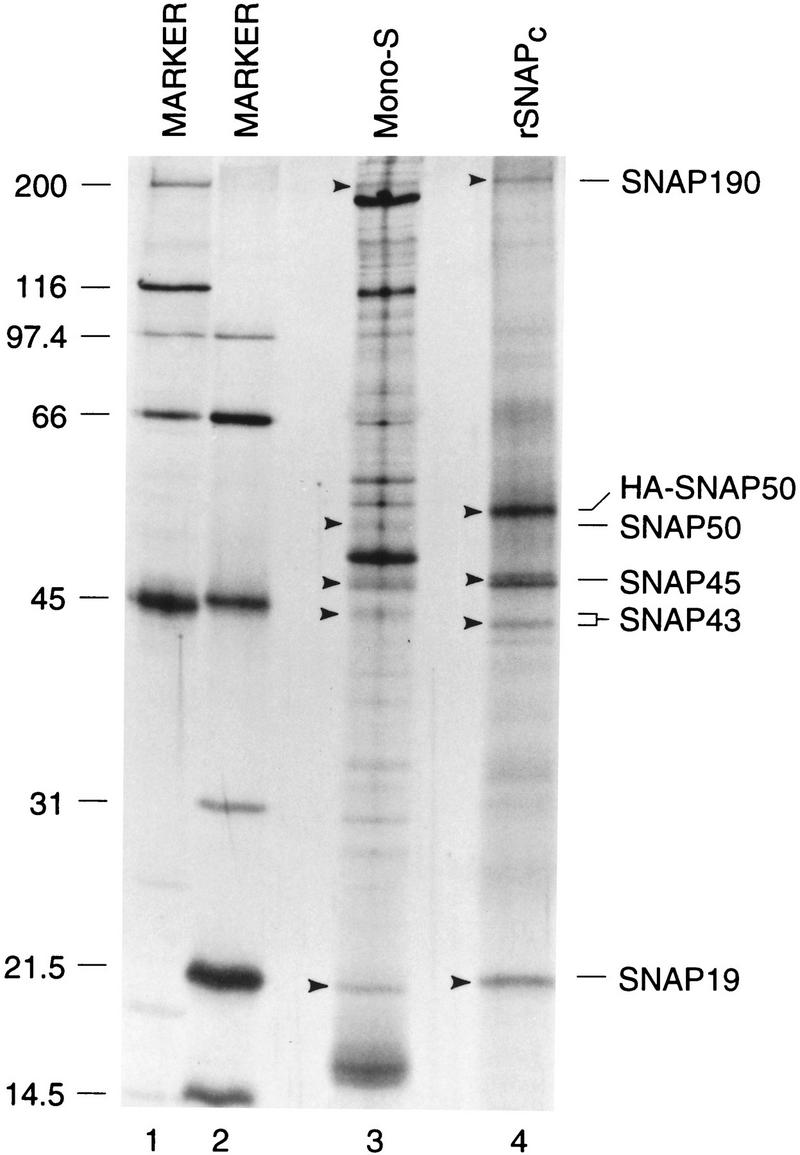
rSNAPc exhibits a similar protein profile as endogenous SNAPc. Endogenous SNAPc (lane 3) was partially purified from HeLa cell S-100 extracts essentially as described (mono-S stage of purification: Henry et al. 1995). rSNAPc (lane 4) containing His-tagged SNAP190 and HA-tagged SNAP50 was assembled in a baculovirus expression system and purified by chromatography over a nickel column followed by immunopurification with anti-HA antibody beads. Bound proteins were eluted with 400 μl of buffer D containing the HA peptide (1 mg/ml). The samples were precipitated with TCA and size fractioned by 15% SDS-PAGE, together with in vitro-translated, [35S]methionine-labeled, individual SNAPc subunits as markers (data not shown). Proteins were then visualized by staining with silver, and the gel was dried and autoradiographed to reveal the location of the radioloabeled SNAPc subunit markers. Arrowheads indicate the location of each SNAPc protein. The identity of each SNAPc subunit is indicated. Lanes 1 and 2 show protein size markers.
Discussion
Reconstitution of a rSNAPc
The identification and cloning of SNAP19 has allowed us to assemble a core SNAPc from recombinant subunits and to describe its architecture. The five SNAPc subunits form a stable complex in which SNAP190 binds strongly and independently to SNAP45 and to SNAP19. The presence of SNAP19 is required for subsequent assembly of SNAP43, which binds to the SNAP190/SNAP19 complex, and SNAP50, which binds to SNAP43. This complex binds specifically to the PSE. In addition, although we cannot exclude the possibility of specific contributions by contaminating insect cell proteins, our results show that the core complex is functional for both RNA polymerase II and III transcription of snRNA genes.
Core-promoter recognition by multiprotein complexes is a common theme in transcription, regardless of RNA polymerase specificity. This is probably because multiprotein complexes can effect the control of transcription via diverse programs of protein–protein interactions. For example, the assembly of complete and partial Drosophila TFIID complexes showed that although partial TFIID subcomplexes were active for basal transcription, they responded only to specific subsets of activators in vitro, consistent with the idea that distinct TFIID TAFs are functionally required for activation in vitro by different activators (Chen et al. 1994). In contrast, for human SL1 and yeast TFIIIB, the same type of experiments showed that all subunits are required to obtain transcriptionally active complexes (Beckmann et al. 1995; Kassavetis et al. 1995; Roberts et al. 1996; Rüth et al. 1996). It is likely that in the case of SNAPc, different subunits perform different functions in RNA polymerase II and III transcription. An exciting possibility is that some subunits are selectively dispensable for transcription by only one of the two RNA polymerases. Because we assembled a rSNAPc from five baculoviruses that each expresses only one of the SNAPc subunits, it will be possible to perform coinfections with subsets of the five viruses and thus assemble partial SNAP complexes that can then be tested for various functions including DNA binding as well as basal and activated RNA polymerase II and III snRNA gene transcription.
How is RNA polymerase specificity determined?
Typically, the basal transcription factors that recognize core promoter elements are dedicated to transcription by distinct RNA polymerases. Thus, the multiprotein complexes SL1, TFIID, TFIIIC, and TFIIIB are specifically required for transcription by RNA polymerases I, II, and III, respectively. Our data presented here suggest that SNAPc is an unusual basal transcription factor in that the very same core complex, consisting of SNAP19, SNAP43, SNAP45, SNAP50, and SNAP190, mediates nucleation of both RNA polymerase II and III snRNA initiation complexes. This raises the question of how RNA polymerase specificity is determined. The answer likely depends on how TATA box binding protein (TBP) is recruited to the promoter. In our original identification of SNAPc, we observed that the complex bound to a PSE could be disrupted with anti-TBP antibodies in an EMSA, suggesting at that time that TBP is a component of SNAPc (Sadowski et al. 1993). We now suspect, however, that this wholesale disruption reflected proteolytic degradation of the complex by contaminants present in the antibody preparations. Nevertheless, both RNA polymerase II and III transcription of the human snRNA genes requires TBP (Lobo et al. 1991; Simmen et al. 1991; Sadowski et al. 1993; Yoon and Roeder 1996), and several lines of evidence suggest that core SNAPc can associate with TBP: First, TBP can interact directly with both SNAP43/PTFδ (Henry et al. 1995; Yoon and Roeder 1996) and SNAP45/PTFγ (Sadowski et al. 1996; Yoon and Roeder 1996); second, significant (but substoichiometric) amounts of TBP copurify extensively with PSE-binding activity (Henry et al. 1995); third, TBP can be coimmunoprecipitated efficiently from a nuclear extract with antibodies directed against SNAP43 (Henry et al. 1995) or PTFβ/SNAP50 (Bai et al. 1996), although in the latter case, the association with TBP was shown to be salt sensitive, suggesting that TBP associates less tightly with SNAPc/PTF than it does with the TFIID TBP-associated factors (TAFs). Because the RNA polymerase II and III snRNA promoters differ by the absence or presence of a TATA box, it seems likely that different modes of TBP recruitment are key to the determination of RNA polymerase specificity.
The core SNAP complex likely plays an important role in TBP recruitment to the RNA polymerase II and III snRNA promoters, and we imagine that it can interact with TBP in a flexible manner that accommodates preinitiation complex assembly for transcription by the two different polymerases. In the case of RNA polymerase III snRNA promoters, which contain a TATA box at a fixed distance downstream of the PSE, SNAPc binding to the PSE directly recruits TBP to the TATA box, and this effect is dependent on the nonconserved amino-terminal domain of TBP (Mittal and Hernandez 1997). Thus, in this case, TBP (perhaps with additional unidentified factors) is probably recruited to the promoter through a combination of protein–DNA interactions with the TATA box involving the TBP core DNA-binding domain, and protein–protein interactions with SNAPc involving the TBP amino-terminal domain. Which subunit of SNAPc is contacted by the amino-terminal domain of TBP remains to be determined. In the case of RNA polymerase II snRNA promoters, which do not have a TATA box, we have shown that in an extract immunodepleted of TBP, partially purified SNAPc fractions (which also contain TBP) restore snRNA gene transcription by RNA polymerase II much more efficiently than recombinant TBP (Sadowski et al. 1993), suggesting that such fractions contain a TBP complex required for RNA polymerase II snRNA gene transcription. Because TBP copurifies with SNAPc and interacts with several of its subunits, we suspect that this putative TBP complex is recruited through protein–protein interactions with core SNAPc. Thus, the absence or presence of a TATA box in snRNA promoters may direct the same core SNAPc to associate with different, RNA polymerase II- or III-specific, TBP-containing complexes.
Materials and methods
Immunodepletions and transcription reactions
The immunoblot in Figure 2A was performed with antibody CS48 (Henry et al. 1995). For the depletions, preimmune or anti-SNAP19 (anti-p19Cpep) antibodies were covalently attached to protein A–Sepharose beads. One hundred microliters of HeLa whole cell extract (∼20 mg/ml) was then incubated with a constant volume of antibody beads (50 μl) containing the various ratios of preimmune to anti-SNAP19 antibody beads specified in the legend to Figure 2. The transcription reactions were performed with 8 μl (AdML), 18 μl (U1), 8 μl (U6), and 4 μl (VAI) of extract as described previously (Sadowski et al. 1993).
Cotranslations and immunoprecipitations
The coding sequences of SNAP190, SNAP50, SNAP45, SNAP43, and SNAP19 were cloned into the pCite2a vector (Invitrogen). These were used for coupled in vitro transcription/translation reactions (50 μl each) in rabbit reticulocyte lysates essentially as described by the manufacturer (Promega). Cotranslation reactions containing all five templates (∼1 μg each) were performed. Alternatively, reactions were done in which each template, in turn, was omitted. Ten microliters of each in vitro cotranslation reaction was diluted to 1 ml in HEMGT-150 buffer (20 mm HEPES at pH 7.9; 0.1 mm EDTA, 12.5 mm MgCl2, 10% glycerol, 0.5% Tween 20; 150 mm KCl) containing protease inhibitors and 3 mm DTT, and were incubated for 1 hr at room temperature with 10 μl of anti-SNAP43 antibody beads. The beads were washed extensively in HEMGT-150 buffer and boiled in Laemmli buffer (Laemmli 1970) to release bound proteins.
Assembly of rSNAPc
The coding sequences of SNAP190, SNAP50, SNAP45, SNAP43, and SNAP19 were cloned into a modified baculovirus transfer vector pAcUW51 (Pharmingen) (a kind gift of Paul Kaufman, University of California, Berkeley). In Figure 5, A and B (top), SNAP50 and SNAP45 were expressed as amino-terminal HA epitope fusion proteins while SNAP19 was expressed as a carboxy-terminal HA epitope fusion protein. In Figure 5B (bottom), none of the subunits was tagged. Like tagged rSNAPc, untagged rSNAPc bound specifically to the PSE and was active for RNA polymerase II snRNA gene transcription (data not shown). Transfer vectors containing each of the SNAPc subunits were used to generate recombinant baculoviruses with a BaculoGold transfection kit (Pharmingen). Positive viruses were plaque purified twice and were amplified to a titer of 1 × 108 PFU/ml. Sf9 cells were infected simultaneously with all five baculoviruses at a multiplicity of infection (m.o.i.) of 10. For the control fraction used in Figure 5 (top), the SNAP190-expressing virus was omitted. Cells were harvested 48 hr postinfection, washed, and incubated in lysis buffer (10 mm Tris-HCl at pH 7.5; 130 mm NaCl, 1% Triton X-100, 10 mm NaF, 10 mm NaPi, 10 mm NaPPi) on ice for 30 min. rSNAPc was purified by immunoaffinity chromatography and peptide elution.
Purification of rSNAPc and silver staining
rSNAP complexes containing either His-tagged SNAP190 and HA-tagged SNAP50 or only untagged subunits were assembled in a baculovirus expression system essentially as described above. Sf9 cells were lysed as described above, 5 ml of lysate was adjusted to 350 mm NaCl and 20 mm imidazole and incubated with Ni–NTA agarose beads (Qiagen) for 2 hr at 4°C. Bound proteins were eluted with the same buffer containing 300 mm imidazole and used directly for immunopurification with a 7:1 (vol/vol) ratio of sample to protein G–Sepharose beads (Boehringer Mannheim) covalently coupled to anti-HA antibodies. Bound proteins were eluted twice with 200 μl of buffer containing 20 mm HEPES (pH 7.5), 15% glycerol, 0.1% Tween 20, 5 mm MgCl2, 100 mm KCl, 0.5 mm PMSF, 2 mm DTT, and 0.7 mg/ml HA peptide. The eluates were pooled and precipitated with TCA. Proteins present in the double-tagged rSNAPc, untagged rSNAPc, and HeLa SNAPc fractions were separated by 15% SDS–polyacrylamide gels alongside in vitro-translated, [35S]methionine-labeled, individual SNAPc subunits. Proteins were visualized by staining with silver, and then the gel was dried and autoradiographed to reveal the location of the radioloabeled SNAPc subunits.
Acknowledgments
We thank MeeWa Wong, Viola Ellison, and Paul Kaufman for help with the baculovirus expression system, Spencer Teplin for oligonucleotide synthesis, and James Duffy, Michael Ockler, and Philip Renna for artwork and photography. We also thank Winship Herr, Cyril Sanders, and Grace Chen for discussion and comments on the manuscript. This work was funded in part by National Institutes of Health grant GM38810. R.W.H. was supported by the Joseph G. Goldring Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hernande@cshl.org; FAX (516) 367-6801.
References
- Bai L, Wang Z, Yoon J-B, Roeder RG. Cloning and characterization of the β subunit of human proximal sequence element-binding transcription factor and its involvement in transcription of small nuclear RNA genes by RNA polymerases II and III. Mol Cell Biol. 1996;16:5419–5426. doi: 10.1128/mcb.16.10.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann H, Chen J-L, O’Brien T, Tjian R. Coactivator and promoter-selective properties of RNA polymerase I TAFs. Science. 1995;270:506–1509. doi: 10.1126/science.270.5241.1506. [DOI] [PubMed] [Google Scholar]
- Chen J-L, Attardi LD, Verrijzer CP, Yokomori K, Tjian R. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- Dahlberg JE, Lund E. The genes and transcription of the major small nuclear RNAs. In: Birnstiel ML, editor. Structure and function of major and minor small nuclear ribonucleoprotein particles. Berlin, Germany: Springer Verlag; 1988. pp. 38–70. [Google Scholar]
- Geiduschek EP, Kassavetis GA. In: RNA polymerase III transcription complexes. Transcriptional regulation. McKnight SL, Yamamoto KR, editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 247–280. [Google Scholar]
- Henry RW, Sadowski CL, Kobayashi R, Hernandez N. A TBP-TAF complex required for transcription of human snRNA genes by RNA polymerases II and III. Nature. 1995;374:653–657. doi: 10.1038/374653a0. [DOI] [PubMed] [Google Scholar]
- Henry RW, Ma B, Sadowski CL, Kobayashi R, Hernandez N. Cloning and characterization of SNAP50, a subunit of the snRNA-activating protein complex SNAPc. EMBO J. 1996;15:7129–7136. [PMC free article] [PubMed] [Google Scholar]
- Kassavetis GA, Nguyen ST, Kobayashi R, Kumar A, Geiduschek EP, Pisano M. Cloning, expression, and function of TFC5, the gene encoding the B″ component of the Saccharomyces cerevisiae RNA polymerase III transcription factor TFIIIB. Proc Natl Acad Sci. 1995;92:9786–9790. doi: 10.1073/pnas.92.21.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli EK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lobo S, Hernandez N. A 7 bp mutation converts a human RNA polymerase II snRNA promoter into an RNA polymerase III promoter. Cell. 1989;58:55–67. doi: 10.1016/0092-8674(89)90402-9. [DOI] [PubMed] [Google Scholar]
- Lobo SM, Lister J, Sullivan ML, Hernandez N. The cloned RNA polymerase II transcription factor IID selects RNA polymerase III to transcribe the human U6 gene in vitro. Genes & Dev. 1991;5:1477–1489. doi: 10.1101/gad.5.8.1477. [DOI] [PubMed] [Google Scholar]
- Mattaj IW, Dathan NA, Parry HW, Carbon P, Krol A. Changing the RNA polymerase specificity of U snRNA gene promoters. Cell. 1988;55:435–442. doi: 10.1016/0092-8674(88)90029-3. [DOI] [PubMed] [Google Scholar]
- Mittal V, Hernandez N. Role for the amino-terminal region of human TBP in U6 snRNA transcription. Science. 1997;275:1136–1140. doi: 10.1126/science.275.5303.1136. [DOI] [PubMed] [Google Scholar]
- Murphy S, Yoon J-B, Gerster T, Roeder RG. Oct-1 and Oct-2 potentiate functional interactions of a transcription factor with the proximal sequence element of small nuclear RNA genes. Mol Cell Biol. 1992;12:3247–3261. doi: 10.1128/mcb.12.7.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes & Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- Roberts S, Miller SJ, Lane WS, Lee S, Hahn S. Cloning and functional characterization of the gene encoding the TFIIIB90 subunit of RNA polymerase III transcription factor TFIIIB. J Biol Chem. 1996;271:14903–14909. doi: 10.1074/jbc.271.25.14903. [DOI] [PubMed] [Google Scholar]
- Rüth J, Conesa C, Dieci G, Lefebvre O, Düsterhöft A, Ottonello S, Sentenac A. A suppressor of mutations in the class III transcription system encodes a component of yeast TFIIIB. EMBO J. 1996;15:1941–1949. [PMC free article] [PubMed] [Google Scholar]
- Sadowski CL, Henry RW, Lobo SM, Hernandez N. Targeting TBP to a non-TATA box cis-regulatory element: A TBP-containing complex activates transcription from snRNA promoters through the PSE. Genes & Dev. 1993;7:1535–1548. doi: 10.1101/gad.7.8.1535. [DOI] [PubMed] [Google Scholar]
- Sadowski CL, Henry RW, Kobayashi R, Hernandez N. The SNAP45 subunit of the small nuclear RNA (snRNA) activating protein complex is required for RNA polymerase II and III snRNA gene transcription and interacts with the TATA box binding protein. Proc Natl Acad Sci. 1996;93:4289–4293. doi: 10.1073/pnas.93.9.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen KA, Bernues J, Parry HD, Stunnenberg HG, Berkenstam A, Cavallini B, Egly J-M, Mattaj IW. TFIID is required for in vitro transcription of the human U6 gene by RNA polymerase III. EMBO J. 1991;10:1853–1862. doi: 10.1002/j.1460-2075.1991.tb07711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MW, Henry RW, Ma B, Kobayashi R, Klages N, Matthias P, Strubin M, Hernandez N. The large subunit of basal transcription factor SNAPc is a Myb domain protein that interacts with Oct-1. Mol Cell Biol. 1998;18:368–377. doi: 10.1128/mcb.18.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-B, Roeder RG. Cloning of two proximal sequence element-binding transcription factor subunits (γ and δ) that are required for transcription of small nuclear RNA genes by RNA polymerases II and III and interact with the TATA-binding protein. Mol Cell Biol. 1996;16:1–9. doi: 10.1128/mcb.16.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-B, Murphy S, Bai L, Wang Z, Roeder RG. Proximal sequence element-binding transcription factor (PTF) is a multisubunit complex required for transcription of both RNA polymerase II- and RNA polymerase III-dependent small nuclear RNA genes. Mol Cell Biol. 1995;15:2019–2027. doi: 10.1128/mcb.15.4.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]