IL-1-induced NFκB and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK) (original) (raw)
Abstract
Mutant I1A cells, lacking IL-1 receptor-associated kinase (IRAK) mRNA and protein, have been used to study the involvement of IRAK in NFκB and c-Jun N-terminal kinase (JNK) activation. A series of IRAK deletion constructs were expressed in I1A cells, which were then tested for their ability to respond to IL-1. Both the N-terminal death domain and the C-terminal region of IRAK are required for IL-1-induced NFκB and JNK activation, whereas the N-proximal undetermined domain is required for the activation of NFκB but not JNK. The phosphorylation and ubiquitination of IRAK deletion mutants correlate tightly with their ability to activate NFκB in response to IL-1, but IRAK can mediate IL-1-induced JNK activation without being phosphorylated. These studies reveal that the IL-1-induced signaling pathways leading to NFκB and JNK activation diverge either at IRAK or at a point nearer to the receptor.
IL-1 is a major inflammatory cytokine, secreted by activated macrophages during infection (1). At low concentrations, IL-1 helps to mediate local inflammation, acting on vesicular endothelial cells to increase the synthesis of cell-surface adhesion molecules, playing an important role in activating leukocytes and in stimulating their recruitment to sites of infection. At higher concentrations, IL-1 enters the bloodstream to exert systemic effects, causing fever and increasing the synthesis of acute-phase proteins by the liver. IL-1 mediates these functions by activating the transcription factors NFκB, AP1, and ATF (2–4), thus stimulating the expression of various genes in different target cells.
The signaling pathways used by IL-1 to activate NFκB, AP1, and ATF have been studied extensively, and the following model has evolved from the work of several laboratories. After IL-1 is bound, a complex is formed between the type 1 receptor and the receptor accessory protein (5–8). The cytosolic proteins MyD88 (9, 10) and Tollip (11) are recruited to this complex, where they function as adaptors, recruiting IL-1 receptor-associated kinase (IRAK) in turn. IRAK, a serine-threonine kinase, is phosphorylated at the receptor complex and then leaves to interact with TRAF6 (8, 12–16). Eventually, phosphorylated IRAK is ubiquitinated and degraded (17). TAK1 and two interacting proteins, TAB1 and TAB2, are also implicated in IL-1 signaling (18, 19). TAB2 is membrane-bound in untreated cells and translocates to the cytosol upon stimulation with IL-1, where it functions as an adaptor, linking TRAF6 to TAK1 and TAB1 (19). Activated TAK1 activates NIK directly (18), leading to the activation of IκB kinases and thus to the phosphorylation and degradation of IκB. NFκB is then translocated to the nucleus to activate transcription (20–30). It is becoming clear that NFκB itself also is phosphorylated in response to IL-1 to achieve full activation, through a pathway involving phosphatidylinositol 3-kinase and Akt (31–33). IL-1-dependent signaling also leads to the activation of c-Jun N-terminal kinase (JNK) and other mitogen-activated protein kinases (15, 16), which results in the phosphorylation and activation of ATF and AP1.
The model described above has been based mostly on results obtained through isolation of specific protein complexes, analysis of protein–protein interactions through yeast two-hybrid screens, and data from gene knockout studies. However, the detailed mechanisms of IL-1-dependent signaling pathways still are not well defined yet. We have isolated four independent IL-1-unresponsive cell lines that lack specific components of the IL-1-signaling pathway (15). Mutant I1A lacks IRAK, whereas the other three (I2A, I3A, and I4A) lack components that have not been identified yet. Characterization of these mutants showed that the component defective in I2A is probably associated with the receptor complex, that I3A is defective in a component located upstream of IRAK but downstream of the receptor complex, and that I4A is defective in a downstream component required for IL-1-induced NFκB but not JNK activation (X.L. and G.R.S., unpublished data).
IRAK-deficient I1A cells have been used successfully as a model system to study the function of IRAK in IL-1 signaling (15). Expression of exogenous IRAK in these cells (I1A-IRAK) restores IL-1-dependent activation of NFκB, JNK, and gene expression. To address the role of the kinase activity of IRAK in IL-1 signaling, its ATP-binding site was mutated (K239A), abolishing kinase activity. In transfected I1A cells, IRAK-K239A was still phosphorylated upon IL-1 stimulation and, surprisingly, still complemented all the defects in I1A mutant cells. Therefore, we concluded that IRAK must be phosphorylated by a different kinase, and that phospho-IRAK must play a role in IL-1-mediated signaling that does not require its kinase activity (15).
We have now studied the function of IRAK more extensively by analyzing a series of deletion mutants for their ability to mediate IL-1-dependent functions in I1A cells. Interestingly, different sets of domains of IRAK are required for IL-1-induced NFκB and JNK activation. Furthermore, IL-1-induced phosphorylation of IRAK is important for activating NFκB but is not required for JNK activation. The IL-1-induced signaling pathways that lead to NFκB and JNK activation appear to diverge at the level of IRAK or nearer to the receptor.
Materials and Methods
Biological Reagents and Cell Culture.
Recombinant human IL-1β was provided by the National Cancer Institute. Antibody TRAF6 was a kind gift of Z. Cao (Tularik, South San Francisco, CA). Anti-pJNK was obtained from New England Biolabs, and anti-IRAK from Santa Cruz Biotechnology. A quantity of 293-TK/Zeo cells (15) was maintained in DMEM, supplemented with 10% (vol/vol) FCS/100 μg/ml penicillin G/100 μg/ml streptomycin.
Recombinant Plasmids.
IRAK deletion fragments were generated by PCR with IRAK-K239A as a template and were cloned into an expression vector driven by the thymidine kinase promoter. pE-selectin-luc, an NFκB-dependent E-selectin-luciferase reporter plasmid, was described by Schindler and Baichwal (34).
Transfection and Reporter Assays.
For stable transfections, 2 × 105 cells were seeded onto a 10-cm plate and cotransfected the following day by the calcium phosphate method with 10 μg of each expression vector and 1 μg of pBabePuro. After 48 h, the cells were selected with 1 μg/ml puromycin until clones appeared. For reporter assays, 2 × 105 cells were transfected by the same procedure with 1 μg of pE-selectin-luc, 1 μg of pSV2-β-gal, and 100 ng of each expression construct. After 48 h, the cells were split onto two 35-mm plates and, the next day, stimulated with IL-1 for 4 h before harvesting. Luciferase and β-galactosidase activities were determined by using the luciferase-assay system and chemiluminescent reagents, respectively, from Promega.
Gel-Shift Assays.
An NFκB-binding site (5′-GAGCAGAGGGAAATTCCGTAACTT-3′) from the IP-10 gene (35) was used as a probe. Complementary oligonucleotides, end-labeled with polynucleotide kinase (Roche Molecular Biochemicals) and γ-32P-labeled ATP, were annealed by slow cooling. Approximately 20,000 cpm of probe were used per assay. Cytoplasmic extracts were prepared as described by Kessler et al. (36). The binding reaction was carried out at room temperature for 20 min in a total volume of 20 μl of 20 mM Hepes buffer, pH 7.0/10 mM KCl/0.1% Nonidet P-40/0.5 mM DTT/0.25 mM phenylmethanesulfonyl fluoride/10% (vol/vol) glycerol.
Coimmunoprecipitation and Immunoblotting.
For coimmunoprecipitations, cell extracts were incubated for 4 h with 1 μl of anti-IRAK or anti-TRAF6 polyclonal antibody (gift of Z. Cao, Tularik) or preimmune serum, followed by a 1-h incubation with 50 μl of protein A-Sepharose beads (prewashed and resuspended in PBS at a 1:1 ratio). After incubations, the beads were washed five times with lysis buffer (8), followed by immunoblotting with anti-IRAK or anti-TRAF6.
Results
Both the N- and C-Terminal Regions of IRAK Contribute to Its Function.
Expression of exogenous IRAK in IRAK-null I1A cells restores their responsiveness to IL-1 (ref. 15 and Fig.1). The kinase-dead IRAK-K239A also complemented all the defects in I1A cells, showing that the kinase activity of IRAK is not required for it to function in IL-1-dependent signaling (15). IRAK is a multidomain protein (Fig. 1, ref. 17), containing an N-terminal DD (residues 1–103), a UD (residues 104–198), a KD (residues 199–522), and C1 and C2 (residues 523–618 and residues 619–712, respectively). To identify functional domains of IRAK, we generated a series of deletion constructs and analyzed their ability to restore IL-1 responsiveness to I1A cells. Because overexpression of wild-type IRAK leads to the autophosphorylation of IRAK, the deletion mutants were generated from kinase-dead IRAK-K239A. The constructs were cotransfected, together with a luciferase reporter driven by the IL-1-responsive E-selectin promoter, into I1A cells, and the fold induction of luciferase activity on IL-1 stimulation was determined (Fig. 1). Both wild-type IRAK (data not shown) and IRAK-K239A (K239A in Fig. 1) restored IL-1-induced E-selectin promoter activity. dDD or dUD abolished IL-1-induced E-selectin promoter activity in the transfected cells, indicating that these two regions play important roles in IL-1-dependent signaling (Fig. 1). However, dKD had no effect on the activity of IRAK, which strongly supports our previous conclusion that the kinase activity of IRAK is not required for it to function (15). dC1 and dC2 (Fig. 1) separately had no effect on IL-1-induced E-selectin promoter activity in the transfected cells, whereas deleting both (dC1dC2 in Fig. 1) abolished this activity. The results show that the C-terminal region of IRAK is required for its function, which can be accomplished by either C1 or C2. Furthermore, the truncated protein containing only the N-terminal DD and UD (DDUD in Fig. 1) failed to confer IL-1-induced promoter activity in I1A cells and has been shown to be a dominant-negative mutant (8). Adding C1 to the truncated protein (DDUDC1 in Fig. 1) made it as fully functional as the wild-type protein, confirming that the kinase activity of IRAK is not required, and that both the N- and C-terminal regions of IRAK contribute to its function.
Figure 1.
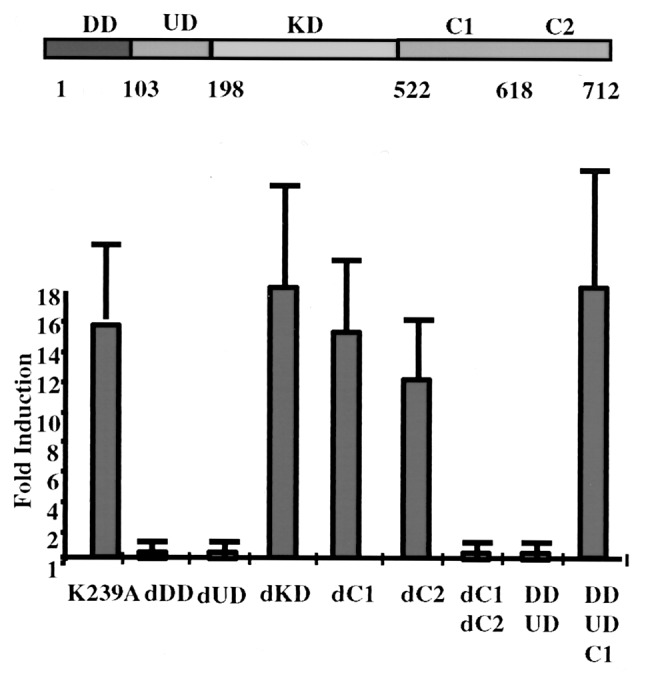
Deletion analysis of IRAK. Constructs encoding the point mutant K239A and the deletion mutants were transiently cotransfected into IRAK-null I1A cells with E-selectin-Luc. The data are presented as fold induction of luciferase activity in IL-1-treated cells compared with the induction in untreated cells. The averages and standard deviations from four independent experiments are shown. DD, death domain; UD, domain of unknown function; KD, kinase domain; dDD, deletion of death domain; dUD, deletion of undetermined domain; dKD, deletion of kinase domain; dC_n_, deletion of C-terminal domain n; C_n_, C-terminal domain n.
The N-Proximal Undetermined Region Is Required for NFκB Activation but Not for JNK Activation.
Neither NFκB nor JNK is activated in IL-1-treated I1A cells, but these responses are restored in I1A-IRAK cells, indicating that IRAK is required for both (15). To identify the domains of IRAK that are responsible, the deletion mutants were stably transfected into I1A cells, which were then examined for IL-1-induced activation of NFκB by gel-shift assay (Fig. 2) and JNK with anti-phospho-JNK (Fig. 3). Mutant IRAK lacking the KD permitted both IL-1-induced NFκB and JNK activation in transfected I1A cells (dKD in Figs. 2 and 3), indicating that none of the KD of IRAK is required for these responses. Deletion of C1 or C2 still allowed IRAK to participate in IL-1-induced NFκB and JNK activation in I1A cells (dC1 or dC2 in Figs. 2 and 3), whereas deletion of the whole C-terminal region abrogated the activation of both pathways completely (dC1C2 in Figs. 2 and 3). Deletion of the N-terminal DD abolished IL-1-induced NFκB and JNK activation (dDD, Figs. 2 and 3). Surprisingly, however, although deletion of the N-proximal undetermined region abolished the ability of IRAK to mediate IL-1-induced NFκB activation (dUD in Fig. 2), it had no effect on IL-1-induced JNK activation (dUD in Fig. 3). Therefore, the regions of IRAK required to mediate IL-1-induced NFκB activation and IL-1-induced JNK activation are separate and distinct, suggesting that the IL-1-induced pathways leading to the activation of NFκB and JNK diverge at or before IRAK.
Figure 2.

Domains of IRAK required for IL-1-induced NFκB activation. IRAK-null I1A cells stably transfected with K239A or the deletion mutants were either untreated (−) or treated with 100 units/ml IL-1 for 15 min (+). Whole-cell extracts were used for gel-shift assays. Wild-type 293 cells (WT) and IRAK-null I1A cells (I1A) were used as positive and negative controls, respectively.
Figure 3.

Domains of IRAK required for IL-1-induced JNK activation. IRAK-null I1A cells stably transfected with IRAK deletion mutants were either untreated (−) or treated with 100 units/ml IL-1 (I) or with 40 J/m2 UV radiation (UV). Cell extracts were analyzed with anti-phospho-JNK after Western transfer. Wild-type 293 cells (WT) and IRAK-null I1A cells (I1A) were used as positive and negative controls, respectively. Data from one of four experiments are shown.
IL-1-Induced Modification of IRAK Correlates with NFκB Activation.
IRAK is phosphorylated, ubiquitinated, and eventually degraded upon IL-1 stimulation (refs. 15 and 17; Fig.4). In transfected I1A cells, the IRAK-K239A mutant protein is still phosphorylated upon stimulation with IL-1 and still complements all the defects in I1A mutant cells (15), revealing that IRAK must be phosphorylated by a different kinase, and that phospho-IRAK must play a role in IL-1-mediated signaling that does not require its kinase activity. To investigate the basis and function of these posttranslational modifications, we examined the IL-1-induced modification of IRAK-deletion mutants in transfected I1A cells. Deletion of the KD of IRAK had no effect on IL-1-induced modification (dKD in Fig. 4), whereas deletion mutants lacking either C1 or C2 exhibited IL-1-dependent phosphorylation and ubiquitination (dC1 or dC2 in Fig. 4); the deletion mutant lacking both C1 and C2 was not modified on stimulation (dC1C2 in Fig. 4). On the other hand, deletion mutants lacking either the N-terminal DD or the N-proximal UD were not modified upon stimulation (dDD and dUD in Fig. 4). When these results are compared with IL-1-induced NFκB activation in I1A cells transfected with the same mutants, it is clear that the IL-1-induced modification of IRAK tightly correlates with its ability to activate NFκB, suggesting that these processes are important for the IL-1-induced activation of NFκB.
Figure 4.

IL-1-induced IRAK modification and degradation. IRAK-null I1A cells transfected with K239A or the deletion mutants were either untreated (−) or treated with IL-1 for the number of minutes indicated. Cell extracts were analyzed with anti-IRAK by the Western procedure. IRAK, unmodified IRAK (arrow); P-IRAK, phosphorylated IRAK; U-IRAK, ubiquitinated IRAK.
IL-1-Induced Phosphorylation of IRAK Is Not Necessary for Signal-Dependent JNK Activation.
The dUD mutant, lacking the N-proximal undetermined region, is not phosphorylated, ubiquitinated, or degraded on IL-1 stimulation (Fig.4). IL-1 did induce JNK activation in transfected I1A cells (dUD in Fig. 3), suggesting that IRAK modification may not be necessary for this process. Nevertheless, because dUD retains a specific IL-1-dependent function, this mutated protein might be phosphorylated slightly upon stimulation, and a low level of phosphorylation might not be detected by Western analysis. To examine the signal-dependent phosphorylation of dUD more sensitively, wild-type 293 cells and I1A cells transfected with dUD were incubated with [32P]orthophosphate. Extracts from the labeled cells were then immunoprecipitated with anti-IRAK and examined by SDS/PAGE. Western analysis showed that similar amounts of IRAK were immunoprecipitated from wild-type cells and from I1A cells transfected with dUD (Fig. 5). Although wild-type IRAK was heavily phosphorylated upon stimulation, no phosphorylation was detected for dUD (Fig. 5). Taken together, the results confirm that IRAK can mediate signal-dependent JNK activation without being phosphorylated.
Figure 5.

IL-1-induced phosphorylation of IRAK. Wild-type (WT) 293 cells or IRAK-null I1A cells transfected with dUD were incubated in phosphate-free medium containing [32P]orthophosphate for 4 h and then either left untreated (−) or treated with 100 units/ml IL-1 for 15 min (+). Cell extracts were then immunoprecipitated with anti-IRAK and separated by SDS/(7%)PAGE. (A) Analysis of phosphorylated IRAK (P-IRAK) by autoradiography. (B) Western analysis of IRAK.
The Deletion of Either DD or UD Abolishes the Binding of IRAK to TRAF6.
KD is not required for IRAK to bind to TRAF6 (Fig.6A). dC1 and dC2 retained IL-1-dependent binding to TRAF6 (Fig. 6 A and B), whereas deletion of the whole C-terminal region abolished binding (Fig.6B). Deletion mutants lacking either the DD or UD failed to interact with TRAF6 (Fig. 6 A and B). The data indicate that both N-terminal domains (DD and UD) and the C-terminal region, all taken together (DD+UD+C1 in Fig. 6), are required for the IL-1-induced binding of IRAK to TRAF6.
Figure 6.

Interaction of IRAK with TRAF6. IRAK-null I1A cells transfected with IRAK deletion mutants were either untreated (−) or treated with 100 units/ml IL-1 for 15 min (+). (A) Extracts from cells transfected with dDD, dUD, dKD, dC1, and dC2; DD + UD + C1 cells were immunoprecipitated with anti-IRAK and probed with anti-TRAF6 and anti-IRAK. (B) Anti-IRAK antibody (Santa Cruz Biotechnology; recognizes 440–712 residues of IRAK) does not immunoprecipitate dC1C2 well, although it does recognize dC1C2 on a Western blot (Fig. 4). Therefore, dC1C2 and a few other IRAK deletion mutants (dUD, dC1, and dC2) were immunoprecipitated with anti-TRAF6 and probed with anti-IRAK and anti-TRAF6. Wild-type 293 cells (WT) were used as a positive control.
Discussion
We have analyzed a series of IRAK deletion mutants for IL-1-mediated functions in IRAK-deficient I1A cells, revealing that both the N-terminal and C-terminal regions of IRAK contribute to IL-1-dependent signaling. However, consistent with the activity of the kinase-dead K239A mutant of IRAK (15), the entire KD of IRAK is dispensable for function. Therefore, IRAK probably mediates IL-1 signaling by interacting with other components in the pathway in a phosphorylation-dependent manner.
Although the C-terminal region is required for both IL-1-induced NFκB and JNK activation, the results from the N-terminal deletion mutants discriminate between these two functions. The N-terminal DD is necessary for both IL-1-induced NFκB and JNK activation, whereas the N-proximal UD is required for NFκB but not for JNK activation. The fact that different regions of IRAK are needed for the activation of NFκB and JNK suggests that these two IL-1-induced pathways diverge at the level of IRAK or further upstream.
IRAK deletion mutants lacking the N-terminal DD and UD and the C-terminal region failed to interact with TRAF6 (Fig. 6). Because the same regions of IRAK are required for IL-1-induced NFκB activation in transfected I1A cells (Fig. 6), the data reveal that the interaction of IRAK with TRAF6 correlates with its ability to activate NFκB. Although I1A cells transfected with dDD and dC1C2 had no IL-1-induced JNK activation (Fig. 3), dUD still conferred JNK activation in these cells (Fig. 3), suggesting that the IRAK–TRAF6 interaction is not necessary for JNK activation. However, previous studies (15, 16) clearly indicate that both IRAK and TRAF6 are required for IL-1-induced JNK activation. The dUD mutant may form a weak complex with TRAF6 that we have not detected and that is sufficient for JNK but not NFκB activation. Alternatively, although the formation of an IRAK–TRAF6 complex is required for NFκB activation, IRAK may mediate the interaction of other signaling components with TRAF6 to activate JNK.
Previous studies have suggested that the N-terminal DD might be involved in the interaction of IRAK with the IL-1 receptor complex after IL-1 stimulation (8, 37, 38). In response to IL-1, a complex is formed between the type 1 receptor and the receptor accessory protein. A Toll domain is present in the cytoplasmic regions of both receptor subunits. MyD88, a cytosolic Toll protein, is then recruited through Toll–Toll interactions to the complex, where it functions as an adaptor, recruiting IRAK in turn. In addition to the Toll domain at the C terminus, MyD88 also contains an N-terminal DD; because IRAK also contains a DD, MyD88 probably recruits IRAK to the receptor complex through the interaction of these two domains. Therefore, dDD of IRAK may prevent its recruitment to the receptor, abolishing all IRAK-mediated downstream signaling, including the activation of NFκB and JNK.
Previous studies have shown that IRAK is phosphorylated, ubiquitinated, and eventually degraded upon IL-1 stimulation (17). IL-1-induced phosphorylation of IRAK might play a role in causing IRAK to leave the receptor complex after activation, because both adaptor molecules MyD88 and Tollip bind only to unphosphorylated IRAK (8, 37, 38). We found that IL-1-induced modification and degradation of the various IRAK deletion mutants correlate tightly with their ability to activate NFκB, suggesting that modification of IRAK might be a necessary step along the pathway leading to NFκB activation. On the other hand, although the dUD mutant was not phosphorylated on stimulation (Figs. 4and 5), it was able to restore IL-1-induced JNK activation in I1A cells (Fig. 3), indicating that the phosphorylation of IRAK is not necessary for JNK activation. If the phosphorylation of IRAK is required for it to leave the receptor complex, the fact that the dUD mutant can activate JNK suggests that the activation may occur before IRAK leaves the receptor complex.
Acknowledgments
We thank Huiqin Nie for technical assistance and Zhaodan Cao for antibodies against IRAK and TRAF6. This work was supported by National Institutes of Health Grant GM 600020 (to X.L.).
Abbreviations
IRAK
IL-1 receptor-associated kinase
JNK
c-Jun N-terminal kinase
DD
N-terminal death domain
UD
domain of unknown function
KD
kinase domain
C_n_
C-terminal domain n
dDD
deletion of death domain
dUD
deletion of undetermined domain
dKD
deletion of kinase domain
dC_n_
deletion of C-terminal domain n
References
- 1.Dinarello C A. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 2.Barnes P J, Karin M. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 3.O'Neill L A J. Biochim Biophys Acta. 1995;1266:31–44. doi: 10.1016/0167-4889(94)00217-3. [DOI] [PubMed] [Google Scholar]
- 4.O'Neill L A J. Biochem Soc Trans. 1997;25:295–302. doi: 10.1042/bst0250295. [DOI] [PubMed] [Google Scholar]
- 5.Greenfeder S A, Nunes P, Kwee L, Labow M, Chizzonite R A, Ju G. J Biol Chem. 1995;270:13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Gao X, Li S, Cao Z. Proc Natl Acad Sci USA. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korherr C, Hofmeister R, Wesche H, Falk W. Eur J Immunol. 1997;27:262–267. doi: 10.1002/eji.1830270139. [DOI] [PubMed] [Google Scholar]
- 8.Wesche H, Henzel W J, Shillinglaw W, Li S, Cao Z. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 9.Lord K A, Hoffman-Liebermann B, Liebermann D A. Oncogene. 1990;5:1095–1097. [PubMed] [Google Scholar]
- 10.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 11.Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, Lewis A, Ray K, Tschopp J, Volpe F. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 12.Cao Z, Henzel W J, Gao X. Science. 1996;271:1126–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 13.Cao Z, Ziong J, Takeuchi M, Kurama T, Goeddel D V. Nature (London) 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 14.Muzio M, Ni J, Feng P, Dixit V M. Science. 1997;278:1612–1615. [Google Scholar]
- 15.Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark G R. Mol Cell Biol. 1999;19:4643–4652. doi: 10.1128/mcb.19.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomaga M A, Yeh W-C, Sarosi I, Duncan G S, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamin T T, Miller D K. J Biol Chem. 1997;272:21540–21547. doi: 10.1074/jbc.272.34.21540. [DOI] [PubMed] [Google Scholar]
- 18.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. Nature (London) 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 19.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H I K, Ninomiya-Tsuji J, Matsumoto K. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 20.DiDonato J A, Hayakawa M, Rothwarf D M, Zandi E, Karin M. Nature (London) 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 21.Mercurio F, Zhu H, Murray B W, Shevchenko A, Bennett B L, Li J, Young D B, Barbosa M, Mann M, Manning A, et al. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 22.Regnier C H, Song H Y, Gao X, Goeddel D V, Cao Z, Rothe M. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 23.Rothwarf D M, Zandi E, Natoli G, Karin M. Nature (London) 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 24.Woronicz J D, Gao X, Cao Z, Rothe M, Goeddel D V. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 25.Yamaoka S, Courtois G, Bessia C, Whiteside S T, Weil R, Agou F, Kirk H E, Kay R J, Israel A. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 26.Zandi E, Rothwarf D M, Delhase M, Hayakawa M, Karin M. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, Lu Q, Hwang J Y, Buscher D, Lee K-F, Izpisua-Belmonte J C, Verma I M. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Estepa G, Memet S, Israel A, Verma I M. Genes Dev. 2000;14:1729–1733. [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 30.Rudolph D, Yeh W-C, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia A J, Mak T W. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 31.Sizemore N, Leung S, Stark G R. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy S A, Huang J H, Liao W S. J Biol Chem. 1997;272:29167–29173. doi: 10.1074/jbc.272.46.29167. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Baldwin A S. J Biol Chem. 1998;273:29411–29416. doi: 10.1074/jbc.273.45.29411. [DOI] [PubMed] [Google Scholar]
- 34.Schindler U, Baichwal V R. Mol Cell Biol. 1994;14:5820–5831. doi: 10.1128/mcb.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majumder S, Zhou L Z H, Chaturvedi R, Babcock G, Aras S, Ransohoff R M. J Neurosci Res. 1998;54:169–180. doi: 10.1002/(SICI)1097-4547(19981015)54:2<169::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 36.Kessler D S, Veals S A, Fu X Y, Levy D E. Genes Dev. 1990;4:1753–1765. doi: 10.1101/gad.4.10.1753. [DOI] [PubMed] [Google Scholar]
- 37.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway C A., Jr Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 38.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer J-L, Di Marco F, French L, Tschopp J. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]