Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation (original) (raw)
Abstract
The disease classification neurodegeneration with brain iron accumulation (NBIA) comprises a clinically and genetically heterogeneous group of progressive neurodegenerative disorders characterized by brain iron deposits in the basal ganglia. For about half of the cases, the molecular basis is currently unknown. We used homozygosity mapping followed by candidate gene sequencing to identify a homozygous 11 bp deletion in the orphan gene C19orf12. Mutation screening of 23 ideopathic NBIA index cases revealed two mutated alleles in 18 of them, and one loss-of-function mutation is the most prevalent. We also identified compound heterozygous missense mutations in a case initially diagnosed with Parkinson disease at age 49. Psychiatric signs, optic atrophy, and motor axonal neuropathy were common findings. Compared to the most prevalent NBIA subtype, pantothenate kinase associated neurodegeneration (PKAN), individuals with two C19orf12 mutations were older at age of onset and the disease progressed more slowly. A polyclonal antibody against the predicted membrane spanning protein showed a mitochondrial localization. A histopathological examination in a single autopsy case detected Lewy bodies, tangles, spheroids, and tau pathology. The mitochondrial localization together with the immunohistopathological findings suggests a pathomechanistic overlap with common forms of neurodegenerative disorders.
Introduction
Brain iron accumulation is a feature of many neurodegenerative disorders, including Friedreich ataxia (MIM 229300), Huntington disease (MIM 143100), and Parkinson disease (PD [MIM 168600]). Pronounced iron deposits in the basal ganglia in the first decades of life are pathognomonic for the diagnosis of neurodegeneration with brain iron accumulation (NBIA), comprising a clinically and genetically heterogeneous group of debilitating neurodegenerative disorders.1 Genetic defects in PANK2 (MIM 606157) are the most common cause of NBIA, followed by mutations in PLA2G6; both genes encode mitochondrial proteins.2–4 PLA2G6 (MIM 603604) provides striking examples of allelic heterogeneity as demonstrated by mutation reports in cases with infantile neuroaxonal dystrophy and adult-onset levodopa-responsive dystonia-parkinsonism.5 Mutations in CP (MIM 117700) and FTL (MIM 134790) have been reported in a small proportion of the remaining NBIA cases with the clearly distinguishable phenotypes aceruloplasminaemia and neuroferritinopathy, respectively. Few reported NBIA families are known to carry mutations in FA2H (MIM 611026),6 a gene previously associated with familial leukodystrophy and spastic paraparesis (MIM 612319).7 However, in about half of NBIA cases the underlying genetic defect remains unknown to date. The identification of additional genetic variation associated with NBIA has the potential to advance our understanding of both rare and common neurodegenerative disease entities.
Subjects and Methods
Cases and Controls
This study was initially conducted in a cohort of 52 NBIA index cases from Warsaw, Poland. Informed consent was obtained from all participants or their guardians, and the study was approved by the institutional review board of the Memorial Children's Health Institute in Warsaw. The diagnosis of NBIA was based on hypointensity in the globus pallidus documented by T2-weighted cranial magnetic resonance imaging and a clinical presentation concordant with NBIA. Detailed clinical information was collected for all individuals. The phenotypic characterization was performed in one clinical center and included a physical examination with an emphasis on neurological and ophthalmologic examination; neuroimaging (a cranial MRI); and electrophysiological (EEG, EMG), neurophysiological, and laboratory testing. The clinical phenotype was documented with a standardized questionnaire. All affected individuals were sequentially screened for mutations in PANK2, PLA2G6, FTL, and CP. We also screened a single case with a NBIA diagnosis where histological samples were available in the German Brain Bank. The clinical details of this case had been collected at the University Hospital, Ludwig Maximilians Universität München.
A group of 676 simplex PD cases from Germany was used for mutation screening of C19orf12. Diagnosis of PD in this group was performed according to the UK PD Brain Bank criteria.8 Mutation screening in controls was performed in samples from 150 healthy individuals from Poland and 600 healthy individuals from Germany.
Homozygosity Mapping and Genetic Screening
DNA was extracted from peripheral leucocytes with standard protocols. We performed a genome-wide linkage analysis in a single family with three affected members by using a genome-wide SNP array (HumanCNV370-Quadv3, Illumina, San Diego, CA). Assuming that the disease allele was identical by descent, we analyzed the data by homozygosity mapping. Frequencies of marker alleles were derived from 1,200 control individuals, and approximately 50,000 evenly distributed polymorphic markers with minimized intermarker linkage disequilibrium (LD) were selected. Homozygous regions were identified with custom scripts. Parametric LOD score calculations were performed under the conservative assumption of a second-cousin marriage. The frequency of the deleterious allele was set to 0.001 and the penetrance to 99% (q = 0.001; f1 = 0.0001; f2 = 0.0001; f3 = 0.99). Data were prepared for linkage analysis with a modified version of Alohomora.9 Multipoint linkage analysis and haplotyping were performed with Merlin (version 1.1.2).10
The ExonPrimer primer design software was used to design primers for PCR amplification of all exons of the 8 transcription units in the linkage interval. PCR products were purified with DNA purification kits (Macherey-Nagel, Düren, Germany). Sequencing of the sense and antisense strands was performed with the same primers as for PCR amplification, ABI BigDye Terminators Version 3, and an ABI 3100 Genetic Analyzer. Primers are listed in Table S1,available online.
Cloning of C19orf12 Transcripts
Total RNA was isolated from human fibroblasts (NDHF-neo, Lonza, Basel, Switzerland) with the Trizol method (Invitrogen, Carlsbad, CA) or from human blood with PAXgene Blood RNA tubes (PreAnalytiX, Hombrechtikon, Switzerland) and the PAXgene Blood RNA kit (QIAGEN). One microgram of RNA was reverse transcribed with M-MLV reverse transcriptase (Promega, Madison, WI) and oligo dT. C19orf12 coding variants 1 and 2 (NM_001031726.2 and NM_031448.3, respectively) were amplified with Thermo-Start Taq DNA Polymerase (ABgene, Epsom, UK).
Cell Culture, Transient Transfection, Stable Transduction, and Immunocytochemistry
NDHF-neo (Lonza) and fibroblast cells were maintained in Dulbecco's Modified Eagle's Medium (PAA, Pasching, Austria) supplemented with 10% fetal bovine serum (PAA), 1% glutamine (GIBCO, Carlsbad, CA), and 1% penicillin plus streptomycin (GIBCO). Cells were transfected with an expression vector containing a C-terminal green fluorescent protein (GFP)-tagged version of C19orf12 variant 1 (Origene, Rockville, MD) with Effectene (QIAGEN, Hamburg, Germany) according to the manufacturer's instructions. After 12-15 hr of culturing, cells were fixed with 4% (wt/vol) paraformaldehyde in PBS for 10 min. Following fixation, cells were permeabilized in PBS, 0.1% NP-40 (Sigma, St. Louis, MO) and blocked with PBS, 2% BSA and 0.1% NP-40 at 37°C. Primary antibody anti-mitochondrial single strand binding protein (mtSSBP) was diluted (1:100) in blocking solution and incubated for 45 min at 37°C. Slides were washed in PBS, 0.1% NP-40 for 30 min. The same incubation and washing procedures were used for the secondary antibody anti-rabbit-Alexa Fluor 568 (Invitrogen). Cells were stably transduced with a lentiviral vector expressing an untagged version of C19orf12 variant 1 (pLenti6.3/V5-TOPO, Invitrogen). Immunocytochemistry was performed with porin (1:1000, MitoSciences) for the detection of mitochondria and C19orf12 (1:500). Anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 568 were used for the secondary detection. Slides were mounted with coverslips with ProLong Gold antifade reagent with 4′,6-diamidin-2′-phenylindol-dihydrochlorid (DAPI, Invitrogen). Images were collected with a Leica TCS SP5 confocal microscope with an 63× oil immersion objective. Mitochondrial morphology was assessed in cell cultures of fibroblasts stained with MitoTracker red (Invitrogen).
Subcellular Localization of C19orf12
For preparation of C19orf12 antibodies, the long isoform of C19orf12 was cloned in an Escherichia coli expression vector with an N-terminal histidine tag (pTrcHis2 TOPO-Invitrogen). After induction of C19orf12 expression with 1 mM isopropyl-β-D-thiogalactopyranosid, the native recombinant protein was purified with Ni-NTA agarose beads according to the manufacturer's protocol (QIAGEN). The purified protein was used for immunization of two rabbits (Pineda).
For preparation of whole cellular lysates, fibroblast cells from cases carrying the Gly69ArgffsX10 in C19orf12 and controls were washed, collected, and resuspended in lysis buffer (0.5 ml/106 cells, 150 mM sodium chloride, 50 mM Tris-HCl [pH 7.4], 0.5% NP-40, 0.25 mM PMSF, 4°C). After 5 s centrifugation at 1000 g to pellet unbroken cells, the supernatant was recovered, and different amounts (5–15 μg each) were separated on a SDS polyacrylamide gel.
To isolate different subcellular fractions, we harvested fibroblasts at 500 g for 3 min; washed them with phosphate-buffered saline; and resuspended them in a solution of 300 mM sucrose, 1 mM EGTA, and 20 mM MOPS (pH 7.4). The cellular suspension was homogenized with a teflon-glass homogenizer with 25 strokes and centrifuged at 600 g for 10 s at 4°C. The pellet representing the unbroken cells and the nuclear fraction was recovered. The mitochondrial-enriched fraction was isolated by centrifugation at 10,000 g for 25 min. The supernatant was centrifuged at 100,000 g for 1 hr to get endoplasmic reticulum (ER) (pellet) and the cytosolic fraction (supernatant). For immune decoration, antisera specific for C19orf12 (1:1000), porin (1:200, Mito Sciences, Eugene, OR), and calnexin (1:2000, Sigma) were used.
Neuropathological Investigations
Histological examination of one case from Germany (Table 1, individual 2) was performed with 4 μm sections from 20 regions of formalin-fixed and paraffin-embedded cerebrum, cerebellum, brainstem and spinal cord. Hematoxylin and eosin and Prussian blue as well as different silver stainings were performed with standard techniques. Immunohistochemistry was performed with antibodies directed against phosphorylated tau (AT8, 1:200, Innogenetics, Ghent, Belgium), α-synuclein (MC42, 1:10, BD Bioscience, Heidelberg, Germany), and amyloid precursor protein (APP) (22C11, 1:100, Chemicon, Temecula, USA).
Table 1.
Clinical Findings
| Familial Cases (NBIA) | Simplex Cases (NBIA) | PD | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family (ID) | 28 | 4 | 21 | 37 | 2a,b | 3 | 5 | 6b | 12 | 20 | 22 | 25 | 27b | 32 | 33b | 34 | 38 | 39 | 41b | 40b | |||||
| II.1 | II.4 | II.5 | II.1 | II.2 | II.1 | II.2 | II.1 | II.2 | |||||||||||||||||
| Sex | m | m | m | f | f | m | f | m | f | m | m | m | m | f | m | m | m | m | m | m | m | f | m | w | M |
| Age of onset | 4 | 12 | 4 | 10 | 9 | 12 | 5 | 10 | 20 | 6 | 8 | 10 | 10 | 10 | 6 | 12 | 12 | 11 | 8 | 12 | 10 | 12c | 8 | 21 | 25 |
| Age at presentation | 12 | 12 | 20 | 25 | 10 | 17 | 15 | 30 | 31 | 21 | 14 | 17 | 18 | 16 | 14 | 15 | 14 | 13 | 13 | 19 | 11 | 14 | 13 | 26 | 59 |
| Pyramidal signsd | + | − | + | − | + | − | ++ | + | ++ | +++ | + | + | ++ | ++ | + | − | +++ | − | +++ | +++ | ++ | − | ++ | ++ | + |
| Extrapyramidal signsd | |||||||||||||||||||||||||
| Oromandibular dystonia | − | − | + | − | − | − | − | ++ | + | − | − | + | + | + | − | − | + | − | − | + | + | − | + | − | − |
| Generalized dystonia | + | − | + | − | − | + | − | ++ | + | − | ++ | ++ | ++ | ++ | − | + | + | − | ++ | + | − | − | + | + | n.a. |
| Parkinsonism | + | − | + | − | − | − | − | + | − | − | − | − | + | − | − | − | − | − | − | + | − | − | − | +++ | +++ |
| Dysarthria | − | − | + | + | − | + | − | ++ | + | +++ | + | + | + | + | − | + | + | − | + | + | + | − | + | +++ | + |
| Wheelchaire | n | n | 18 | 21 | n | n | n | 20 | 31 | 19 | n | 18 | 18 | n | n | n | n | n | n | n | n | n | n | 25 | n |
| Optic atrophyf | yg | yg | yg | y | y | yg | yg | y | y | y | y | yg | y | y | yg | y | yg | n | yg | y | yg | n | y | n | n.a. |
| Motor axonal neuropathy | n | n | y | y | n | n | n | y | y | y | y | y | n.a. | n | n | y | n | y | y | n | n | n | n | n.a | n.a. |
| Psychiatric signsh | n | n | n | n | n | y | y | y | n | n | n | n | n | n | n | y | y | n | n | n | n | n | n | y | Y |
| Cognitive score | 82i | 90i | P7j | 60k | 73k | P25j | P25j | 80i | 80i | n.a. | 117k | 57k | 79i | 85k | 50i | 65i | P29j | 77i | P25j | 73k | 87i | 90i | 96i | n.a. | 22l |
Results
Genetics
We screened a cohort of 52 NBIA index cases from Poland for sequence variants in PANK2, PLA2G6, FTL, and CP and identified 28 individuals carrying mutations in PANK2. Twenty-two of these individuals with PANK2 mutations had been described previously.11 High-density genome-wide SNP genotyping in one of the 24 families that lacked PANK2 mutations and had three affected members (Figure 1) revealed a disease-segregating 1.2 Mb region at 19q12 containing eight transcriptional units (Table S2). Sequence analysis of all exons of these transcripts identified a single homozygous mutation, c.204_214del11 (Gly69ArgfsX10), in the orphan gene C19orf12 (Figure 2). This 11 bp deletion predicts a frameshift and a premature stop codon leading to a predicted truncation of more than 50% of the protein sequence. Sanger sequencing of the gene in the remaining 23 index cases revealed a homozygous 11 bp deletion in 12 of them. Haplotype analysis suggests that the 11 bp deletion derives from a common founder at least 50 to 100 generations ago (Figure S1). Three index cases carried the deletion in combination with different missense mutations (p.Gly65Glu, p.Gly53Arg, and p.Thr11Met) in the compound heterozygous state, one case harbored two missense mutations (p.Gly69Arg and p.Lys142Glu), and two cases carried homozygous missense mutations (p.Tyr11Met and p.Gly69Arg). All investigated healthy parents (n = 30) of individuals with C19orf12 mutations were found to be heterozygous carriers. Five (5/52) index cases remained without a molecular diagnosis. The protein encoded by C19orf12 is highly conserved in evolution with 115 out of 141 amino acids sequences being identical between the human and mouse (Figure 3). In humans it codes for two protein isoforms originating from two alternative first exons. In silico analysis revealed a transmembrane region in all investigated C19orf12 orthologs. Three missense mutations (p.Gly53Arg, p.Gly65Glu, and p.Gly69Arg) change highly conserved glycines in the predicted transmembrane region to a charged amino acid. The mutation p.Thr11Met exclusively affects the longer predicted version of the protein (Figure 2). The p.Lys142Glu missense mutation changes a conserved positive lysine residue to a negative charged glutamate. Although C19orf12 RNA levels in blood remained unaffected by the c.204_214del11 deletion, the protein is absent in fibroblasts according to immunoblot analysis (Figure S2). The discovered mutations were absent from 750 control chromosomes.
Figure 1.
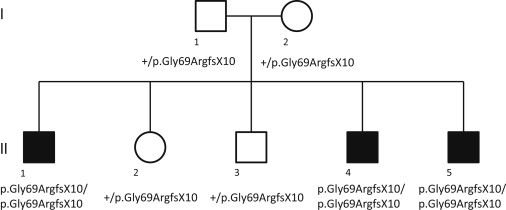
Pedigree of the NBIA Family Used for Homozygosity Mapping
Mutated and nonmutated alleles are indicated.
Figure 2.
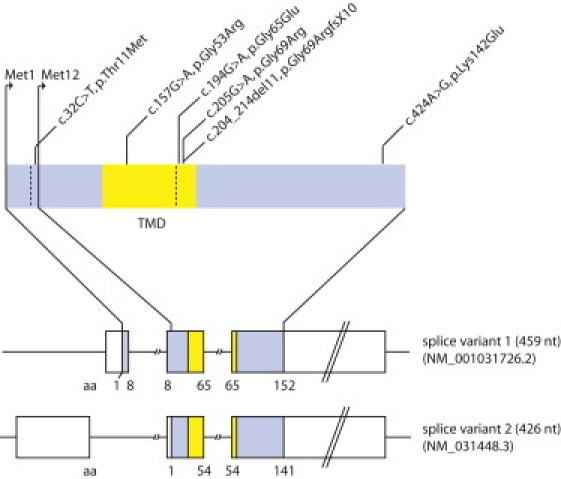
Gene Structure and Identified Disease Alleles
Gene structure of the two isoforms of C19orf12 with the identified mutations. The predicted transmembrane domain is marked in yellow. Mutation nomenclature of the C19orf12 gene is based on splice variant 1 (NM_001031726.2). RefSeq accession number of splice variant 2 is NM_031448.3.
Figure 3.

Evolutionary Conservation of C19orf12
Multiple sequences of C19orf12 orthologs were aligned with CLUSTALW2. Amino acids are indicated as identical (red), highly similar (green), and similar (blue). The predicted transmembrane domain is marked in yellow.
Clinical Findings
Detailed clinical information was available for all 24 Polish NBIA cases with two C19orf12 mutations (Table 1). Eighteen of these are homozygous for the 11 bp deletion in C19orf12 and have a mean age at onset of 9.2 ± 3.7 years (a range of 4 to 20 years), 13 presented with clinical signs of neurodegeneration before the age of 11. The most common presenting symptoms were speech and gait difficulties, which occurred in 10 out of 18 and 12out of 18 of cases, respectively. The predominant neurologic features were extrapyramidal and included oromandibular and generalized dystonia, and parkinsonism (13/18). Involvement of the corticospinal tract leading to spasticity, hyperreflexia, and Babinski sign was frequent (14/18) and progressive. Most of the affected individuals were still able to walk as they entered adulthood, but in some the course of the disease progressed to loss of independent ambulation by the mean (±SD) age of 21.1 ± 4.5 years (n = 5; range of 18 to 31). An unexpected finding was that electrophysiological examination revealed a motor axonal neuropathy in 44% of cases (8/18). Although there was no clinical or electroretinographic evidence for a retinopathy, a feature always present was optic atrophy, which was observed in all 18 cases with two loss-of-function alleles. The severity of the optic atrophy varied but did not result in blindness in any of the cases of our cohort.
Six (6/24) cases of the Polish cohort carried at least one missense mutation. A trend toward a later onset of the disease (13.2 ± 4.4 years) did not reach significance when compared to patients carrying two C19orf12 loss-of-function alleles. Optic atrophy was present in three out of six cases, and motor axonal neuropathy in two out of four cases investigated. The course of the disease was remarkably mild in individuals 27 and 38, which did not display any pyramidal or extrapyramidal signs. Individual 27 suffered from a motor axonal neuropathy causing mild gait impairment at the age of 13 years, and individual 38 presented with a mild affection of fine motor skills only at the age of 14 years.
Psychiatric signs were common in cases with C19orf12 mutations (6/24), including impulsive or compulsive behavior, depression, and emotional lability. No seizures were reported.
T2-weighted MRI revealed hypointensities in the globus pallidus and substantia nigra in all 24 C19orf12 cases, whereas PKAN-typical hyperintensity in the central anteromedial region of the globus pallidus, also referred to as “eye of the tiger” sign, was only seen in one (Figure 4).
Figure 4.
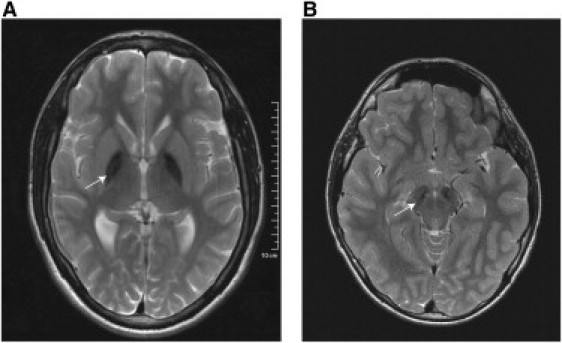
Magnetic Resonance Images of Cases with the Homozygous C19orf12 Deletion c.204_214del11, p.Gly69ArgfsX10
Axial T2-weighted magnetic resonance imaging (1.5 T) shows a bilateral hypointensity of the globus pallidus (A) and substantia nigra (B).
Neuropathological Findings
Brain pathology was available from a German case (Table 1, individual 2) previously diagnosed with idiopathic NBIA. Sequence analysis of C19orf12 identified a homozygous p.Gly69Arg mutation. This person had presented at school enrollment (at the age of 6) with clumsiness and fatigue. Optic atrophy had been diagnosed at the age of 10. Progressive gait spasticity had become evident from the age of 14 on, leading to a loss of gait and wheelchair dependence by the age of 18. Examination at age 21 had shown spastic tetraparesis, ataxia, marked dysarthria, axonal motor neuropathy, and cognitive decline. A T2-weighted MRI had revealed hypointensities in the globus pallidus. The person had died at the age 23 from respiratory insufficiency. Histopathological examination showed iron-containing deposits, axonal spheroids, numerous α-synuclein-positive Lewy bodies, and Lewy body-like inclusions, sparse Lewy neurites and hyperphosphorylated tau-containing neuronal inclusions in various regions of the brain (Figure 5). Iron-containing deposits were concentrated in the globus pallidus and the substantia nigra. The hippocampus showed only a small number of α-synuclein-containing deposits but numerous tau-positive pyramidal cells. Loss of myelin was seen in the pyramidal tracts of the spinal cord and optic nerve and was most pronounced in the optic tract.
Figure 5.
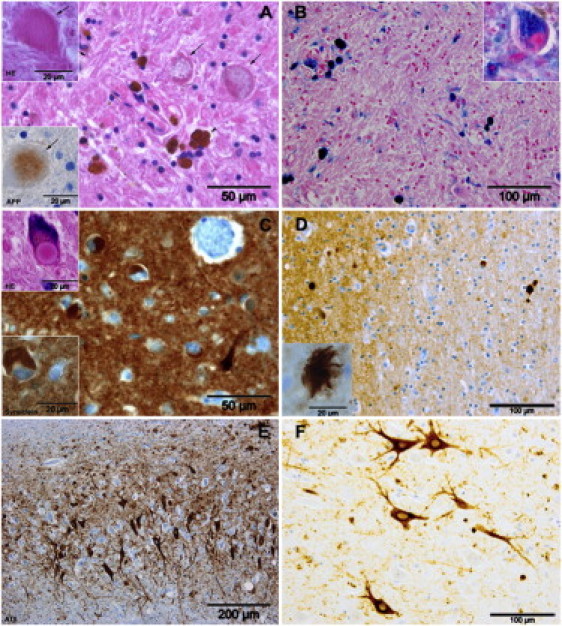
Brain Histopathology of an Individual with the Homozygous C19orf12 Missense Mutation c.205G>A, p.Gly69Arg, p.Gly69Arg
(A) Hematoxylin and eosin staining and (B) Prussian blue staining of the globus pallidus showed iron-containing deposits in neurons (insert in B) that are more diffusely distributed in astrocytes and very dense in perivascular macrophages (arrowhead in A) as well as many axonal spheroids (arrows in A) that were stained faintly by antibodies against APP (insert in B). Spheroids were found in the globus pallidus, putamen, and caudate nucleus, and at a lower density in the thalamus, internal capsule, brainstem, cerebral cortex, dentate nucleus, and spinal cord. Numerous α-synuclein-positive Lewy bodies, Lewy body-like inclusions, and some Lewy neurites were detected in the frontal cortex (C) and other neocortical areas; the hippocampus was less severely affected. Similar histopathology with Lewy bodies and Lewy body-like inclusions was found in the brain stem, including the substantia nigra, basal ganglia, cortex, and spinal cord (not shown). Some seemingly extracellular α-synuclein-positive deposits were noted in various areas (D, showing the external capsule and claustrum). Hyperphosphorylated tau-containing neuronal inclusions were identified in the hippocampus (E). The tau protein often seemed densely packed around pyramidal cell nuclei as is sometimes observed in tauopathies (F). Only a very small number of tau-positive inclusions stained with silver staining methods (Bodian, Bielschowsky, Gallyas) but did not show fibrils. Tufted astrocytes or coiled bodies were not seen. No neurofibrillary tangels (NFT) were seen in the globus pallidus or the substantia nigra. However, tau-positive extracellular punctuate and globular or irregularly shaped structures were found widely distributed in the hippocampus and the neocortex. In some instances these structures seemed to be associated with astrocytes.
Mutation Screen in Cases with Parkinson Disease
Intrigued by the abundant presence of Lewy bodies, the neuropathological hallmark of ideopathic Parkinson disease,12 we performed a mutation screen of C19orf12 in 676 simplex PD cases. This analysis led to the identification of one sample (Table 1, individual 40) with two compound heterozygous mutations (p.Gly69Arg and p.Lys142Glu). Both compound heterozygous mutations have been also identified in a patient of the Polish cohort (Table 1, individual 38) presenting with a notably mild course of the disease that consisted of only an impairment of fine motor skills at the age of 14. In fact, the MRI performed at the age of 12 was conducted because of a pituitary adenome, and the brain iron accumulation was an accidental finding.
Evaluation of medical reports revealed that individual 40 presented with paranoid hallucinations at the age of 25. By the age 49, he was diagnosed PD, and a treatment with L-Dopa was started. The latter was effective in preventing akinesia, but a strong fluctuation of the symptoms was described 11 years later when he was included in a Parkinson study. Typical PD signs such as rigidity, akinesia, and a mild tremor at rest were documented. Further, axial signs, off-dystonia of the legs with muscle cramps, hypophonia, hypomimia, vivid dreams, sleep disturbance, and optic hallucinations were described. The Unified Parkinson Disease Rating Score (UPDRS, version III) at that time was 11 and 37, with and without medication, respectively. The mini-mental status test (MMST) was 22, indicating a cognitive decline. A cranial computed tomography conducted at the age of 58 showed a marked generalized cerebral atrophy. An MRI has not been performed, and the person was not available for follow-up to resolve the question of brain iron deposits.
Functional Investigations
We expressed a C19orf12-GFP fusion protein in fibroblast cell lines where the C19orf12 fusion protein colocalized with mitochondria (Figure 6). This finding was confirmed by subcellular fractionation experiments (Figure 7) and in vitro import of the radiolabelled gene product into mouse mitochondria (Figure S3).
Figure 6.
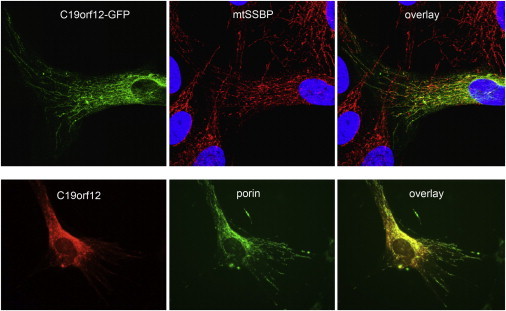
Subcellular Localization of C19orf12
A C19orf12-GFP fusion protein (upper α-synuclein-positive deposits were noted in various areas [E]) and C19orf12 untagged protein (lower panel) exhibit a mitochondrial localization in transiently transfected and in stably transduced fibroblasts, respectively. Mitochondria were stained with mitochondrial single strand binding protein (anti-mtSSBP; red) or anti-porin (green). Nuclear DNA was stained with DAPI (blue).
Figure 7.
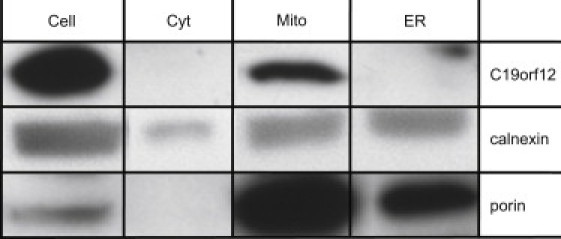
Subcellular Localization of C19orf12 in Mitochondria
NDHF-neo cells were harvested to obtain mitochondria, ER, and cytosol fractions. Equal amount of proteins (20 μg) from each fraction were resolved by SDS-PAGE and immunoblotted for C19orf12 with an antibody against the whole protein. Porin and calnexin were used as a loading control of inner mitochondrial membrane proteins and endoplasmatic reticulum, respectively.
On the basis of these findings, we propose the acronym MPAN (_m_itochondrial membrane _p_rotein _a_ssociated _n_eurodegeneration) to name the clinical subtype of NBIA caused by C19orf12 mutations.
Although biochemical analysis of respiratory chain complexes and mitochondrial morphology in fibroblasts was inconspicuous (data not shown), transcription profiles in blood and adipocytes provided indirect evidence of the functional role of C19orf12. Its expression is prominent in the brain, blood cells, and adipocytes according to public databases (Figure S4). Analysis of _C19orf12_-coregulated genes in whole blood13 (n = 380) of a general population sample (KORA) revealed the most significant enrichment of genes involved in the two pathways fatty acid biogenesis (p = 4.6 × 10−6) and valine, leucine, and isoleucine degradation (p = 5.8 × 10−4, Table S3). These two pathways predominantly represent mitochondrial processes related to CoA metabolism. Expression profiles in whole blood of NBIA cases compared with controls (n = 7) showed a significant enrichment of differentially expressed genes within the pathway category mitochondrial dysfunction both in the MPAN cases (n = 7; p = 2.5 × 10−3) and the PKAN cases (n = 6; p = 2.7 × 10−3). Finally, we analyzed C19orf12 expression levels in relation to transcriptome profiles in an in vitro preadipocyte differentiation model. During adipocyte differentiation, we found C19orf12 upregulated with genes involved in the two categories valine, leucine and isoleucine degradation (p = 3.46 × 10−7) and fatty acid metabolism (p = 2.5 ×10−0.6; Figure S5).
Discussion
Of the approximately 25,000 genes in the human genome, 1,241 are still tagged with an open reading frame symbol.14 We identified mutations in a recessive form of NBIA, we named MPAN, in one such open reading frame on chromosome 19. C19orf12 is a small gene that spans less than 17 kb of the genomic sequence and codes for two protein isoforms of 141 and 152 amino acid residues in humans. The protein is highly conserved because almost 60% of the amino acid residues are identical between the human and zebrafish. In contrast, the presence of two protein isoforms is only documented for three species. In the human, chimpanzee, and chicken, the first exon allows both isoforms to be produced, whereas in mice and fish for instance, only a single isoform (the short one) is coded. This has implications for the future establishment of animal models for MPAN.
Genotype-phenotype correlations covering the mutational spectrum observed allow several conclusions. A single case carries a mutation situated in front of the start codon of the short isoform (Thr11Met). This identifies the long isoform as the one associated with disease pathology. The majority of cases investigated (18/24) carry the homozygous frameshift mutation. Because C19orf12 could not be detected in a cell culture of one of these cases, we conclude that this mutation exerts its effect by loss of function. The age of onset in this group is between 4 and 20 years. A loss of ambulance occurs 8 to > 24 years later. In comparison cases with loss-of-function PANK2 mutations have earlier age of onset, a faster disease progression, a different ocular phenotype (RP), and no signs of axonal neuropathy.11 In addition to the frameshift mutation in C19orf12, we identified three missense mutations changing conserved glycines within the predicted transmembrane domain to charged amino acids. The phenotype associated with these mutations is within the clinical spectrum caused by the frameshift mutation. The allelic series observed in MPAN cases is completed by the c.424A>G (p.Lys142Glu) missense mutation, which is predicted to exert a milder effect (PolyPhen2).15 Two cases carry this mutation together with the p.Gly65Glu mutation. One of them is now 19 years old and suffers only from impaired fine motor skills, the other one was diagnosed at the age of 49 with Parkinson disease. No other diagnosis of a neurodegenerative disorder was made previously in these persons. The molecular diagnosis of the C19orf12 mutations is facilitated by the small size of the gene. Because of the founder mutation observed in the Polish NBIA cohort, the proportion of NBIA cases with C19orf12 mutation might be overestimated compared with other populations. However, the number of different disease alleles discovered in our study argues that a considerable proportion of NBIA cases worldwide are due to mutations in this gene.
Using both GFP-tagged protein and a C19orf12-specific antibody, we showed that the protein localizes predominantly to mitochondria. So far at least five genes are known in which mutations cause a NBIA phenotype with similarities to MPAN cases. At least two of these genes (PANK2 and PLA2G6) encode proteins that share with C19orf12 a mitochondrial localization, and they are suggested to play a role in lipid homeostasis. Preliminary evidence for a role for C19orf12 in lipid homeostasis is provided by two experimental observations. First, there are high C19orf12 expression levels observed in adipose tissue, which is upregulated during adipocyte differentiation. Second, _C19orf12_-coregulated genes are predominately those involved in fatty acid metabolism.
C19orf12 is ubitquitously expressed. Nevertheless, both cells and entire organisms with two loss-of-function C19orf12 alleles are viable. However, over an extended period of time, degeneration occurs preferentially in neurons. The final stage of neurodegeneration—as demonstrated by the autopsy case described—is characterized by a variegated histopathological picture in basal ganglia and cortex regions of the brain including Lewy bodies and tau inclusions as well as spheroids and iron deposits. This observation has two main implications. First, it shows—as has been shown before for familial cases of Parkinson disease—that distinct histopathological entities associated with neurodegeneration can occur as a consequence of a single gene defect. Second, it confirms the view that the histological end stage of the disease is far detached from the initial pathophysiological disturbances. A detailed functional investigation of both normal and mutated C19orf12 forms has to tell us the details.
Acknowledgments
We are indebted to all individuals and their families donating samples and clinical datasets. We thank I. Gromek, A. Kostera-Pruszczyk, B. Lojszczyk, and B. Chipczynska for their efforts in clinical phenotyping. We gratefully acknowledge the support of R. Hellinger and A. Löschner in genotyping; B. Schmick, K. Junghans, and B. Siegel in expression analysis; and S. Heinisch in bioinformatics analysis. T.M. and H.P. were supported by the Impulse and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Health in an Ageing Society (HA-215), the German National Research Network (NGFNplus #01GS08134), the German Network for Mitochondrial Disorders (mitoNET 01GM0867), and Systems Biology of Metabotypes (SysMBo #0315494A). T.K. is supported by the German Network for Mitochondrial Disorders (mitoNET #01GM0862). J.W. and K.O. are supported by a grant from the Deutsche Forschungsgemeinschaft (WI 1820/4-1). The Brain-Net is supported by the Federal Ministry of Education and Research (BMBF). Work on primary human adipocytes was supported by the seventh framework program of the European-Union-funded LipidomicNet (proposal 202272). S.D. is supported by Italian Foundation AISNAF (Associazione Italiana Sindromi Neurodegenerative Accumulo di Ferro). The financial support of Mariani Foundation of Milan (grant R-10-84 to V.T.) is gratefully acknowledged.
Supplemental Data
Document S1. Five Figures and Four Tables
Web Resources
The URLs for data presented herein are as follows:
- BioGPS database, http://biogps.gnf.org
- Brain-Net, http://www.brain-net.net
- CLUSTALW2, http://www.ebi.ac.uk/clustalw/
- ExonPrimer, http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html
- Ingenuity pathway analysis, http://www.ingenuity.com
- OMIM, http://www.omim.org
- TMHMM, http://www.cbs.dtu.dk/services/TMHMM/
- UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Gregory A., Hayflick S.J. Genetics of neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2011;11:254–261. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou B., Westaway S.K., Levinson B., Johnson M.A., Gitschier J., Hayflick S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 3.Hörtnagel K., Prokisch H., Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum. Mol. Genet. 2003;12:321–327. doi: 10.1093/hmg/ddg026. [DOI] [PubMed] [Google Scholar]
- 4.Morgan N.V., Westaway S.K., Morton J.E., Gregory A., Gissen P., Sonek S., Cangul H., Coryell J., Canham N., Nardocci N. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paisan-Ruiz C., Bhatia K.P., Li A., Hernandez D., Davis M., Wood N.W., Hardy J., Houlden H., Singleton A., Schneider S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruer M.C., Paisán-Ruiz C., Boddaert N., Yoon M.Y., Hama H., Gregory A., Malandrini A., Woltjer R.L., Munnich A., Gobin S. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA) Ann. Neurol. 2010;68:611–618. doi: 10.1002/ana.22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edvardson S., Hama H., Shaag A., Gomori J.M., Berger I., Soffer D., Korman S.H., Taustein I., Saada A., Elpeleg O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am. J. Hum. Genet. 2008;83:643–648. doi: 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes A.J., Daniel S.E., Kilford L., Lees A.J. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindstrom M.J., Bates D.M. Newton-Raphson and EM Algorithms for Linear Mixed-Effects Models for Repeated-Measures Data. J. Am. Stat. Assoc. 1988;83:1014–1022. [Google Scholar]
- 10.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 11.Hartig M.B., Hörtnagel K., Garavaglia B., Zorzi G., Kmiec T., Klopstock T., Rostasy K., Svetel M., Kostic V.S., Schuelke M. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann. Neurol. 2006;59:248–256. doi: 10.1002/ana.20771. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meisinger C., Prokisch H., Gieger C., Soranzo N., Mehta D., Rosskopf D., Lichtner P., Klopp N., Stephens J., Watkins N.A. A genome-wide association study identifies three loci associated with mean platelet volume. Am. J. Hum. Genet. 2009;84:66–71. doi: 10.1016/j.ajhg.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pruitt K.D., Tatusova T., Klimke W., Maglott D.R. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37(Database issue):D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Five Figures and Four Tables