Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration (original) (raw)
. Author manuscript; available in PMC: 2011 Oct 12.
Abstract
Microtubule associated protein (MAP) tau is abnormally hyperphosphorylated in Alzheimer’s disease (AD) and related tauopathies; in this form it is the major protein subunit of paired helical filaments (PHF)/neurofibrillary tangles. However, the nature of protein kinases and phosphatases and tau sites involved in this lesion has been elusive. We investigated self-assembly and microtubule assembly promoting activities of hyperphosphorylated tau isolated from Alzheimer disease brain cytosol, the AD abnormally hyperphosphorylated tau (AD P-tau) before and after dephosphorylation by phosphoseryl/phosphothreonyl protein phosphatase-2A (PP-2A), and then rephosphorylation by cyclic AMP-dependent protein kinase (PKA), calcium, calmodulin-dependent protein kinase II (CaMKII), glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent protein kinase 5 (cdk5) in different kinase combinations. We found that (i) dephosphorylation of AD P-tau by PP-2A inhibits its polymerization into PHF/straight filaments (SF) and restores its binding and ability to promote assembly of tubulin into microtubules; (ii) rephosphorylation of PP-2A-dephosphorylated AD P-tau by sequential phosphorylation by PKA, CaMKII and GSK-3β or cdk5, and as well as by cdk5 and GSK-3β, promotes its self-assembly into tangles of PHF similar to those seen in Alzheimer brain, and (iii) phosphorylation of tau sites required for this pathology are Thr231 and Ser262, along with several sites flanking the microtubule binding repeat region. Phosphorylation of recombinant human brain tau441 yielded similar results as the PP-2A dephosphorylated AD P-tau, except that mostly SF were formed. The conditions for the abnormal hyperphosphorylation of tau that promoted its self-assembly also induced the microtubule assembly inhibitory activity. These findings suggest that activation of PP-2A or inhibition of either both GSK-3β and cdk5 or one of these two kinases plus PKA or CaMKII might be required to inhibit Alzheimer neurofibrillary degeneration.
Keywords: abnormal hyperphosphorylation of tau, Alzheimer’s disease, paired helical filaments, protein phosphatase-2A, tauopathies
Introduction
Neurofibrillary degeneration of abnormally hyperphosphorylated tau is a hallmark of Alzheimer’s disease (AD) and related tauopathies (Braak et al., 1986; Grundke-Iqbal et al., 1986a; Tolnay & Probst, 1999). The hyperphosphorylated tau is seen both as polymerized into tangles of paired helical filaments (PHF) admixed with straight filaments (SF) and up to 40% as the cytosolic protein in AD brain (Grundke-Iqbal et al., 1986b; Iqbal et al., 1986; Kopke et al., 1993). Unlike normal microtubule associated protein (MAP) tau, which promotes assembly and stabilizes microtubules (Weingarten et al., 1975), the cytosolic AD abnormally hyperphosphorylated tau (AD P-tau) sequesters normal tau, MAP1 and MAP2, and inhibits assembly and depolymerizes microtubules (Alonso et al., 1994).
Tau in PHF/SF has been found to be phosphorylated at over 30 serine/threonine residues (Hanger et al., 1998) and approximately half of these are canonical sites for proline directed protein kinases (PDPKs), suggesting the role of PDPKs and serine/threonine protein phosphatase(s) in the abnormal hyperphosphorylation of tau.
Phosphoseryl/phosphothreonyl protein phosphatase-2A (PP-2A), which is colocalized with tau and microtubules in the brain (Sontag et al., 1999) is apparently the most active enzyme in dephosphorylating the abnormal tau to a normal-like state (Wang et al., 1995; Wang et al., 1996b). In AD brain, both the activity and the mRNA of PP-2A are decreased (Gong et al., 1993; Gong et al., 1995; Vogelsberg-Ragaglia et al., 2001). Inhibition of the brain PP-2A activity results in the abnormal hyperphosphorylation of tau (Bennecib et al., 2000; Gong et al., 2000; Planel et al., 2001; Sun et al., 2003; Tian et al., 2004), not only by a decrease in the dephosphorylating activity but probably also by up-regulating the activities of calcium, calmodulin-dependent protein kinase II (CaM-KII), cyclic AMP-dependent protein kinase (PKA), and several members of the mitogen activated protein kinase (MAPK) family (Bennecib et al., 2001; Kins et al., 2003; Pei et al., 2003; Li et al., 2004).
Tau is phosphorylated by several protein kinases (Pei et al., 2003; Liu et al., 2004; Iqbal et al., 2005). PKA (Jicha et al., 1999), CaMKII (Xiao et al., 1996), glycogen synthase kinase-3β (GSK-3β) (Yamaguchi et al., 1996; Pei et al., 1997; Pei et al., 1999; Ferrer et al., 2001), and cyclin-dependent protein kinase 5 (cdk5) and its activator p25 (Pei et al., 1998) have been shown to be associated with tangles in AD brain. Both cdk5 and GSK-3β are associated with microtubules in the brain (Ishiguro et al., 1992; Sobue et al., 2000) and phosphorylation of tau by cdk5 promotes its subsequent phosphorylation by GSK-3β (Sengupta et al., 1997).
Recombinant human brain taus, in vitro hyperphosphorylated with brain extract as a source of protein kinases, and AD P-tau isolated from AD brain, self-assemble into tangles of PHF and SF (Alonso et al., 2001). However, the exact nature of the protein kinases and phosphatases and the critical tau sites involved have been elusive. The present study shows, for the first time, the exact nature of the protein kinases and phosphatases and the tau sites involved in the self-assembly of tau into PHF/SF.
Materials and methods
Materials
Catalytic subunit of PKA, purified from bovine heart, was purchased from Sigma (St Louis, MO), and rat brain CaMKII and recombinant GSK-3β were purchased from CalBiochem (San Diego, CA). Recombinant cdk5/P25 was a kind gift from Dr J. Wang (Hong Kong University, HK). PP-2A was purified from bovine brain as described (Cohen et al., 1988).
Isolation of recombinant human brain tau441 and AD P-tau
Recombinant tau441 was purified as described (Singh et al., 1995b) except that the perchloric acid extraction was avoided. The phosphocellulose-purified tau was further purified by a Sephacyl 300 column chromatography at 4 °C. AD P-tau was purified from histopathologically confirmed and autopsied frozen AD brains (obtained within 6 h postmortem) as described previously (Kopke et al., 1993). The use of human tissue for these studies was approved by our Institutional Review Board. Briefly, a 27 000–200 000 × g pellet fraction from AD cerebral cortex was extracted in 8 M urea, dialysed first against 50 mM Tris, pH 7.0, and then against three changes of 25 mM Mes, pH 6.4, containing 0.5 mM MgCl2/1 mM DTT, followed by phosphocellulose column chromatography. The column fractions were analysed by Western blots developed with Tau-1 with or without previous dephosphorylation of the protein on the PVDF membrane by alkaline phosphatase. The AD P-tau eluted at a salt concentration of ~0.2 M NaCl. The tau peak was brought to 2 M NaCl by adding solid salt, and it was loaded onto a Phenyl Sepharose high performance column (Amersham Pharmacia, Arlington, IL), employing 0.2 mL bed volume/g of starting material. Tau was detected in the unbound fraction. This fraction was concentrated by embedding the sample in the dialysis bag in 20-kDa polyethylene glycol powder. Once concentrated, the sample was dialysed against 5 mM Mes buffer, pH 6.8, containing 0.05 mM EGTA, lyophilized and kept at −75 °C until used.
Dephosphorylation of AD P-tau by PP-2A
Dephosphorylation was carried out at 37 °C for 6 h by incubating AD P-tau (0.2 mg/mL) with PP-2A (4 μg/mL) in 25 mM Tris-HCl buffer (pH 7.0) containing 2 mM MnCl2, 1 mM EGTA, 10 mM β-mercaptoethanol (β-ME), 0.5 mM PMSF. The reaction was terminated by boiling the sample for 3 min. The mixture was then bath sonicated for 15 min and used for rephosphorylation.
Rephosphorylation of PP-2A-AD P-tau by various kinases, sites phosphorylated and the effect on polymerization of tau into filaments
Rephosphorylation of PP-2A-AD P-tau was performed by adding to the above dephosphorylated sample, a mixture of 30 mM Hepes (pH 6.8) with PKA, or 30 mM Hepes (pH 7.5) with CaMKII, GSK-3β or cdk5, and 10 mM β-mercaptoethanol (β-ME), 2 mM ATP. In the case of CaMKII, 0.4 mM CaCl2 and 40 μg/mL calmodulin were also included. The rephosphorylation was carried out at 30 °C for 4 h totally. For sequential phosphorylation of the two kinases, the substrate was exposed to the first kinase for 1 h, then the second kinase for 3 h. For the three-kinase combination, the phosphorylation reaction was carried out by the first kinase for 1 h, then the second kinase for 1 h, and finally the third kinase for 2 h, or all the three kinases added to the reaction mixture simultaneously. Unless otherwise stated, the amounts of kinases used in this study for AD P-tau were 40 μg/mL for PKA, 4 μg/mL for CaMKII, 100 units/mL for GSK-3β and 40 μg/mL for cdk5/P25. The concentration of AD P-tau was 0.2 mg/mL. The amount of the kinases used for phosphorylation of recombinant tau441 was cut into half of the amount used for AD P-tau because tau441 was more easily phosphorylated by the kinases than AD P-tau. (γ32P)-ATP was employed to determine the stoichiometry of the phosphorylation. The phosphorylation sites were mapped by Western blots as described (Grundke-Iqbal et al., 1986a). The blots were developed by alkaline phosphatase-conjugated secondary antibodies by using 5-bromo-4-chloro-3-indoyl phosphate _p_-toluidine salt (BCIP) and _p_-nitro blue tetrazolium chloride (NBT) as a substrate. Negative stain electron microscopy (EM) and Congo-red birefringence light microscopy were used to study the critical kinase(s) involved in conversion of PP-2A-AD P-tau into AD-like PHF/tangles (Iqbal et al., 1984; Wisniewski et al., 1984). Each experiment for the effect of rephosphorylation on the polymerization of PP-2A-AD P-tau and for the effect of phosphorylation on recombinant tau441 was carried out 2–3 times and more than 10 negative stain electron microscopy (EM) grids were screened for each set of the samples.
Effect of hyperphosphorylated tau on microtubule assembly
For biological activity assay, after phosphorylation of tau441 or rephosphorylation of PP-2A-AD P-tau, the samples were boiled for 3–5 min to terminate the reaction. The samples were then dialysed against four changes of the dialysis buffer (5.0 mM Mes, pH 6.7 containing 0.05 mM EGTA). Dialysed samples were then bath-sonicated for 30 min and centrifuged 1–2 min (Capsule HF-120, Tokyo, Japan). The supernatants, which were free from any filaments as determined by negative stained electron microscopy, were lyophilized in a Speed Vac Concentrator (Savant Instruments, Inc., Farmingdale, NY) and reconstituted in microtubule assembly buffer (100 mM Mes, pH 6.7, 1 mM EGTA and 1 mM MgCl2).
Tubulin was prepared from brains of one-month-old rats by one and a half microtubule assembly disassembly cycle, and purified by phosphocellulose column as described previously (Alonso et al., 1994). The use of rat brains for these studies was approved by our Institute Animal Welfare Committee. The in vitro microtubule assembly assay was carried out by incubating tau (10–30 μg/mL for tau441; 300 μg/mL for AD P-tau) with purified rat brain tubulin (3.0 mg/mL) in 1 cm quartz microcuvette at 32 °C. The reaction of assembly was monitored up to 10 min by recording light scattering changes at 350 nm. The microtubule assembly disruption was assayed by adding rephosphorylated or untreated AD P-tau to the assembly reaction promoted by tau441.
Binding of tau to taxol-stabilized microtubules
For the preparation of taxol-stabilized microtubules for the binding assay, 1 μL of 10 mM taxol (in DMSO) was added to 1.75 mg phosphocellulose-purified tubulin in a final volume of 500 μL, incubated at 37 °C for 30 min, then centrifuged at 80 000 g (TL45, Beckman) for 30 min at 30 °C, and the pellets containing microtubules were stored at −75 °C until used. The microtubule binding assay was carried out by incubating 6 μg AD P-tau (or 0.5 μg tau441) and 30 μg microtubules (or 20 μg for tau441) in a total volume of 20 μL containing 100 mM Mes pH 6.7, 1 mM EGTA, 1 mM MgCl2, 1 mM PMSF, at 37 °C° for 30 min. Then the suspension was centrifuged at 40 000 g for 30 min at 30 °C through a 0.25 M sucrose cushion; the collected supernatant and pellet were analysed by (125I)Western blots developed with polyclonal tau antibody 92e (Grundke-Iqbal et al., 1988). The binding activity of tau to microtubules was calculated from the amounts of tau found in the pellet and the supernatant.
Results
Dephosphorylation of AD P-tau by PP-2A
AD P-tau has been previously shown to self-assemble into PHF/tangles (Alonso et al., 2001). PP-2A has been implicated most in the abnormal hyperphosphorylation of tau but the identity of the sites, dephosphorylation of which by PP-2A inhibits its self assembly into PHF, was not known. We investigated the sites of AD P-tau dephosphorylated by PP-2A by Western blots developed with various site-specific and phosphorylation-dependent antibodies (Table 1). We found that PP-2A dephosphorylated AD P-tau at all the sites studied except that dephosphorylation of Thr205, Thr212, Thr217 and Ser396 was unremarkable (Fig. 1; also see below, Fig. 3A and B, lanes 1 and 2), and the dephosphorylated protein did not aggregate into filaments (see below, Fig. 4B). These findings are consistent with recent studies that showed that tau at Thr212 and Ser396 is preferentially dephosphorylated by PP-1 and PP-2B, respectively (Rahman et al., 2005, 2006).
Table 1.
Tau antibodies employed and their properties
| Antibody | Dilution | Type* | Specificity† | Phosphorylation sites‡ |
|---|---|---|---|---|
| Tau-1 | 1 : 30 000 | Mono | unP | Ser198/Ser199/Ser202 |
| PHF-1 | 1 : 500 | Mono | P | Ser396/Ser404 |
| 12E8 | 1 : 500 | Mono | P | Ser262/Ser356 |
| AT100 | 1 : 500 | Mono | P | Thr212/Ser214 |
| R145d | 1 : 500 | Poly | P | Ser422 |
| 92e | 1 : 3000 | Poly | P + unP | Total tau |
| P-181 | 1 : 1000 | Poly | P | Thr181 |
| P-199 | 1 : 1000 | Poly | P | Ser199 |
| P-202 | 1 : 1000 | Poly | P | Ser202 |
| P-205 | 1 : 1000 | Poly | P | Thr205 |
| P-212 | 1 : 1000 | Poly | P | Thr212 |
| P-217 | 1 : 1000 | Poly | P | Thr217 |
| P-231 | 1 : 1000 | Poly | P | Thr231 |
| P-235 | 1 : 1000 | Poly | P | Ser235 |
| P-262 | 1 : 1000 | Poly | P | Ser262 |
| P-396 | 1 : 1000 | Poly | P | Ser396 |
| P-403 | 1 : 1000 | Poly | P | Thr403 |
| P-404 | 1 : 1000 | Poly | P | Ser404 |
| P-409 | 1 : 1000 | Poly | P | Ser409 |
| P-413 | 1 : 1000 | Poly | P | Ser413 |
| P-422 | 1 : 1000 | Poly | P | Ser422 |
Fig. 1.

A schematic showing the AD abnormally hyperphosphorylated tau sites, the sites studied and sites found dephosphorylated by PP-2A. PP-2A effectively dephosphorylated AD P-tau at all the sites studied, but to varying degrees; dephosphorylation at Thr205, Thr212, Thr217 and Ser396 was less remarkable.
Fig. 3.

Western blots showing site-specific dephosphorylation of AD P-tau by PP-2A and its rephosphorylation (A and B), and phosphorylation of tau441 (C and D) by PKA, CaMKII, GSK-3β and cdk5 individually and in different combinations. (A) AD P-tau isolated from AD brain (lane 1) was dephosphorylated by PP-2A (lane 2), and then rephosphorylated by PKA (lane 3), or CaMKII (lane 4), or GSK-3β (lane 5) or PKA, CaMKII and GSK-3β (lane 6); or (B) cdk5 (lane 7), PKA plus cdk5 (lane 8), or CaMKII plus cdk5 (lane 9), or cdk5 and GSK-3β (lane 10) or PKA, CaMKII and GSK-3β (lane 11). (C) Tau441 (lane 12) was phosphorylated by PKA (lane 13), or CaMKII (lane 14), or GSK-3β (lane 15), or PKA, CaMKII and GSK-3β (lane 16) or (D) cdk5 (lane 17), or PKA plus cdk5 (lane 18) or CaMKII plus cdk5 (lane 19), or cdk5 plus GSK-3β (lane 20) or PKA, CaMKII and GSK-3β (lane 21). Western blots were probed with antibodies as indicated below each blot.
Fig. 4.
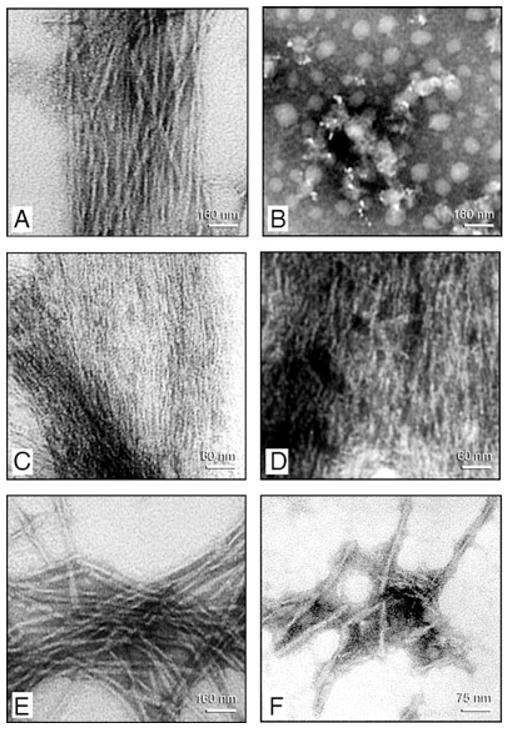
Negative stain electron microscopy of products of self assembly of AD P-tau and of this protein after its dephosphorylation with PP-2A and rephosphorylation with different combination of kinases. Incubation of untreated AD P-tau in rephosphorylation buffer for 4 h converted it into classic PHF/tangles (panel A). Dephosphorylation of AD P-tau by PP-2A abolished its self polymerization and no specific fibrous structures could be detected (panel B). Rephosphorylation of PP-2A-AD P-tau with CaMKII plus GSK-3 (panel C) induced formation of filaments with ~2.4 nm (panel C) and ~10 nm diameter (panel D). Rephosphorylation of PP-2A-AD P-tau with PKA, CaMKII and GSK-3β (panel E) or cdk5 plus GSK-3β (panel F) induced formation of ~22 nm PHF.
Rephosphorylation of PP-2A-AD P-tau and phosphorylation of tau441 by combination of protein kinases
Hyperphosphorylation has been previously shown to promote self assembly of tau into filaments but the exact identity of the protein kinases involved was not known. We have previously shown that phosphorylation of tau by PKA, CaMKII or cdk5 primes tau by subsequent phosphorylation by GSK-3β, and PKA and CaMKII prime tau for subsequent phosphorylation by cdk5 (Singh et al., 1995a; Sengupta et al., 1997; Sengupta et al., 1998; Liu et al., 2004). Based on these studies, we postulated that phosphorylation of tau by more than one protein kinase and more than one combination of kinases are required for its self assembly into filaments. We therefore investigated the phosphorylation of tau by PKA, CaMKII, cdk5 and GSK-3β. Employing tau441, we first standardized the conditions for phosphorylation. We found that PKA, CaMKII and GSK-3β phosphorylated tau441 in a dose-dependent manner (Fig. 2A–E). From these findings, we selected the optimal concentration of each kinase and compared the levels of phosphorylation obtained by these kinases between tau441 and PP-2A-AD P-tau (Fig. 2E). We found that PKA, CaMKII and GSK-3β phosphorylated PP-2A-AD P-tau with different stoichiometry and the maximal phosphorylation was obtained by sequential phosphorylation of tau by PKA, CaMKII and GSK-3β. Tau441 became phosphorylated at higher stoichiometry than the PP-2A-AD P-tau. Rephosphorylation of PP-2A-AD P-tau by PKA, CaMKII plus GSK-3β resulted in a slower moving tau band as compared with rephosphorylation by GSK-3β alone on SDS-PAGE (Fig. 2D).
Fig. 2.
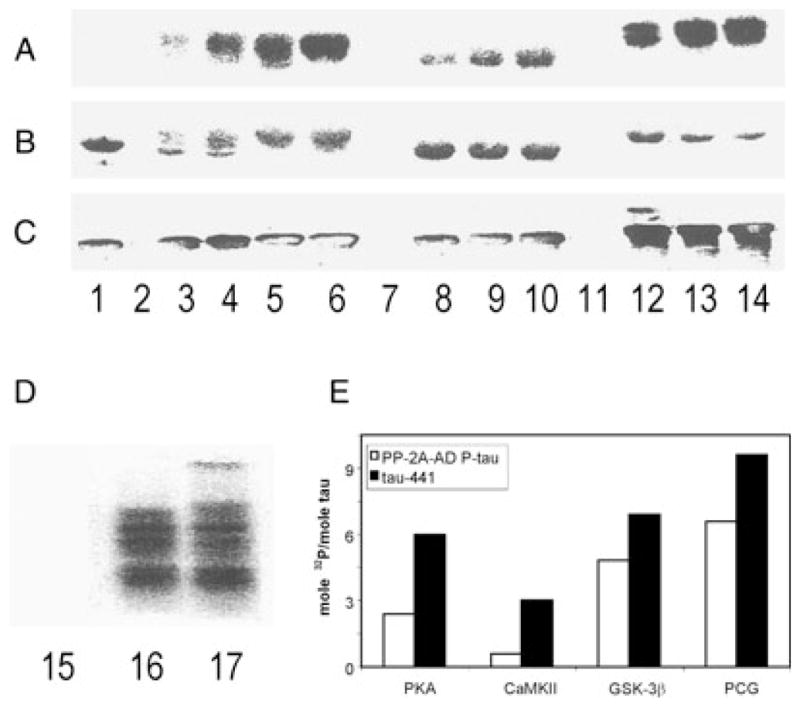
Phosphorylation of tau441 and PP-2A-AD P-tau by PKA, CaMKII, GSK-3β, and the combination of the three kinases. (A) 32P autoradiography; (B) Coomassie blue; (C) Western blots developed with tau antibody 92e to total tau; lane 1, no kinase; lanes 2, 7 and 11, with kinase but no tau; lanes 3–6, with tau and 2.5 μg/mL, 5.0 μg/mL, 10 μg/mL and 20 μg/mL of PKA; lanes 8–10, 0.5 μg/mL, 1 μg/mL, and 2 μg/mL of CaMKII, lanes 12–14, 12.5 U/mL, 25 U/mL and 50 U/mL of GSK-3β. (D) Rephosphorylation (32P autoradiography) of PP-2A-AD P-tau; lane 15, no-kinase control; lane 16, 100 U/mL GSK-3β; lane 17, 40 μg/mL, 4 μg/mL and 100 U/mL of PKA, CaMKII and GSK-3β, respectively. Phosphorylation of PP-2A-AD P-tau generated an upward shifted band. (E) Stoichiometry of the phosphorylation of tau441 by 20 μg/mL of PKA, 2 μg/mL of CaMKII, 50 U/mL of GSK-3β or all these three kinases (PCG), and phosphorylation of PP-2A-AD P-tau by double the amount of the kinases used for tau441. PKA, CaMKII and GSK-3β phosphorylated tau441 dose dependently, and phosphorylation of tau by PKA and GSK-3β led to a significant upwards mobility shift of tau in SDS-PAGE.
Employing these conditions of optimal phosphorylation, we carried out the rephosphorylation of PP-2A-AD P-tau and the phosphorylation of tau441 by PKA, CaMKII, cdk5 and GSK-3β alone, or sequentially by PKA or CaMKII and GSK-3β or cdk5, or by PKA, CaMKII and GSK-3β or cdk5 and mapped the specific sites phosphorylated by Western blots developed with site-specific phosphorylation-dependent tau antibodies. A rabbit antibody to total tau, 92e, was employed to confirm equal loading of the sample in each gel lane. We found that PKA, CaMKII and GSK-3β individually rephosphorylated several but not all the sites of AD P-tau which were dephosphorylated by PP-2A, whereas a combination of PKA, CaMKII and GSK-3β or cdk5 and cdk5 plus GSK-3β were most effective in rephosphorylation of the sites dephosphorylated by PP-2A (Fig. 3A and B; Table 2). Similarly, the most robust phosphorylation of tau441 was achieved with a combination of PKA, CaMKII and GSK-3β or cdk5 and as well as by cdk5 and GSK-3β (Fig. 3C and D; Table 3). Taken together, these data suggested that phosphorylation of tau at multiple sites, covering the amino terminal proline-rich region, the microtubule binding repeats region and as well as the carboxy terminal regions are required for its conversion into an AD-like state and that a combination of more than one tau kinase is required for its abnormal hyperphosphorylation.
Table 2.
Dephosphorylation of AD P-tau by PP-2A and rephosphorylation by different kinases
| Sites dephosphorylated | |
|---|---|
| Dephosphorylation of AD P-tau by PP-2A | |
| Phosphatase | |
| PP-2A | Thr181, Ser199, Ser202, Thr205, Thr212, Ser214, Thr217, Thr231, Ser262, Ser396, Thr403, Ser404, Ser422, Ser198/Ser199/Ser202*, Ser262/Ser356*, Ser396/Ser404*, Thr212/Ser214* |
| Rephosphorylation of AD P-tau Dephosphorylated with PP-2A (PP-2A-AD P-tau) | |
| Kinases | |
| PKA | Thr181, Ser199, Thr212, Ser214, Ser262, Thr403, Ser404, Ser422, Thr212/Ser214*, Ser262/Ser356*, Ser396/Ser404* |
| CaMKII | Thr181, Ser199, Thr212, Ser262, Thr403, Ser404, Ser262/Ser356*, Ser396/Ser404* |
| GSK-3β | Thr181, Ser199, Ser202, Thr212, Thr217, Thr231, Ser396, Thr403, Ser404, Ser422, Ser198/Ser199/Ser202*, Thr212/Ser214*, Ser396/Ser404* |
| PKA/cdk5/GSK-3β | Ser198/Ser199/Ser202*, Thr212/Ser214*, Ser262/Ser356*, Ser396/Ser404* Thr181, Ser199, Ser202, Thr212, Ser214, Thr217, Thr231, Ser262, Ser396, Thr403, Ser404, |
| cdk5 | Thr181, Ser199, Ser202, Thr205, Thr231, Ser396, Ser403 |
| cdk5/GSK-3β | Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Ser262, Ser396, Thr403, Ser404, Ser422 |
| PKA/CaMKII/cdk5 | Thr181, Ser199, Ser202, Thr205, Thr217, Thr231, Ser262, Ser396, Thr403, Ser404, Ser422 |
Table 3.
Phosphorylation sites of tau441
| Kinases | Sites phosphorylated |
|---|---|
| PKA | Thr212, Ser214, Thr217, Ser262, Ser404, Ser409, Ser413 |
| CaMKII | Ser214, Thr217, Ser262, Ser404 |
| GSK-3β | Thr181, Ser199, Ser202, Thr205, Thr212, Ser214, Thr217, Thr231, Ser396, Ser404, Ser409, Ser413, Ser422, Ser198/Ser199/Ser202*, Ser396/Ser404* |
| PKA/CaMKII/GSK-3β | Thr181, Ser199, Ser202, Thr205, Thr212, Ser214, Thr217, Thr231, Ser262, Ser396, Thr403, Ser404, Ser409, Ser413, Ser422, Ser198/Ser199/Ser202*, Ser396/Ser404* |
| cdk5 | Ser199, Ser202, Thr205, Thr212, Thr231, Ser404 |
| PKA/cdk5 | Thr181, Ser199, Ser202, Thr205, Thr212, Thr231, Ser235, Ser262, Ser396, Ser404 |
| CaMKII/cdk5 | Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Ser235, Ser262, Ser396, Thr403, Ser404 |
| cdk5/GSK-3β | Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Ser235, Ser396, Thr403, Ser404 |
| PKA/CaMKII/cdk5 | Thr181, Ser199, Ser202, Thr205, Thr212, Thr231, Ser235, Ser262, Ser396, Thr403 Ser404 |
Formation of tangles of PHF/SF by abnormal hyperphosphorylation of tau by various combinations of protein kinases
To determine the involvement of PP-2A and to identify the protein kinases and the sites involved in the assembly of tau into tangles of PHF/SF, we investigated the effect of rephosphorylation of PP-2A-AD P-tau on its aggregation. We found that rephosphorylation of PP-2A-AD P-tau by PKA, CaMKII and GSK-3β or cdk5 and as well as by cdk5 plus GSK-3β resulted in the formation of tangles of PHF (Fig. 4). PHF formed both by AD P-tau and as well as PP-2A-AD P-tau rephosphorylated by the combination of the three kinases were ~22 nm wide with every ~80 nm narrowing to ~10 nm and appeared the same as seen in AD brain (Fig. 4A, E and F). Rephosphorylation of PP-2A-AD P-tau sequentially by CaMKII and GSK-3β also resulted in tangles of filaments but these filaments were straight and ~2.4 nm and ~10 nm in diameter (Fig. 4C and D). Thin filaments with diameter ~2.4 nm to ~8.3 nm (mostly ~6 nm) and tight sheets were seen in the PP-2A-AD P-tau samples which were rephosphorylated by PKA plus GSK-3β, PKA plus cdk5 and CaMKII plus cdk5, and only amorphous structures were seen by PKA plus CaMKII, or by PKA, CaMKII, GSK-3β and cdk5 individually (figures not shown).
Phosphorylation of tau441 yielded similar results as the rephosphorylation of PP-2A-AD P-tau, except that the filaments formed from the former were mostly SF, which were admixed with PHF (Fig. 5). The most robust assembly of tau441 into filaments was observed when the protein was phosphorylated with PKA, CaMKII and GSK-3β or cdk5 or by cdk5 plus GSK-3β (Fig. 5A–C and E). Incubation of tau441 in the phosphorylation buffer without any kinase resulted only in short filaments admixed with amorphous structures.
Fig. 5.
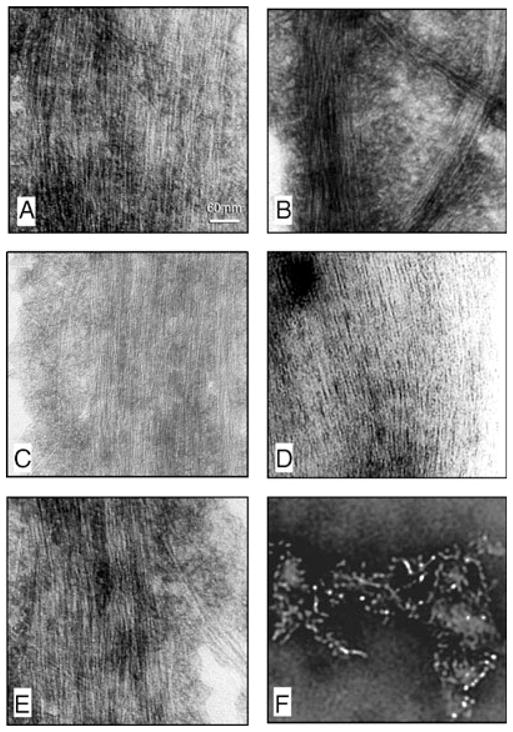
Negative stain electron microscopy of products of self assembly of tau441 before and after phosphorylation with different combination of kinases. Phosphorylation of tau441 with PKA, CaMKII and GSK-3β (panel A), PKA, CaMKII and cdk5 (panels B and C), CAMKII plus GSK-3β (panel D) and cdk5 plus GSK-3β (panel E) induced formation of straight filaments with mostly 2.4 nm and occasionally 10 nm (panel C) diameter filaments. Incubation of tau441 without kinase showed short filaments (panel F).
Congo-red birefringence of in vitro produced tangles of PHF
A histochemical characteristic of AD PHF/tangles is the Congo-red birefringence, which occurs only when this dye binds to β-pleated sheets. To confirm the nature of the polymerized structures, we employed Congo-red staining birefringence light microscopy technique. Numerous structures with apple green birefringence were observed when PP-2A-AD P-tau was rephosphorylated by PKA, CaMKII and GSK-3β or cdk5, or by cdk5 plus GSK-3β (Fig. 6A–C). On the other hand, rephosphorylation of PP-2A-AD P-tau by these kinases individually or by PKA or CaMKII plus GSK-3β or cdk5 or cdk5 plus CaMKII did not result in Congophilic products. Phosphorylation of tau441 with the combination of only PKA, CaMKII and GSK-3β resulted in Congo-red birefringence producing tangles of filaments (Fig. 6D); none of the other combinations of the kinases that were used above for PP-2A-AD P-tau or phosphorylation with individual kinases produced Congo-red birefringent products.
Fig. 6.
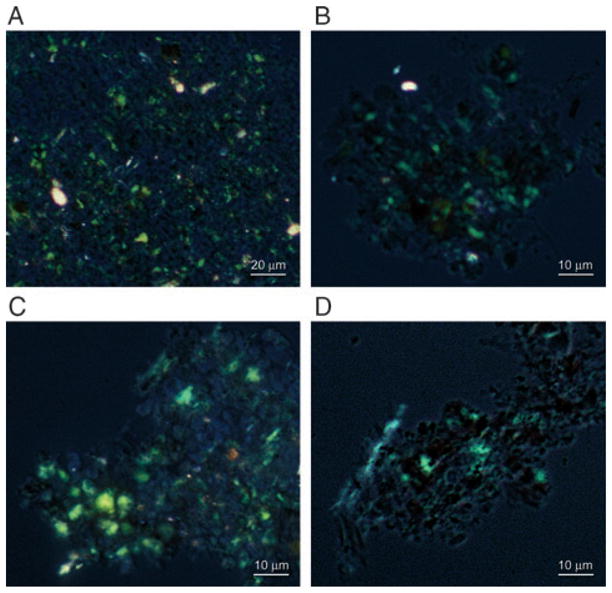
Congo-red birefringence of PP-2A-AD P-tau and tau441 after self assembly into filaments induced by phosphorylation with different kinase combinations. PP-2A-AD P-tau (panels A–C) and tau441 (panel D) were phosphorylated by PKA, CaMKII and GSK-3β (panels A and D), PKA, CaMKII and cdk5 (panel B), or by cdk5 plus GSK-3β (panel C). The products of self assembly were stained by Congo-red and birefringence was viewed and photographed by a Nikon Biophot microscope.
Effect of rephosphorylation of PP-2A-AD-P-tau on its binding to microtubules and on microtubule assembly
The biological activity of tau is known to be regulated by its degree of phosphorylation (Lindwall & Cole, 1984; Alonso et al., 1994). Unlike normal tau, which binds to microtubules and promotes assembly and stabilizes microtubules (Weingarten et al., 1975), the AD P-tau has been previously shown to sequester normal tau, MAP1 and MAP2 and inhibits assembly and depolymerizes microtubules (Alonso et al., 1994; Alonso et al., 1997). To learn whether the combination of protein kinases that induced self assembly of tau into tangles of filaments induced AD P-tau-like activity, we investigated the effect of phosphorylation of PP-2A-AD P-tau and tau441 on binding to microtubules and on microtubule assembly. We found that the dephosphorylation of AD P-tau by PP-2A increased its binding to microtubules from 27.5% to 82.5%, which was similar to the binding of untreated tau441 to microtubules (80%) (Table 4). Rephosphorylation of PP-2A-AD P-tau by different combinations of protein kinases inhibited its binding to microtubules to different degrees and the maximal inhibition was observed in the case of the sequential phosphorylation of tau by PKA, CaMKII and GSK-3β. Interestingly, when a mixture of these three protein kinases was employed for the rephosphorylation, the binding to microtubules was inhibited to a lesser degree and at a level similar to rephosphorylation by GSK-3β alone. In principle, similar effects were observed when tau441 was phosphorylated by the different kinase combinations. A notable exception, however, was a minimal effect on the binding to microtubules of PP-2A-AD P-tau rephosphorylated with PKA. The PKA-phosphorylated tau441 showed a decrease in the microtubule binding from a maximal of 80% for the untreated to 53% for the treated protein.
Table 4.
Effect of rephosphorylation on the binding of PP-2A-AD P-tau on taxol-stabilized microtubules*
| Treatment | Bound (%) | |
|---|---|---|
| AD P-tau | Tau441† | |
| Untreated | 27.5 ± 4.5 | 80.0 ± 5.3 |
| PP-2A | 82.5 ± 7.4 | NA |
| PP-2A/PKA | 80.0 ± 5.5 | 53.0 ± 3.5† |
| PP-2A/CaMKII | 63.5 ± 8.2 | 32.6 ± 2.1† |
| PP-2A/GSK-3β | 43.0 ± 5.8 | 45.5 ± 3.2† |
| PP-2A/PKA/CaMKII/GSK-3β | 30.0 ± 5.3 | 20.0 ± 3.8† |
| PP-2A/(PKA + CaMKII + GSK-3β)‡ | 42.6 ± 6.5 | 35.5 ± 4.7† |
The in vitro microtubule assembly measured by turbidimetric changes at 350 nm revealed that, unlike tau441, the AD P-tau had no detectable microtubule assembly promoting activity (Fig. 7A, curves 1 and 4). The dephosphorylation of AD P-tau by PP-2A completely restored its ability to promote microtubule assembly (Fig. 7A, curve 2) and sequential rephosphorylation by PKA, CaMKII and GSK-3β practically completely inhibited this activity (Fig. 7A, curve 3). For microtubule assembly studies only, disaggregated hyperphosphorylated tau was employed as the tau polymerized in fibrils is inert, as shown by us previously (Alonso et al., 2006). Similarly, phosphorylation of tau441 by a sequential phosphorylation by PKA, CaMKII and GSK-3β completely inhibited its microtubule assembly promoting activity (Fig. 7B, curve 5). Phosphorylation of tau441 by GSK-3β, PKA and CaMKII individually inhibited the microtubule assembly promoting activity only partially and the order of this inhibitory activity was CaMKII > PKA > GSK-3β (Fig. 7B, curves 2–4).
Fig. 7.

In vitro assembly of microtubules measured by light scattering at 350 nm at 32 °C. (A) Microtubule assembly promoting activity of PP-2A-AD P-tau before and after rephosphorylation. AD P-tau did not promote microtubule assembly (curve 4); dephosphorylation of AD P-tau with PP-2A restored its biological activity (curve 2); rephosphorylation of PP-2A-AD P-tau by PKA, CaMKII and GSK-3β completely suppressed its ability to promote microtubule assembly (curve 3). Curve 1 shows the assembly activity of tau441 (10 μg/mL) and curve 5 of tubulin 3 mg/mL used as positive and negative controls, respectively. (B) Comparison of the microtubule assembly activity of tau441 after phosphorylation with various kinases. Phosphorylation of tau441 (30 μg/mL) with GSK-3β (curve 7), PKA (curve 8) or CaMKII (curve 9) inhibited microtubule assembly in an increasing order; the most potent inhibition was observed on phosphorylation of tau with PKA, CaMKII and GSK-3β (curve 10), which reached the same level as untreated AD P-tau (curve 11). Curve 6 is the assembly with unphosphorylated tau441 and curve 12 is with tubulin alone without addition of any tau. (C) Disruption of tau441 assembled microtubules with PP-2A-AD P-tau before and after rephosphorylation by PKA, CaMKII and GSK-3β. Microtubule assembly was initiated with tubulin (3 mg/mL) and tau441 (20 μg/mL). After 5 min (↓) of the assembly reaction, the addition of assembly buffer (100 mM MES, 1 mM EGTA, 1 mM MgCl2 and 1 mM GTP, curve 13 or PP-2A-AD P-tau in the same buffer (curve 14) did not have any significant effect on microtubule assembly. On the other hand, addition of untreated AD P-tau (curve 15) or PP-2A-AD P-tau rephosphorylated with PKA, CaMKII and GSK-3β (curve 16) disrupted the tau441 assembled microtubules. Curve 17 is a tubulin control. Rest of the details are the same as in panels A and B.
The abnormally hyperphosphorylated taus were found not only to inhibit microtubule assembly but also depolymerize the preformed microtubules (Fig. 7C). Like AD P-tau, the PP-2A-AD P-tau rephosphorylated sequentially with PKA, CaMKII and GSK-3β depolymerized microtubules assembled from tubulin and tau441 (Fig. 7C, curves 3 and 4); PP-2A-AD P-tau used under identical conditions as a control did not disrupt microtubules (Fig. 7C, curve 2).
Discussion
In 1986, microtubule associated protein tau was discovered to be abnormally hyperphosphorylated in AD brain and to be the major protein subunit of PHF/tangles (Grundke-Iqbal et al., 1986a; Grundke-Iqbal et al., 1986b; Iqbal et al., 1986). Since this discovery, the hyperphosphorylation of tau has been increasingly recognized as a major denominator underlying the pathological aggregation and dysfunction of tau in the brain of patients with AD and with other tauopathies (Tolnay & Probst, 1999; Iqbal et al., 2005), and a dysregulation of the phosphorylation system is believed to be involved in this tau pathology (Grundke-Iqbal et al., 1986b; Iqbal et al., 1986). Therefore, the identification of protein kinases involved in the abnormal hyperphosphorylation and self assembly of tau into PHF/tangles has been a major goal of research on the pathogenesis of AD and other tauopathies. The present study shows that: (i) a combination of PKA and CaMKII with GSK-3β or cdk5, or cdk5 plus GSK-3β, can produce Alzheimer neurofibrillary degeneration and (ii) PP-2A can inhibit this pathology by dephosphorylating the critical sites required for tau’s self assembly into PHF/tangles, and for the inhibition of its binding to microtubules and promoting assembly and stabilizing microtubules.
AD P-tau was phosphorylated at multiple sites and PP-2A dephosphorylated, to varying degrees, practically all the sites studied. Unlike the untreated AD P-tau, the PP-2A-AD P-tau bound to taxol-stabilized microtubules, stimulated assembly of tubulin into microtubules and could not self-assemble into filaments. Rephosphorylation of PP-2A-AD P-tau individually with PKA, CaMKII, GSK-3β or cdk5 failed to rephosphorylate tau at all of the sites dephosphorylated with PP-2A and could not convert it back into an AD P-tau-like state in terms of self assembly and microtubule binding and assembly inhibitory activities. These findings suggested that phosphorylation of tau by more than one kinase was required to convert it into the pathological protein. A sequential rephosphorylation of PP-2A-AD P-tau by PKA, CaMKII and GSK-3β or cdk5, or by cdk5 and GSK-3β, resulted in the phosphorylation of tau at most of the sites dephosphorylated by PP-2A, and converted it back into the pathological protein which self-assembled into PHF/tangles, did not bind to taxol-stabilized microtubules, and which, as disaggregated protein, inhibited microtubule assembly and depolymerized preassembled microtubules.
Phosphorylation of recombinant tau441 by different combination of kinases yielded similar results as those with the rephosphorylation of PP-2A-AD P-tau, except the filaments formed employing the former were mostly SF, while those using the latter protein were mostly PHF. These differences could be either due to the fact that, like in situ, the AD P-tau is a mixture of all six tau isoforms or it has other post-translational modifications such as _N_-glycosylation (Wang et al., 1996a) in addition to hyperphosphorylation, or both.
The PHF formed from AD P-tau and from rephosphorylated PP-2A-AD P-tau appeared the same as those seen in AD brain and, like the in situ tangles, were congophilic and produced green birefringence under polarized light. A comparison of the sites phosphorylated by various combination of protein kinases, which resulted in the formation of PHF/tangles and the microtubule inhibitory activities, and which did not result in those characters, revealed that the phosphorylation of Thr231 and or Ser262 along with Thr181, Ser199, Ser202, Thr212, Ser396, Thr403 and Ser404 appeared to be required for the assembly of tau into filaments.
GSK-3β alone phosphorylated tau at almost all the sites that were phosphorylated by the sequential phosphorylation with PKA, CaMKII and GSK-3β or cdk5 except at Ser262, and did not induce self assembly of either PP-2A-AD P-tau or tau441 into filaments. Similarly, PKA alone could phosphorylate tau at multiple sites except Ser202 and Thr231 and was not sufficient to induce self assembly of tau. All the kinase combinations, i.e. PKA, CaMKII and GSK-3β or cdk5, cdk5 and GSK-3β, PKA or CaMKII and cdk5 or GSK-3β, which induced self assembly of tau into filaments, had resulted in the phosphorylation of tau at Thr231 and Ser262 along with Thr181, Ser199, Ser202, Thr212, Ser396, Thr403 and Ser404. Thus, it appears that phosphorylation of tau at Thr231 and Ser262 is critically involved in its self-assembly into PHF.
The abnormal hyperphosphorylation of tau appears to be the most deleterious step in neurofibrillary degeneration. Both in situ (Iqbal et al., 1986; Cash et al., 2003) and in vitro studies (Alonso et al., 2006) have demonstrated that the cytosolic abnormally hyperphosphorylated tau and not its aggregation into PHF/SF are apparently involved in the breakdown of the neuronal microtubule network. Thus, identification of the kinases and phosphatases and of critical sites, phosphorylation of which converts normal functional tau into an inhibitory protein, is crucial for the development of drugs to inhibit this lesion in AD and related tauopathies.
In conclusion, the present study shows for the first time the protein kinases and their combinations that can abnormally hyperphosphorylate tau and induce its self assembly into tangles of PHF similar to those seen in AD. This study also demonstrates that PP-2A alone can dephosphorylate practically all the tau sites, phosphorylation of which appears to be required for its self assembly. The PHF formed appeared indistinguishable from those seen in AD brain and, like the in situ tangles, the in vitro generated tangles were congophilic, which exhibited green birefringence under polarized light. Phosphorylation of tau by sequential treatment with PKA, CaMKII and GSK-3β or cdk5 or by cdk5 and GSK-3β, converts normal tau into AD P-tau-like protein, which self-assembles into PHF/tangles and inhibits microtubule assembly. PP-2A can dephosphorylate AD P-tau at the sites required for its self assembly and microtubule inhibitory activity. The phosphorylation of tau at multiple sites appears to be required to convert it into the pathological protein, i.e. AD P-tau-like state. These sites include four clusters amino terminal to the microtubule binding domains, i.e. Thr181, Ser199-Thr205, Thr212-Thr217 and Thr231-Ser235 regions, one cluster in the microtubule binding repeat region, i.e. Ser262 and one cluster C-terminal to the repeat region, i.e. Ser396–Ser422. Phosphorylation of Thr231 and Ser262 appears to be necessary for the conversion of normal tau to pathological tau. These findings suggest more than one therapeutic strategy is likely to be effective for the inhibition of Alzheimer neurofibrillary degeneration. These strategies include the restoration of the PP-2A activity that is known to be compromised in AD brain and inhibition of activities of either GSK-3β and cdk5, or of PKA or CaMKII and GSK-3β or cdk5.
Acknowledgments
We thank Dr Ezzat El-Akkad for providing recombinant human brain tau, Ms Tanweer Zaidi for AD P-tau, Dr M. Goedert for tau plasmids, and Drs L.I. Binder, P. Davies and D. Schenk for antibodies Tau-1, PHF-1 and 12E8, respectively. We are very grateful to Dr Robert Freedland for his help in the preparation of all figures, and Ms Janet Murphy for secretarial assistance in the preparation of the manuscript. AD autopsied brain specimens used for the isolation of AD P-tau were provided by the Brain Tissue Resource Center (Public Health Service Grant MN/NS31862), McLean Hospital, Belmont, MA. These studies were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities and by National Institutes of Health Grant AG019158, and a grant from the Alzheimer’s Association, Chicago, and the grants from the National Natural Science Foundation of China (30430270) and from the Science and Technology Committee of China (2006CB500703).
Abbreviations
AD
Alzheimer’s disease
AD P-tau
Alzheimer abnormally hyperphosphorylated tau
CaMKII
calcium, calmodulin-dependent protein kinase II
PKA
cyclic AMP-dependent protein kinase
cdk5
cyclin-dependent protein kinase 5
GSK-3β
glycogen synthase kinase-3β
MAP
microtubule associated protein
PHF
paired helical filaments
PP-2A
phosphoseryl/phosphothreonyl protein phosphatase-2A
SF
straight filaments
References
- Alonso A, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci USA. 2006;23:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000;485:87–93. doi: 10.1016/s0014-5793(00)02203-1. [DOI] [PubMed] [Google Scholar]
- Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K. Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser262/356. FEBS Lett. 2001;490:15–22. doi: 10.1016/s0014-5793(01)02127-5. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Alemany S, Hemmings BA, Resink TJ, Stralfors P, Tung HY. Protein phosphatase-1 and protein phosphatase-2A from rabbit skeletal muscle. Meth Enzymol. 1988;159:390–408. doi: 10.1016/0076-6879(88)59039-0. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M. Differential expression of active, phosphorylation-dependent MAP kinases, MAPK/ERK, SAPK/JNK and p38, and specific transcription factor substrates following quinolinic acid excitotoxicity in the rat. Brain Res Mol Brain Res. 2001;94:48–58. doi: 10.1016/s0169-328x(01)00198-x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem. 2000;275:5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986a;261:6084–6089. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986b;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Vorbrodt AW, Iqbal K, Tung YC, Wang GP, Wisniewski HM. Microtubule-associated polypeptides tau are altered in Alzheimer paired helical filaments. Brain Res. 1988;464:43–52. doi: 10.1016/0169-328x(88)90017-4. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, Wisniewski HM, Alafuzoff I, Winblad B. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–426. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Zaidi T, Thompson CH, Merz PA, Wisniewski HM. Alzheimer paired helical filaments: bulk isolation, solubility, and protein composition. Acta Neuropathol (Berl) 1984;62:167–177. doi: 10.1007/BF00691849. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Omori A, Takamatsu M, Sato K, Arioka M, Uchida T, Imahori K. Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci Lett. 1992;148:202–206. doi: 10.1016/0304-3940(92)90839-y. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kins S, Kurosinski P, Nitsch RM, Gotz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol. 2003;163:833–843. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–269. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5305. [PubMed] [Google Scholar]
- Liu SJ, Zhang JY, Li HL, Fang ZY, Wang Q, Deng HM, Gong CX, Grundke-Iqbal I, Iqbal K, Wang JZ. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem. 2004;279:50078–50088. doi: 10.1074/jbc.M406109200. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Gong CX, An WL, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70, S6, similar to that in Alzheimer’s disease. Am J Pathol. 2003;163:845–858. doi: 10.1016/S0002-9440(10)63445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 1998;797:267–277. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Rahman A, Grundke-Iqbal I, Iqbal K. Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem Res. 2005;30:277–287. doi: 10.1007/s11064-005-2483-9. [DOI] [PubMed] [Google Scholar]
- Rahman A, Grundke-Iqbal I, Iqbal K. PP2B isolated from human brain preferentially dephosphorylates Ser-262 and Ser-396 of the Alzheimer disease abnormally hyperphosphorylated tau. J Neural Transm. 2006;113:219–230. doi: 10.1007/s00702-005-0313-5. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol Cell Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J Neurochem. 1995a;64:1420–1423. doi: 10.1046/j.1471-4159.1995.64031420.x. [DOI] [PubMed] [Google Scholar]
- Singh TJ, Zaidi T, Grundke-Iqbal I, Iqbal K. Modulation of GSK-3-catalyzed phosphorylation of microtubule-associated protein tau by non-proline-dependent protein kinases. FEBS Lett. 1995b;358:4–8. doi: 10.1016/0014-5793(94)01383-c. [DOI] [PubMed] [Google Scholar]
- Sobue K, Agarwal-Mawal A, Li W, Sun W, Miura Y, Paudel HK. Interaction of neuronal Cdc2-like protein kinase with microtubule-associated protein tau. J Biol Chem. 2000;275:16673–16680. doi: 10.1074/jbc.M000784200. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, 3rd, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. 2003;118:1175–1182. doi: 10.1016/s0306-4522(02)00697-8. [DOI] [PubMed] [Google Scholar]
- Tian Q, Lin ZQ, Wang XC, Chen J, Wang Q, Gong CX, Wang JZ. Injection of okadaic acid into the meynert nucleus basalis of rat brain induces decreased acetylcholine level and spatial memory deficit. Neuroscience. 2004;126:277–284. doi: 10.1016/j.neuroscience.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Tolnay M, Probst A. REVIEW: tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol. 1999;25:171–187. doi: 10.1046/j.1365-2990.1999.00182.x. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol. 2001;168:402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nature Med. 1996a;2:871–875. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A-2B and -1. Brain Res Mol Brain Res. 1996b;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Merz PA, Iqbal K. Ultrastructure of paired helical filaments of Alzheimer’s neurofibrillary tangle. J Neuropathol Exp Neurol. 1984;43:643–656. doi: 10.1097/00005072-198411000-00008. [DOI] [PubMed] [Google Scholar]
- Xiao J, Perry G, Troncoso J, Monteiro MJ. Alpha-calcium-calmodulin-dependent kinase II is associated with paired helical filaments of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:954–963. doi: 10.1097/00005072-199609000-00002. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol (Berl) 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]