Conformational transformation and selection of synthetic prion strains (original) (raw)
. Author manuscript; available in PMC: 2012 Oct 28.
Published in final edited form as: J Mol Biol. 2011 Aug 4;413(3):527–542. doi: 10.1016/j.jmb.2011.07.021
Abstract
The prion protein (PrP) is capable of folding into multiple self-replicating prion strains that produce phenotypically distinct neurological disorders. Although prion strains often breed true upon passage, they can also transform or “mutate” despite being devoid of nucleic acids. To dissect the mechanism of prion strain transformation, we studied the physicochemical evolution of a synthetic prion strain (MoSP1) after repeated passage in mice and cultured cells. We show that MoSP1 gradually adopted shorter incubation times and lower conformational stabilities. These changes were accompanied by a structural transformation as indicated by a shift in the molecular mass of the protease-resistant core of MoSP1 from approximately 19 kDa [MoSP1(2)] to 21 kDa [MoSP1(1)]. We show that MoSP1(1) and MoSP1(2) can breed with fidelity when cloned in cells, but when present as a mixture, MoSP1(1) preferentially proliferated, leading to the disappearance of MoSP1(2). In culture, the rate of this transformation process can be influenced by the nature of the culture media and the presence of polyamidoamines. Our findings demonstrate that prions can exist as a conformationally diverse population of strains, each capable of replicating with high fidelity. Rare conformational conversion, followed by competitive selection among the resulting pool of conformers, provides a mechanism for the adaptation of the prion population to their host environment.
INTRODUCTION
In prion diseases, including Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle, an alternatively folded conformer of the prion protein (PrP) propagates by catalyzing a posttranslational structural transition, utilizing endogenous cellular PrP (PrPC) as a substrate 1; 2. This transition converts the α-helix–rich PrPC into pathogenic, aggregation-prone, β-sheet–rich conformers, termed PrPSc 3; 4; 5. This conversion can occur spontaneously or be induced by autosomal dominant mutations in the PrP gene (PRNP) 6; 7. Alternatively, prion disease can result from exposure to exogenous PrPSc 5.
The pioneering work of Pattison, Dickinson and colleagues indicated that the sheep scrapie agent can propagate as multiple phenotypically distinct strains 8; 9; 10. More recent work has identified the existence of strains within prions derived from other species 11; 12. Prion strains can be distinguished based on their pathogenic properties (incubation times, neuropathology, ability to infect cell lines) and biochemical characteristics (electrophoretic mobility, glycoform ratio, conformational stability, exposure of antibody epitopes, affinity towards conjugated polymers) 13; 14; 15; 16. Evidence suggests that these strain-specific properties are enciphered in the conformation of PrPSc 11; 14; 17. The unique biological and biochemical signatures of strains can propagate faithfully in animals, cell culture and in-vitro amplification experiments 5; 12; 18; 19; 20.
Although natural prion strains can breed true upon serial passage, several observations suggest that they are capable of altering their molecular properties in order to adapt to new hosts and environments, in a process referred to as “strain adaptation”. This phenomenon is frequently observed when prions are transmitted to a different host species. The interspecies transmission of prions is typically an inefficient process characterized by long incubation times and low infection rates 21; 22; 23; 24. Evidence suggests that this species barrier is a result of incompatibilities between the conformations of the infecting prion strain and host PrPC, due in part to differences in the amino acid sequences 23; 25. However, upon repeated passage, the prion conformation changes in its new host and the incubation period gradually shortens 23.
Prion adaptation has also been observed in the form of dynamic interconversions of strains derived from the same organism. Passaging of biologicially cloned transmissible mink encephalopathy (TME) prions into Syrian golden hamsters can result in two phenotypically distinct strains: a long-incubation-period strain named drowsy (DY) and a short-incubation-period strain named hyper (HY) 26. Upon initial infection of hamsters with biologically cloned TME prions, the DY strain predominated. However, continuous serial passage of DY prions in hamsters resulted in the gradual selection of the HY strain 20; 27; 28.
A similar phenomenon was observed in mice infected with variant CJD strains. Transgenic (Tg) mice expressing chimeric human/mouse PrP inoculated with vCJD prions can harbor two distinct strains of prions 29. In mice that expressed a mixture of the two strains, the faster-replicating strain became dominant on subsequent passage30. More recent experiments have shown that prions can also evolve in response to selective environmental pressures. The presence of the antiprion drug quinacrine 31 or the glycoside hydrolase inhibitor swainsonine 32 result in the selection of prion conformations that are resistant to the respective drug’s actions in cell-culture models. Although these observations suggest that prion strains are dynamic pathogens capable of adaptation to novel environments, the mechanism of prion strain evolution remains unknown and subject to speculation 33.
Prion strain diversity has recently been augmented by the creation of infectious synthetic prions formed exclusively from bacterially derived recombinant PrP. The first synthetic prion strain (termed MoSP1) was created by refolding recombinant, truncated mouse PrP(Δ23–88) into β-sheet–rich amyloid fibrils followed by intracerebral inoculation into Tg mice expressing the same truncated PrP sequence (Tg9949) 34. Although aged Tg9949 mice are prone to neurologic disease, they do not spontaneously accumulate infectious prions 35. However, when inoculated with MoSP1 fibrils, Tg9949 mice amassed protease-resistant infectious prions (rPrPSc) 34. Passage of MoSP1 in wild-type (wt) inbred FVB or Tg mice overexpressing full-length PrP (Tg4053) led to diverse incubation times, suggesting that MoSP1 may be comprised of multiple strains 36; 37. Further inoculations of wt and Tg mice with various PrP amyloid preparations led to the generation of additional, distinct synthetic prion strains 35; 38; 39.
Compared to prions that are endemic in natural populations (such as scrapie), newly created synthetic prion strains have not been exposed to extended periods of natural selection. We therefore reasoned that synthetic strains reflect unoptimized prion states that may be prone to conformational transformation and selection upon repeated passage. We serially passaged MoSP1 in mice and cultured cell lines, and monitored changes in its biochemical and biological properties. Our data indicate that MoSP1 gradually adopted shorter incubation periods and a defined set of biochemical and neuropathological properties. We demonstrate that the observed MoSP1 transformation resulted from rare conversion events followed by competition and selection among a heterogeneous pool of prions.
RESULTS
Generation and serial passage of MoSP1 in mice
Previously, we demonstrated that inoculation of β-sheet–rich amyloid fibrils prepared from truncated (residues 89–230), recombinant mouse PrP (recMoPrP) into Tg mice expressing a similarly truncated construct (Tg9949 mice 40) led to the generation of synthetic prions 34. Although Tg9949 mice inoculated with phosphate-buffered saline (PBS) or bovine serum albumin (BSA) developed late-onset ataxia after 600 days, neither infectious prions nor related neuropathology were detectable in their brains 38. Conversely, Tg9949 mice inoculated with PrP amyloid fibrils developed neurologic disease between 380 and 608 days postinoculation 34, which was characterized by the presence of vacuoles and astrocytic gliosis, and biochemical analysis indicated the presence of proteinase K (PK)–resistant PrPSc 34. The resulting synthetic prion isolate was termed MoSP1.
Brain homogenates from MoSP1-infected mice were serially passaged in Tg9949, Tg4053, and wt FVB mice. After each passage, extracts from multiple infected brains were used as inocula for additional rounds of infections (Tables S1–S3). Upon serial passaging, the incubation period for MoSP1 decreased in a stochastic fashion in all three mouse models (Fig. 1A, Tables S1–S3; 37).
Fig. 1.

Propagation of MoSP1 in mice. (A) MoSP1 amyloid fibers, derived from purified recMoPrP, were inoculated into Tg9949 mice (black, 1° infection), then passaged in Tg9949 (black, 2° passage) and FVB (gray, 2° passage) mice. Codes (e.g., K4985) indicate individual mice that were subsequently analyzed. The incubation period for each inoculated mouse, in days postinoculation (dpi), is indicated in parentheses. (B) Western blot analysis of a select group of MoSP1-infected Tg9949 (top) and FVB (bottom) mice. Brain homogenates were subjected to immunoblotting after limited digestion with proteinase K (PK). Blots were probed with D18 or 12B2 Abs as indicated. The number in parentheses indicates the passage number. Red and blue arrows denote the unglycosylated, protease-resistant bands of PrPSc migrating to 19 kDa [MoSP1(2)] and 21 kDa [MoSP1(1)], respectively. Molecular masses of protein standards are indicated in kilodaltons. (C) As an example of a typical conformational-stability assay, Western blot of PK-digested N70149 brain homogenate after incubation with increasing concentrations of GdnHCl (left) and the corresponding densitometry analysis (right) of MoSP1(1) (blue) and MoSP1(2) (red) bands are shown. (D) Conformational stabilities of passaged MoSP1. GdnHCl1/2 values were measured for each brain homogenate as shown in (C). Stabilities for MoSP1(1) and MoSP1(2) bands are plotted in blue and red, respectively. The stability of RML is plotted for comparison. (E) Quantified changes in the incubation period, banding pattern, and conformational stability of MoSP1 passaged in Tg9949 mice. Circles represent MoSP1(2) (red) and MoSP1(1) (blue) strains for the indicated mouse and passage. In mice showing a mixture of MoSP1(1) and MoSP1(2), solid lines connect the circles corresponding to MoSP1(1) and MoSP1(2), with the areas of the circles representing the relative ratios of the bands, as measured by densitometry.
Biochemical characterization of passaged MoSP1
Brain extracts from selected MoSP1-infected mice (Fig. 1A) were digested with PK and protease-resistant PrPSc bands were detected on Western blots using the D18 antibody41 (Fig. 1B). The epitope of D18 lies between residues 132 and 157 of mouse PrP. Tg9949 mice that succumbed to disease after primary infection with recombinant MoSP1 amyloid fibers had PrPSc banding patterns that were distinct from those of mouse-passaged, Rocky Mountain Laboratory (RML) scrapie strain in Western blots. In three Tg9949 mice (K4974, K4985 and K4973) with incubation periods of >500 days after infection with MoSP1, the unglycosylated PrPSc bands had apparent molecular weights of 19 kDa (Fig. 1B, red arrow). In comparison, Tg9949 mice infected with RML prions had an unglycosylated PrPSc band of 21 kDa (Fig. 1B, blue arrow). In one Tg9949 mouse (K4977) that was infected with recMoPrP(89–230) amyloid and developed signs of neurologic dysfunction after 380 days, PrPSc bands of both molecular weights were observed. These results contend that MoSP1-infected mice can accumulate at least two biochemically distinct prion isoforms. Analogous variations in PK-resistant PrPSc banding patterns have been observed in CJD cases 42. “Type 1” prions, found in most sporadic (s) CJD patients homozygous for methionine at PrP residue 129, have an unglycosylated PrPSc band of 21 kDa, whereas “type 2” prions, commonly found in variant (v) CJD patients, have an unglycosylated PrPSc band of 19 kDa. We refer to the MoSP1 isoform with the 21-kDa, unglycosylated protease-resistant fragment as MoSP1(1) and the MoSP1 isoform with the 19-kDa, unglycosylated protease-resistant fragment as MoSP1(2).
Serial passage of MoSP1 in Tg9949 and FVB mice resulted in the accumulation of MoSP1(1) or a mixture of MoSP1(1) and MoSP1(2). Mice with shorter incubations periods (e.g., M6050, M72698 and O7303) accumulated MoSP1(1) alone whereas mice with longer incubation periods had a mixture of the two strains (Fig. 1B). The 12B2 antibody, raised against the N-terminal domain of PrP (residues 86 to 104) 43 can detect the protease-resistant core of MoSP1(1) but not of MoSP1(2) (Fig. 1B). The result supports the proposition that MoSP1(1) and MoSP1(2) are conformationally distinct strains.
We next measured the conformational stability of passaged MoSP1 strains by determining the GdnHCl concentrations required to expose half of the PrPSc population to limited proteolysis (GdnHCl1/2) (Fig. 1C, D). In general, MoSP1(1) had lower conformational stabilities compared to MoSP1(2). GdnHCl1/2 values ranged from 1. 8 M to 3. 2 M for MoSP1(1), and from 3. 8 M to 5 M for MoSP1(2). In passages resulting in a mixture of MoSP1(1) and MoSP1(2), differences between the stabilities of two coexisting strains were detectable in the same brain extract and could be separately measured by analyzing the disappearance of the corresponding 19-kDa or 21-kDa band (e. g., Fig. 1C, sample N70149).
Figure 1E summarizes the quantitative changes in the incubation period, banding patterns, and conformational stability of MoSP1 upon three passages in Tg9949 mice. Together, the data indicate that serial passage of MoSP1 in mice results in: (i) shortening of the incubation period; (ii) accumulation of MoSP1(1); (iii) depletion of MoSP1(2); and (iv) a decrease in the conformational stability of both strains.
Infection of a neuroblastoma (N2a) cell line with MoSP1
We next attempted to recapitulate the adaptation of MoSP1 that occurred in animals in a cell-culture system. N2a cells were infected with brain extracts from four MoSP1-infected FVB mice (N70116, N70118, N70149 and M72698). Protease-resistant PrPSc was detectable after 6 passages following infection by all four extracts (Fig. 2A). Cells infected with the M72698 extract retained the MoSP1(1) banding pattern (Fig. 1B and 2A) and were detectable by the 12B2 antibody. Infection with extracts N70116, N70118, and N70149 resulted in the cellular accumulation of MoSP1(2) alone, which was undetectable by 12B2. The distinct banding patterns of the infected cultures were retained after two passages (passages 6–8 postinfection). Interestingly, cells infected with M72698 accumulated much higher levels of PrPSc compared to those infected with N70116, N70118, or N70149 (Fig. 2A). The greater PrPSc accumulation may reflect a faster rate of formation for MoSP1(1) compared to MoSP1(2).
Fig. 2.
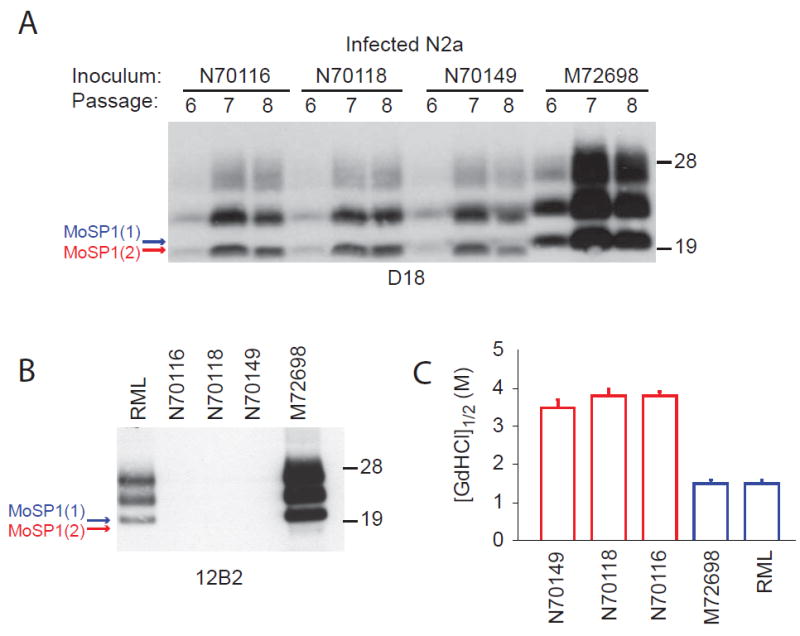
Infection of N2a cells with MoSP1. (A, B) N2a cells were infected with the indicated brain homogenates. Western blots of PK-digested lysates indicate the presence of protease-resistant PrPSc. Lysates were taken after passages 6–8 (A) and after passage 8 (B). Red and blue arrows denote the unglycosylated, protease-resistant bands of PrPSc migrating to 19 kDa [MoSP1(2)] and 21 kDa [MoSP1(1)], respectively. Blots were probed with D18 (A) or 12B2 (B). Molecular masses of protein standards are indicated in kilodaltons. (C) The conformational stabilities of MoSP1 in infected N2a cells. Lysates from passage 8 were analyzed. Stabilities for MoSP1(1) and MoSP1(2) bands are plotted in blue and red, respectively. The stability of RML is plotted for comparison.
GdnHCl denaturation assays indicated that the distinct conformational stabilities of MoSP1(2) and MoSP1(1) were faithfully transferred to cells (Fig. 2C). The measured GdnHCl1/2 values of PrPSc in cells mirrored those of the corresponding brain-derived inocula (compare Fig. 1D and 2C).
Gradual transformation of MoSP1 in culture
N2a cells infected with N70116 and harboring MoSP1(2) were continuously propagated for 32 weekly passages in dextrose-rich DMEM media (Fig. 3A). As a control, the same N2a subclone was infected with RML prions and propagated for the same number of passages (Fig. 3B). During the first 24 passages, the accumulation of MoSP1(2) progressively increased over time. At passage 23, the accumulation of MoSP1(1) was detectable. Between passages 24 and 27, MoSP1(1) levels steadily increased and MoSP1(2) levels declined. After passage 27, MoSP1(2) was undetectable. Interestingly, the culture media influenced the relative proportions of MoSP1(1) and MoSP(2) between passages 24–29 (Fig. 3C). When cells were switched to MEM at passage 24, MoSP(1) did not accumulate as quickly compared to levels observed with DMEM. Furthermore, MoSP1(2) persisted at passage 29 in MEM, whereas it was depleted at passage 29 in DMEM. The rates of cell division in MEM and DMEM were 0. 02 h-1 and 0. 04 h-1, respectively.
Fig. 3.
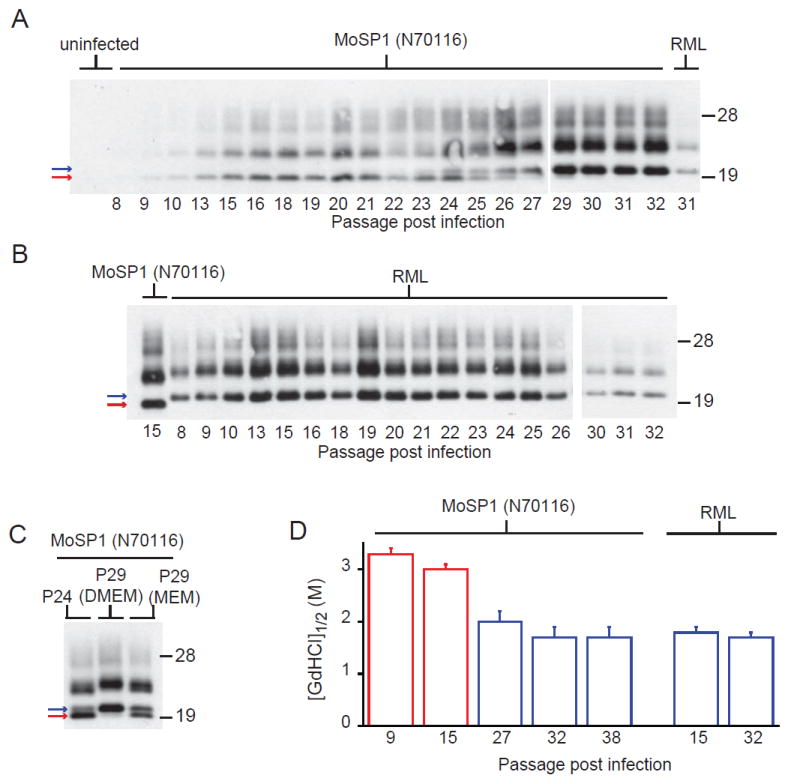
Long-term propagation of MoSP1 in cell culture. N2a cells infected with brain homogenate from mouse N70116 (A) or RML (B) were propagated for 32 weekly passages in DMEM. After each passage (as indicated by the number below each lane), lysates were collected, treated with PK, and analyzed by Western blots. The red and blue arrows indicate MoSP1(2) and MoSP1(1) bands, respectively. Starting from passage 24, cells were also propagated in MEM (C), indicating a slower rate of transformation from MoSP1(2) to MoSP1(1) between passages 24 and 29, compared to cells propagated in DMEM. (D) Conformational stability of MoSP1 and RML after passage in N2a cells. Lysates were taken after the indicated number of passages postinfection. Stabilities for MoSP1(1) and MoSP1(2) bands are plotted in blue and red, respectively. Upon serial passage, the conformational stability of MoSP1 decreased. For the Western blots, molecular masses of protein standards are indicated in kilodaltons. Blots were probed with the D18 antibody.
As the PrPSc population shifted from MoSP1(2) to MoSP1(1) with passage, the conformational stability of PrPSc in the infected N2a cells decreased (Fig. 3D). At passage 9, PrPSc from N70116-infected cells had a GdnHCl1/2 value of 3. 2 M. By passage 32, the strain exhibited a GdnHCl1/2 value of 1. 7 M. By comparison, the conformational stability of RML-infected cells was 1. 7 M.
Sensitivity of MoSP1 strains to antiprion drugs
In order to provide further evidence that MoSP1(2) and MoSP1(1) represent discrete conformational states with distinct biological properties, we assessed their sensitivities to known antiprion compounds. Positive polyamidoamines (PAMAM) and quinacrine have been shown to reduce PrPSc levels in RML-infected cells 44; 45 through poorly understood mechanisms of action. We added increasing concentrations of PAMAM and quinacrine to N2a cells infected with MoSP1 (from mouse N70116) or RML prions, at varying timepoints (passage numbers) postinfection. The cells were treated for 7 days, and the lysates were digested with PK and analyzed by Western blots (Fig. 4). At passage 15, RML-infected cells were cleared of PrPSc by 1 μg/mL PAMAM whereas N70116-infected cells, harboring MoSP1(2), were resistant to its antiprion activity. In N70116-infected cells at passage 26, both MoSP1(2) and MoSP1(1) were present; 1 μg/mL PAMAM completely abolished the MoSP1(1) population whereas MoSP(2) remained. By passage 38, 1 μg/mL PAMAM cleared all PrPSc from N70116-infected cells. Unlike PAMAM, 1 μM of quinacrine cleared PrPSc from both RML- and MoSP1-infected cells, abolishing both MoSP1(1) and MoSP(2) from the cultures (Fig. 4). These results indicate that MoSP1(1) and MoSP1(2) have differential sensitivities to PAMAM but are equally susceptible to quinacrine.
Fig. 4.
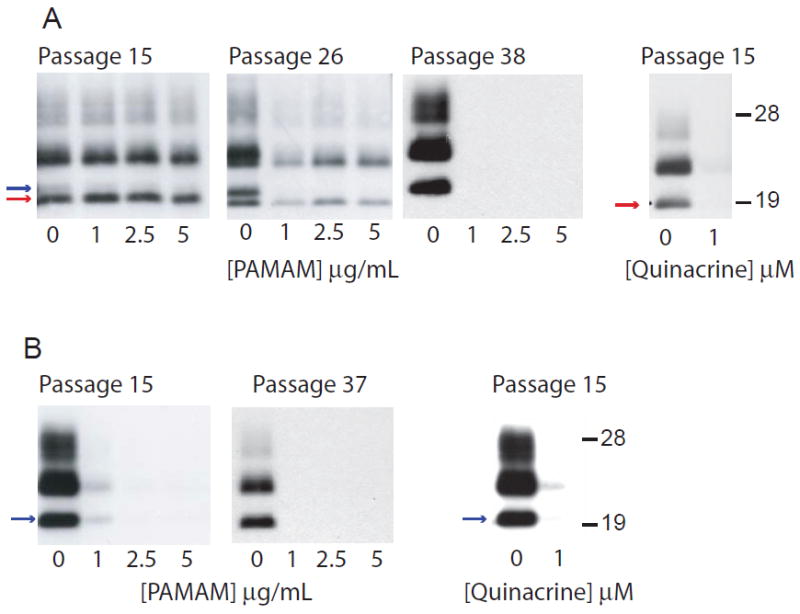
Sensitivity of MoSP1 strains (A) and RML (B) to antiprion drugs after passage in N2a cells. PAMAM or quinacrine was added at the indicated concentrations and cells were treated for 7 days; lysates were taken, digested with PK, and subjected to Western immunoblotting. The red and blue arrows indicate MoSP1(2) and MoSP1(1) bands, respectively. PAMAM selectively depleted MoSP1(1) but not MoSP1(2) in infected N2a cells, whereas quinacrine depleted all prion strains. Molecular masses of protein standards are indicated in kilodaltons. Blots were probed with the D18 antibody.
Serial passage of MoSP1 results in prions with shorter incubation periods
We next assessed the infectivity of MoSP1 at various passage numbers in culture. Cell extracts were collected at passages 9, 15, 27 and 38 for MoSP1-infected cells, and passages 15 and 24 for RML-infected cells. The extracts were intracerebrally inoculated in Tg4053 mice overexpressing full-length MoPrP. Mice inoculated with RML-infected cell lysates developed signs of neurologic dysfunction after ~60 days (Fig. 5A). This incubation period is similar to mice infected with serially passaged RML brain homogenate (for example, see 46). In comparison, lysates derived from MoSP1-infected cells caused signs of neurologic dysfunction with progressively shorter incubation periods as a function of passage number (Fig. 5A). Mice inoculated with MoSP1 lysate at passage 9 showed signs of neurologic dysfunction after ~180 days, whereas those infected with passage 38 cells became ill after ~80 days. Mice inoculated with lysate from uninfected N2a cells remained healthy for 400 days postinoculation, at which point the experiment was terminated.
Fig. 5.
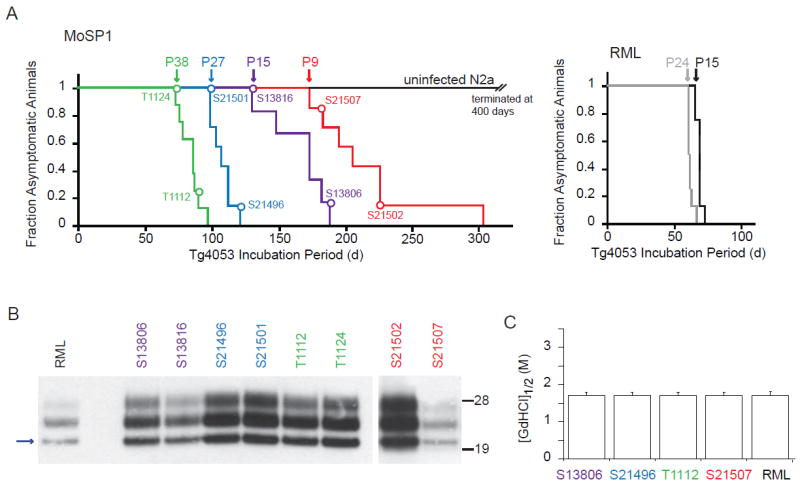
Bioassays of MoSP1 in Tg4053 mice after propagation in culture. (A) Survival curves of mice inoculated with cell-passaged MoSP1 (left graph) and cell-passaged RML (right graph). Inoculation with uninfected N2a cells is shown as control. Cell extracts were collected at passages 9, 15, 27 and 38 for MoSP1-infected cells, and passages 15 and 24 for RML-infected cells; extracts were then intracerebrally inoculated in Tg4053 mice overexpressing full-length MoPrP. Colored curves indicate the different inocula; colored circles indicate the corresponding ill mice (also given unique ID codes) that were biochemically analyzed by Western immunoblotting (B) and the conformational-stability assay (C). For the Western blots, molecular masses of protein standards are indicated in kilodaltons, and PrP probed with the D18 antibody. The blue arrow indicates the MoSP1(1) band.
The banding patterns and conformational stabilities of PrPSc derived from a number of terminal mice were subsequently analyzed (Fig. 5B, C). The results indicated that, at the time of death, all mice harbored the MoSP1(1) strain with a conformational stability of ~1. 7 M. Thus, during the course of the bioassay, prions derived from MoSP1-infected cells at passage 9 and passage 15 had undergone a transition from a high-stability MoSP1(2) strain (see Fig. 3A) to a low-stability MoSP1(1) strain. The results suggest that the course of MoSP1 adaptation initiated in cell culture continued along the same trajectory when inoculated into the brains of mice.
Cloning of MoSP1(2) and MoSP1(1)
The foregoing data indicate that a gradual shift in the prion population towards MoSP1(1) occurred in infected cultures. Such a shift is likely to occur because MoSP1(1) has a faster rate of replication in comparison to MoSP1(2). However, the origin of the MoSP1(1) strain in the infected cells remains unclear. In theory, MoSP1(1) that existed in the brain-derived inocula could have been passed on to cells at levels that were initially undetectable, but gradually accumulated upon serial passage, competing out the MoSP1(2) strain (see Discussion below). Alternatively, MoSP1(1) may be formed by direct conformational conversion from MoSP1(2) over time. In order to distinguish between these two possibilities, we sought to clone MoSP1(1) and MoSP1(2) in culture and assess their fidelity of replication. At passage 24, the cells were subcloned by limiting dilution and 16 individual clones were obtained. Upon expansion, 11 of the 16 clones contained detectable levels of PK-resistant PrPSc by Western blot (Fig. 6A). Interestingly, most clones contained either MoSP1(2) or MoSP1(1), but not both. This finding is consistent with the observation that intercell prion propagation occurs more slowly than intracell PrPSc formation 47. Although the rate of cell-to-cell transmission of MoSP1(1) is likely to be relatively slow, once a cell acquires small levels of MoSP1(1), the strain rapidly propagates within that cell, outcompeting the MoSP1(2) population. Thus, within an adapting culture, the transition from MoSP1(2) to MoSP1(1) appears to occur in a quantized manner, and cells harboring detectable levels of both strains (e.g., as seen in subclone 4) are rare.
Fig. 6.
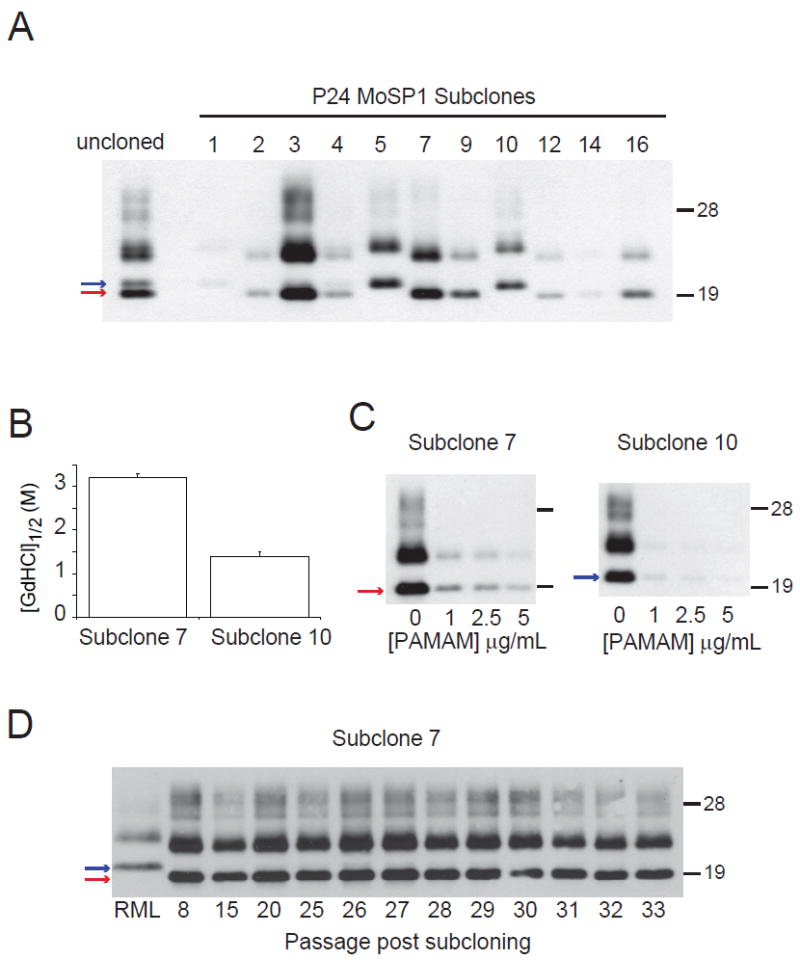
Cloning and propagation of MoSP1(2) and MoSP1(1). At passage 24, N70116-infected N2a cells were subcloned by limiting dilution. After expansion, subclones were lysed, treated with PK, and analyzed by Western blot (A). Subclones 7 and 10 were subsequently analyzed by the conformational-stability assay (B) and tested for sensitivity to PAMAM (C). Subclone 7 was propagated for 33 additional passages and the banding pattern was analyzed by Western blot (D). The results indicate that upon cloning, MoSP1(2) can stably propagate without conversion to MoSP1(1). The red and blue arrows indicate MoSP1(2) and MoSP1(1) bands, respectively. Molecular masses of protein standards are indicated in kilodaltons. Blots were probed with anti-PrP antibody D18.
Subclone 7 that harbored only MoSP1(2) and subclone 10 that harbored only MoSP1(1) had the respective conformational stabilities and PAMAM sensitivities of the MoSP1(2) and MoSP1(1) in the uncloned cultures (Fig. 6B, C). MoSP1(2) in subclone 7 remained in the culture for at least 32 subsequent passages (Fig. 6D). Conversely, in the uncloned N70116-infected culture containing a mixture of MoSP1(2) and MoSP1(1), MoSP1(2) was depleted after 5 passages (from passage 24 to 29) and replaced by MoSP1(1) (Fig. 3A). Thus, when cloned, MoSP1(2) can replicate with fidelity in culture. This result suggests that the rapid depletion of MoSP1(2) that was seen in the uncloned culture (Fig. 3A) required the competitive presence of previously formed MoSP1(1) that was introduced to the culture during infection.
Bioassays of cloned MoSP1(1) and MoSP1(2)
Extracts from cloned cells harboring MoSP1(1) and MoSP1(2) (subclone 10 and subclone 7, respectively), as well as RML, were collected at passage 8. The extracts were normalized based on the detectable levels of PrPSc on Western blots (see Methods) and intracerebrally inoculated into Tg4053 mice. MoSP1(1), MoSP1(2) and RML resulted in incubation periods of ~120 d, ~220 d and ~60 d, respectively (Fig. 7A). Brains of Tg4053 mice at the terminal stage of disease were collected for biochemical analysis. As expected, protease-resistant PrPSc in mice infected with RML and MoSP1(1) had a type 1 banding pattern and a GdnHCl1/2 value of ~1. 7 M (Fig. 7B, C). Conversely, protease-resistant PrPSc in mice infected with MoSP1(2) primarily showed a type 2–banding pattern and had relatively high conformational stability (GdnHCl1/2 value of ~4 M). However, it is important to note that in all three MoSP1(2)-infected mice analyzed, small levels of MoSP1(1) were detectable in the brain (Fig. 7B). Thus, although cloned MoSP1(2) could replicate with high fidelity in culture (Fig. 6D), during the course of its incubation in the brain, it is capable of conformationally converting to MoSP1(1) (see Discussion below).
Fig. 7.

Bioassays of cloned MoSP1(1) and MoSP1(2) in Tg4053 mice. (A) Survival curves of mice inoculated with cell lysates containing cloned MoSP1(1), MoSP1(2) and RML. Colored curves indicate the different inocula; colored circles indicate the corresponding ill mice (also given unique ID codes) that were biochemically analyzed by Western immunoblotting (B) and the conformational-stability assay (C). For the Western blots, the red and blue arrows indicate MoSP1(2) and MoSP1(1) bands, respectively. Molecular masses of protein standards are indicated in kilodaltons, and PrP probed with the D18 antibody. The red and blue bars in (C) indicate the conformational stability of MoSP1(2) and MoSP1(1), respectively.
Neuropathological profiles of MoSP1(1) and MoSP1(2)
We compared the neuropathological profiles of terminal Tg4053 mice infected with RML, MoSP1(1) and MoSP1(2) lysates by vacuolation score histograms and neurohistology (Fig. 8). Vacuolation scores are the estimated area of a brain region covered by vacuoles. The vacuolation scores of MoSP1(1) and MoSP1(2) were essentially the same, except with regard to the cerebellar cortex. MoSP1(2) infection resulted in vacuoles in the cerebellar molecular layer whereas MoSP1(1) infection did not. In contrast, the vacuolation histogram of RML prions was distinct from the two synthetic strains (Fig. 8A).
Fig. 8.
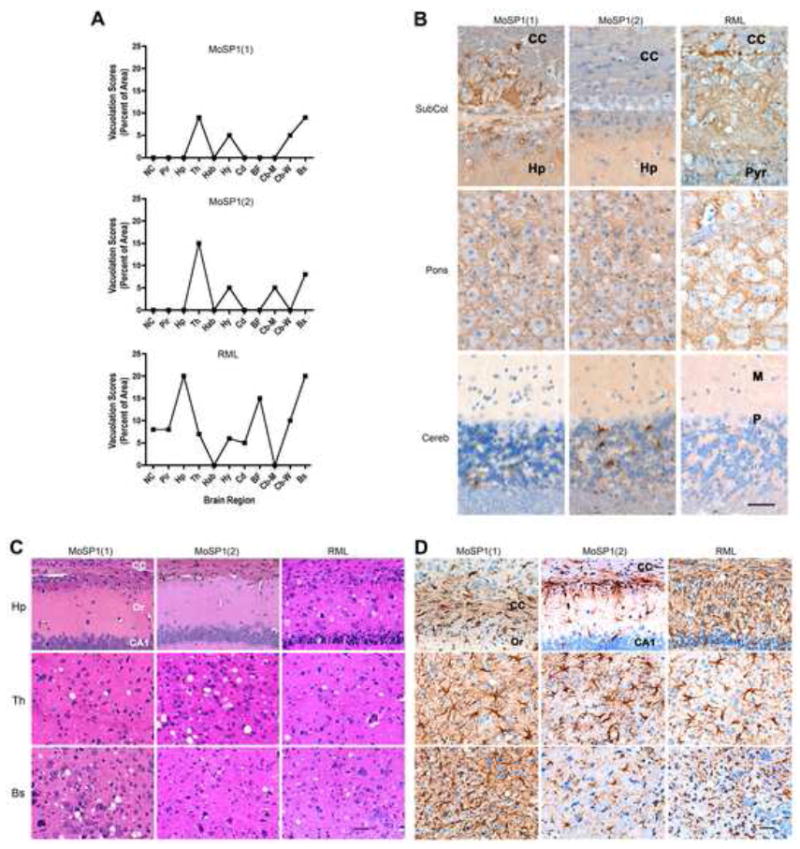
MoSP1(1), MoSP1(2), and RML produced three different neuropathologic phenotypes in Tg4053 mice. Brains from T1159, S24838 and S24827 mice were analyzed. (A) Vacuolation score histograms of MoSP1(1) (top), MoSP1(2) (middle), and RML (bottom) prions. The vacuolation score is the estimated area of a slide occupied by vacuoles. MoSP1(1) and MoSP1(2) histograms were very similar except the MoSP1(2) histogram showed mildly intense vacuolation in the cerebellar molecular layer and MoSP1(1) did not. The RML histogram was characterized by a moderate degree of vacuolation in many more brain regions than MoSP1(1) or MoSP1(2). (B) PrPSc immunohistochemistry in mice following inoculation with MoSP1(1) (left column), MoSP1(2) (middle column), and RML (right column). MoSP1(1) prions resulted in large PrPSc plaque-like deposits in the corpus callosum and subcallosal region. In contrast, no PrPSc was found in the corpus callosum or subcallosal region with MoSP1(2) prions. With RML prions, finely granular and small plaque-like deposits were located in the subcallosal hippocampus and small plaque-like deposits were found in the corpus callosum. For all three inocula, finely granular PrPSc deposits were present in the motor trigeminal nucleus of the brainstem. Sparse numbers of PrPSc deposits were found in the cerebellar granule cell layer, but none in the molecular layer with MoSP1(1) prions. In contrast, MoSP1(2) prions were associated with many coarsely granular deposits in the granule cell layer and sparse numbers of finely granular deposits in the molecular layer. No PrPSc deposits were seen in the cerebellar cortex with RML prions. (C) H&E staining showed no vacuolar degeneration of neurons with MoSP1(1) (left column) and MoSP1(2) prions (middle column) in the hippocampus whereas RML prions (right column) resulted in vacuolar degeneration and nerve cell loss in the CA1 region. (D) Reactive astrocytic gliosis following infection with MoSP1(1) (left column), MoSP1(2) (middle column), and RML (right column) prions. (Top row) A moderate degree of white matter reactive astrocytic gliosis was found in the corpus callosum in association with the PrPSc plaque-like deposits with MoSP1(1) prions but was absent from the corpus callosum with MoSP1(2) prions. RML prions were associated with very intense astrocytic gliosis in the hippocampus. In the thalamus (middle row), reactive astrocytic gliosis was severe with MoSP1(1) prions, moderately intense with MoSP1(2) prions, and mildly to moderately intense with RML prions. In the brainstem (bottom row), a very intense reactive astrocytic gliosis was found with MoSP1(1) prions, mild with MoSP1(2) prions, and moderately intense with RML prions. BF, basal forebrain; Bs, brainstem; CA1, region of the hippocampus; Cb-M, cerebellar molecular layer; Cb-W; cerebellar white matter; cc, corpus callosum; Cd; caudate nucleus; G, cerebellar granular cell layer; Hab, habenula; Hp, hippocampus; Hy, hypothalamus; M, molecular layer of the cerebellar cortex; NC, neocortex; Or, stratum oriens of the CA1 region; P, Purkinje cell layer of the cerebellar cortex; Pir, piriform cortex; Pyr, pyramidal cell layer of the CA1 region of the hippocampus; Th, thalamus. Bars represent 50 μm and apply to all photomicrographs in the same panel.
The neurohistology resulting from MoSP1(1) and MoSP1(2) inoculations also differed substantially (Fig. 8B–D). MoSP1(1) prions formed large plaque-like deposits of PrPSc in the corpus callosum while MoSP1(2) did not. In the cerebellar granule cell layer, very little PrPSc was deposited with MoSP1(1) prions (Fig. 8B, bottom row), but a large amount of PrPSc was found with MoSP1(2) (Fig. 8B, bottom row). The accumulation of PrPSc in the cerebellum with MoSP1(2) was also associated with vacuolation of the molecular layer (Fig. 8A). In the brainstem, MoSP1(1), MoSP1(2), and RML showed similar changes: finely granular PrPSc deposits tended to be localized the neuropil between nerve cells and not in the nerve cell bodies (Fig. 8B, middle row). H&E staining (Fig. 8C) did not show vacuoles in the hippocampus with either MoSP1(1) or MoSP1(2) prions. Vacuolation was less intense in the thalamus with MoSP1(1) than MoSP1(2), but more intense in the brainstem with MoSP1(1) than MoSP1(2). RML differed in that vacuolation was mostly distributed throughout the CNS.
Reactive astrocytic gliosis also varied for the three prion strains (Fig. 8D). With MoSP1(1) and MoSP1(2) prions, no reactive astrocytic gliosis was seen in the hippocampus proper. However, the large plaques associated with MoSP1(1) prions evoked a moderately intense reactive astrocytic gliosis in the corpus callosum. MoSP1(2) prions were associated with no plaque-like PrPSc deposits in the callosal region, and astrocytes were of normal size and shape. In contrast, RML was associated with vacuolation of the hippocampus proper, small amyloid plaques in the subcallosal region, and fine granular PrPSc in the hippocampus proper; as a result, intense reactive astrocytic gliosis was present in the hippocampus. Infection with MoSP1(1), MoSP1(2), and RML resulted in a similar, moderately severe astrocytic gliosis in the thalamus. In the brainstem, the relatively intense vacuolation seen with MoSP1(1) prions (Fig. 8A) was associated with a very intense degree of reactive astrocytic gliosis (Fig. 8D, bottom row). In comparison, the relatively low vacuolation in the brainstem with MoSP1(2) prions was associated with a relatively low degree of reactive astrocytic gliosis. RML was associated with a moderate degree of vacuolation and the reactive astrocytic gliosis was moderate. Together, the neuropathological data argue that each of the three inocula [MoSP1(1), MoSP1(2) and RML] represents a distinct prion strain.
DISCUSSION
Our studies demonstrate that MoSP1 undergoes profound conformational transformations during serial passage in both mice and cultured cells. These conformational changes were accompanied by marked decreases in incubation periods and changes in neuropathological profiles. Importantly, we were able to infect N2a cells with MoSP1 prions and recapitulate its conformational transformation in culture. Recent experiments have shown that mammalian 31;32 and yeast 48; 49 prions can evolve conformationally in response to environmental pressures. By isolating and characterizing conformationally distinct subtypes of MoSP1, we were able to investigate the molecular mechanism of prion strain adaptation. The salient observations were as follows:
- Based on electrophoretic mobility of protease-resistant bands, two distinct MoSP1 strains can be defined in vivo: MoSP1(1) and MoSP1(2).
- When cloned in cultured cells, MoSP1(1) and MoSP1(2) can each propagate with fidelity.
- When MoSP1(1) and MoSP1(2) co-exist in cells, MoSP1(1) preferentially propagates and MoSP1(2) is depleted.
- MoSP1(1) and MoSP1(2) are independently capable of biological transformation and selection. For example, uncloned MoSP1-infected cells at passage 9 and 15 contained only MoSP1(2), yet had different GdnHCl1/2 values and incubation periods (Fig. 4). This observation highlights the limitations of using Western blot banding patterns to define prion strains and suggests that MoSP1(1) and MoSP1(2) likely encompass a spectrum of conformations.
- The inoculation of mice with cloned MoSP1(2) resulted in the formation and accumulation of small amounts of MoSP1(1) (Fig. 6).
Based on these observations, we propose a model for the transformation of MoSP1 (Fig. 9). After inoculation with MoSP1(2), low levels of MoSP1(1) can be formed in the brain by rare conversion events. It is not clear why this conversion does not occur in cloned MoSP1(2) in cultured cells. Perhaps extended in vivo incubation periods are required for MoSP1(1) to form initially. However, once MoSP1(1) is formed in the brain, its selective accumulation occurs by a process of competitive selection. Upon serial passage, MoSP1(1) accumulates faster than MoSP1(2), leading to the extinction of the latter. As they propagate, both MoSP1(1) and MoSP1(2) become conformationally less stable. However, MoSP1(1) can access lower conformational stabilities and cause disease in shorter incubation periods.
Fig. 9.
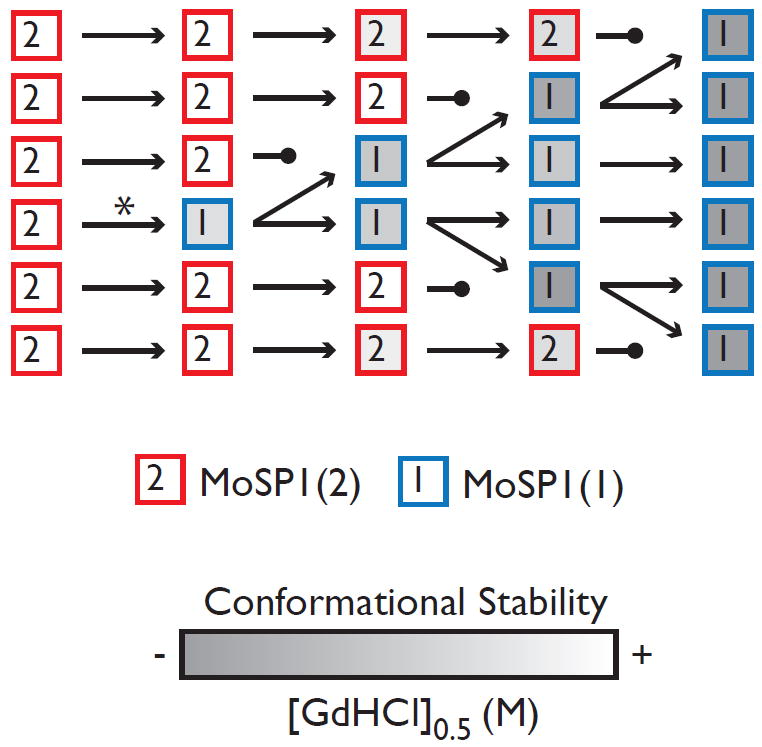
Proposed model for the transformation of MoSP1 prions through a process of competitive selection. MoSP1(1) (squares labeled “1”) is initially created by a low-frequency conversion event (*) and exists at low levels in a heterogeneous prion population. Upon serial passage, MoSP1(1) replicates faster than MoSP1(2) (squares labeled “2”), leading to the extinction of the latter. As they propagate, both MoSP1(1) and MoSP1(2) become conformationally less stable (shades of gray). However, MoSP1(1) can access lower conformational stabilities and cause disease with shorter incubation periods.
What provides the selective pressure in favor of MoSP1(1)? It is evident from our data that MoSP1(1) has a faster rate of formation and causes disease with shorter incubation times in mice. Thus, during serial passage in mice, lethality provides a clear bias for the gradual accumulation of MoSP1(1). Because of its faster rate of formation, MoSP1(1) can reach lethal levels in a shorter period of time. Thus, mice inoculated with a mixture of MoSP1(1) and MoSP1(2) succumb to disease before the slower MoSP1(2) strain can accumulate. MoSP1(2) levels gradually decline as infected brains are used as inocula for further rounds of passage. However, a similar strain competition was observed in cultured cells. Given that neither MoSP1(2) nor MoSP1(1) is lethal to or alters the rate of growth in N2a cells, lethality cannot be the only source of selective pressure. It is therefore likely that the presence of MoSP1(1) causes the depletion of MoSP1(2) by outcompeting it for a limited resource. As a mixture, both strains are competing for the same pool of PrPC substrate and other auxiliary factors required for PrPSc formation. The faster replicating MoSP1(1) strain may be more effective at interacting with PrPC or other auxiliary factors, denying MoSP1(2) the resources required for its propagation 20; 50; 51.
In our studies, increases in the molecular weight of the deglycosylated band (from 19 kDa to 21 kDa) as well as decreases in the conformational stability of PrPSc correlated with shorter incubation periods. It is not clear whether these two biochemical properties produce faster rates of formation or are independent correlates of more infectious conformations. However, it is important to note that a direct correlation between conformational stability and incubation period has been previously noted for several natural and synthetic mouse prion strains 37. Likewise, in two independent models of prion strain diversity, shifts in PrPSc banding patterns, from 19 kDa to 21 kDa, have been associated with shorter incubation periods. In human CJD cases, two distinct biochemical subtypes of PrPSc have been identified 42: 21-kDa “type 1” prions and 19-kDa “type 2” prions (described above). Upon infection of Tg mice with vCJD prions, type 2 prions accumulated in the brain after an extended incubation period 29. However, after serial passage into two different lines of Tg mice, the incubation period decreased and occasionally the banding pattern shifted to type 1 PrPSc 30. Similarly, infection of Syrian golden hamsters with TME prions predominantly resulted in the formation of DY prions, a long-incubation-period strain with a 19-kDa banding pattern. Upon serial passage, a transformation to the HY strain occurred, with shorter incubation periods and a 21-kDa band 27. It remains to be determined whether the 21-kDa banding pattern is a general biochemical signature of increased infectivity in synthetic prion strains.
Our findings with MoSP1 propagation in both cell culture and mice indicate that the MoSP1(1) prion strain has a selective advantage over MoSP1(2). In cell culture, environmental conditions can influence the relative advantage of the two strains. Growth in minimal media slows the rate of MoSP1(1) selection while growth in dextrose-rich media increases the rate of N2a cell division and MoSP1(1) selection. These data suggest that cell growth acts as a constant dilution force, decreasing the cellular levels of intracellular prions after each round of division 47. Thus, in a rapidly dividing culture, a prion strain with a slow rate of formation, such as MoSP1(2), may be at a particular disadvantage as it may not be able reach steady-state levels prior to the next round of division. In addition to the culture media, the presence of biologically active chemicals also influenced the relative abundance of MoSP1(1) and MoSP1(2). PAMAM reversed the natural selection of MoSP1 prions by inducing the clearance of MoSP1(1) while sparing MoSP1(2). Other antiprion compounds, such as swainsonine and quinacrine, have also demonstrated strain-specific efficacy 31; 32.
Among the neurodegenerative disorders, infectivity was previously thought to be a unique feature of PrP-associated prion diseases. However, recent experiments have demonstrated that aggregates of proteins causing other neurodegenerative diseases such as Aβ 52; 53,α-synuclein 54; 55, tau 56; 57, and huntingtin 58 can stimulate their own modified states under certain conditions. It now seems clear that protein aggregates can have a spectrum of self-replicative properties. The protracted nature of Alzheimer’s disease and Parkinson’s disease may reflect a relatively inefficient aggregation process. Conversely, in endemic prion diseases such as scrapie and chronic wasting disease in which horizontal transmission is common 59; 60, PrPSc has likely undergone many rounds of conformational selection and evolved into a highly infectious state. The conformational selection observed for MoSP1 may represent a general mechanism by which protein aggregates can transform into highly infectious prions.
MATERIALS AND METHODS
Ethics statement
All animal experiments followed “The Guide for the Care and Use of Laboratory Animals,” National Research Council publication, 1996. All operations and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at UCSF.
Production of recMoPrP(89–230) and amyloid fibers
The expression and purification of recMoPrP(89–230), and the formation of amyloid fibers have been previously described 61. The molecular weight and purity of recMoPrP(89–230) were confirmed by laser desorption and electrospray mass spectrometry. Amyloid fibrils were formed by incubating recMoPrP(89–230) at 37 °C in 3 M urea; 0. 2 M NaCl; 50 mM sodium acetate buffer, pH 5. 0, as previously described61.
Mouse bioassays
Tg9949 and Tg4053 mice have been previously described 40; 62. Amyloid fibers were prepared for inoculation by dialysis in sterile PBS without calcium or magnesium, pH 7. 2 over 2 days. The concentration of recMoPrP(89–230) in the inocula was ~0. 5 mg/ml. Serial passage was conducted by inoculation with brain homogenates in sterile PBS without calcium or magnesium; brain homogenates were prepared by repeated extrusion through syringe needles of successively smaller size, from 18- to 22-gauge. All work was carried out in laminar flow hoods to avoid cross contamination. Mice of either sex, aged 7 to 10 weeks, were inoculated intracerebrally with 30 μl of recMoPrP(89–230) amyloid fibrils, control PBS, or 1% brain homogenate or 5 mg of PTA-precipitated PrPSc from cell lysates in calcium- and magnesium-free PBS plus 5 mg/ml BSA. Inoculation was carried out with a 27-gauge disposable hypodermic needle inserted into the right parietal lobe. After inoculation, mice were examined daily for neurologic dysfunction. Standard diagnostic criteria were used to identify animals exhibiting signs of prion disease 63; 64. The indicated mice were sacrificed, and their brains were removed for biochemical analysis.
Cell culture
The N2a cell line was purchased from American Tissue Culture Collection. Cells were infected by exposure to the indicated brain homogenates as previously described 65. Cells were maintained at 37 °C in 10 mL of MEM or DMEM supplemented with 10% FBS, 1% penicillin-streptomycin and 1% GlutaMAX (Invitrogen). Media were refreshed every 2 days. Cells were propagated in 100-mm plates and allowed to grow to 95% confluence before dissociation with 1 mL of enzyme-free cell dissociation buffer. Cells were then replated at 10% confluence for further propagation. To collect cell lysates, cells were rinsed with PBS (3 × 10 mL) and lysed with 1 mL cold lysis buffer (10 mM TrisHCl, pH 8. 0; 150 mM NaCl; 0. 5% Nonidet P-40; 0. 5% sodium deoxychloate). Lysates were centrifuged for 3 min at 10,000 x g to remove cell debris and the total protein concentration was measured in the supernatant using the bicinchoninic acid assay (BCA; Pierce, Rockford, IL). Aliquots containing 500 μg of total protein were titrated by adding lysis buffer to achieve a final protein concentration of 1 mg/mL, then stored at -20 °C until further analysis. Cloning was conducted by limiting dilution as previously described 65. For drug treatment studies, stock solutions of 10 mg/ml PAMAM in PBS and 2 mM quinacrine in PBS were prepared prior to adding to the culture to obtain the indicated concentrations. Cultures were exposed to drugs for 7 days while being replenished every 2 days during feeding.
Western blots, conformational-stability assay, and densitometry
Nuclei and debris were removed from brain homogenates and cell lysates by centrifugation at 1000 x g for 10 min. Cleared extracts were adjusted to 1 mg of protein per ml in 100 mM NaCl–1 mM EDTA–2% Sarkosyl–50 mM Tris-HCl (pH 7. 5). PK (20 μg/mL; Boehringer Mannheim) was added to 0. 5 ml of adjusted homogenate to achieve a protein:enzyme ratio of 50:1. After incubation at 37 °C for 1 h, proteolytic digestion was terminated by the addition of 8 ml of 0. 5 M phenylmethylsulfonyl fluoride in absolute ethanol. Both PK-digested and undigested samples were prepared for sodium dodecyl sulfate–12% polyacrylamide gel electrophoresis by mixing equal volumes of adjusted homogenate and 2x sample buffer. Following electrophoresis, Western blotting was performed as previously described 65. Membranes were blocked with 5% nonfat milk protein in calcium- and magnesium-free PBS plus 0. 1% Tween 20 (PBST) for 1 h at room temperature. Blocked membranes were incubated with primary PrP-specific recFab HuM-D18 at 1 μg/mL in PBST for 1 h at 4 °C 41. After incubation with primary Fab, membranes were washed 3x for 10 min in PBST, incubated with horseradish peroxidase–labeled, anti-human Fab secondary antibody (ICN), diluted 1:5000 in PBST for 25 min at room temperature, and washed again 3x for 10 min in PBST. After chemiluminescent development with an ECL reagent (Amersham) for 1 to 5 min, blots were sealed in plastic covers and exposed to ECL Hypermax film (Amersham).
The conformational-stability assay has been previously described 13. Briefly, 50 μL of 10% brain homogenate or 1 mg/mL cell lysate were mixed with 50 μL of GdnHCl, varying from 0 to 6 M and incubated for 1 h at room temperature. For GdnHCl concentrations greater than 4 M, less volume of extract was used. Prior to PK digestion, all samples were diluted in lysis buffer to obtain GdnHCl concentrations of 0. 4 M. Densitometry on the appropriate bands was performed using a CCD camera (FluorChem 8800, Alpha Innotech). Measurements were normalized with respect to the highest intensity band in the denaturation curve. The GdnHCl1/2 values were obtained by least-square fitting to the following sigmoidal equation: Y = 1/(1+(X/GdnHCl1/2)ˆHC), for which Y represents the measured intensities, X represents the GdnHCl concentrations, and HC represents the Hill coefficient.
Neuropathology
Brains were removed rapidly at the time of sacrifice, immersion-fixed in 10% buffered formalin, and embedded in paraffin. Eight-micrometer-thick sections were stained with hematoxylin and eosin for evaluation of prion disease. Peroxidase immunohistochemistry with antibodies to glial fibrillary acidic protein was used to evaluate the degree of reactive astrocytic gliosis. Immunohistochemistry of PrPSc was performed by the hydrated autoclaving method using the PrP-specific, human-mouse R2 recombinant monoclonal antibody fragment (Fab) 66.
Supplementary Material
01
02
03
Acknowledgments
This work was supported by grants from the National Institutes of Health (AG02132, AG10770, and AG021601) as well as by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation. The authors thank Dr. Jan Langeveld for providing the 12B2 antibody.
Abbreviations
CJD
Creutzfeldt-Jakob disease
sCJD
sporadic Creutzfeldt-Jakob disease
vCJD
variant Creutzfeldt-Jakob disease
PrP
prion protein
MoSP
mouse synthetic prion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 3.Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46:417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 4.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prusiner SB. Shattuck Lecture — Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 6.Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338:342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB. Biology of prions. In: Rosenberg RN, Prusiner SB, DiMauro S, Barchi RL, editors. The Molecular and Genetic Basis of Neurological Disease. 2. Butterworth Heinemann; Stoneham, MA: 1997. pp. 103–143. [Google Scholar]
- 8.Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec. 1967;80:1–8. doi: 10.1136/vr.80.1.2. [DOI] [PubMed] [Google Scholar]
- 9.Dickinson AG, Meikle VMH. A comparison of some biological characteristics of the mouse-passaged scrapie agents, 22A and ME7. Genet Res. 1969;13:213–225. doi: 10.1017/s0016672300002895. [DOI] [PubMed] [Google Scholar]
- 10.Bruce ME, Dickinson AG. Biological stability of different classes of scrapie agent. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Diseases of the Nervous System. Vol. 2. Academic Press; New York: 1979. pp. 71–86. [Google Scholar]
- 11.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angers RC, Kang HE, Napier D, Browning S, Seward T, Mathiason C, Balachandran A, McKenzie D, Castilla J, Soto C, Jewell J, Graham C, Hoover EA, Telling GC. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328:1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peretz D, Scott M, Groth D, Williamson A, Burton D, Cohen FE, Prusiner SB. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 15.Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Polymenidou M, Schwarz P, Leclerc M, Hammarstrom P, Wüthrich K, Aguzzi A. Prion strain discrimination using luminescent conjugated polymers. Nat Methods. 2007;4:1023–1030. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- 16.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA. 2007;104:20908–20913. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 18.Solassol J, Crozet C, Lehmann S. Prion propagation in cultured cells. Br Med Bull. 2003;66:87–97. doi: 10.1093/bmb/66.1.87. [DOI] [PubMed] [Google Scholar]
- 19.Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. Coinfecting prion strains compete for a limiting cellular resource. J Virol. 2010;84:5706–5714. doi: 10.1128/JVI.00243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattison IH. Experiments with scrapie with special reference to the nature of the agent and the pathology of the disease. In: Gajdusek DC, Gibbs CJ Jr, Alpers MP, editors. Slow, Latent and Temperate Virus Infections, NINDB Monograph 2. U.S. Government Printing; Washington, D.C.: 1965. pp. 249–257. [Google Scholar]
- 22.Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol. 1986;67:255–263. doi: 10.1099/0022-1317-67-2-255. [DOI] [PubMed] [Google Scholar]
- 23.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Wälchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 24.Prusiner SB, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson GA, Hoppe PC, Westaway D, DeArmond SJ. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 25.Scott M, Groth D, Foster D, Torchia M, Yang S-L, DeArmond SJ, Prusiner SB. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell. 1993;73:979–988. doi: 10.1016/0092-8674(93)90275-u. [DOI] [PubMed] [Google Scholar]
- 26.Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73:329–334. doi: 10.1099/0022-1317-73-2-329. [DOI] [PubMed] [Google Scholar]
- 27.Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol. 2000;74:5542–5547. doi: 10.1128/jvi.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schutt CR, Bartz JC. Prion interference with multiple prion isolates. Prion. 2008;2:61–63. doi: 10.4161/pri.2.2.6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, DeArmond SJ, Prusiner SB. Abbreviated incubation times for human prions in mice expressing a chimeric mouse—human prion protein transgene. Proc Natl Acad Sci U S A. 2003;100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. Human prion strain selection in transgenic mice. Ann Neurol. 2010;68:151–161. doi: 10.1002/ana.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5:e1000673. doi: 10.1371/journal.ppat.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 34.Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 35.Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen H-OB, Lemus A, Cohen FE, DeArmond SJ, Prusiner SB. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Legname G, Nguyen H-OB, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legname G, Nguyen H-OB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, Prusiner SB. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA. 2009;106:20417–20422. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. Identification of two prion protein regions that modify scrapie incubation time. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. Mapping the prion protein using recombinant antibodies. J Virol. 1998;72:9413–9418. doi: 10.1128/jvi.72.11.9413-9418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AAF, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 43.Langeveld JP, Jacobs JG, Erkens JH, Bossers A, van Zijderveld FG, van Keulen LJ. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res. 2006;2:19. doi: 10.1186/1746-6148-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Supattapone S, Nguyen H-OB, Cohen FE, Prusiner SB, Scott MR. Elimination of prions by branched polyamines and implications for therapeutics. Proc Natl Acad Sci U S A. 1999;96:14529–14534. doi: 10.1073/pnas.96.25.14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001;98:9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlson GA, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci U S A. 1994;91:5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, Prusiner SB. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci USA. 2007;104:17971–17976. doi: 10.1073/pnas.0708372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruce KL, Chernoff YO. Sequence specificity and fidelity of prion transmission in yeast. Semin Cell Dev Biol. doi: 10.1016/j.semcdb.2011.03.005. In press. [DOI] [PubMed] [Google Scholar]
- 49.Chen B, Bruce KL, Newnam GP, Gyoneva S, Romanyuk AV, Chernoff YO. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol Microbiol. 2010;76:1483–1499. doi: 10.1111/j.1365-2958.2010.07177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruce ME, Fraser H, McBride PA, Scott JR, Dickinson AG. The basis of strain variation in scrapie. In: Prusiner SB, Collinge J, Powell J, Anderton B, editors. Prion Diseases in Human and Animals. Ellis Horwood; London: 1992. pp. 497–508. [Google Scholar]
- 51.Kimberlin RH, Walker CA. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology. 1985;23:74–81. doi: 10.1159/000149588. [DOI] [PubMed] [Google Scholar]
- 52.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 53.Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral β-amyloidosis: Intracerebral versus systemic Aβ inoculation. Proc Natl Acad Sci USA. 2009;106:12926–12931. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 55.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–136. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- 60.Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. Asymptomatic deer excrete infectious prions in faeces. Nature. 2009;461:529–532. doi: 10.1038/nature08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 62.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 1996;10:1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 63.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol. 1982;11:353–358. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 64.Carlson GA, Kingsbury DT, Goodman PA, Coleman S, Marshall ST, DeArmond S, Westaway D, Prusiner SB. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 65.Ghaemmaghami S, Ullman J, Ahn M, St Martin S, Prusiner SB. Chemical induction of misfolded prion protein conformers in cell culture. J Biol Chem. 2010;285:10415–10423. doi: 10.1074/jbc.M109.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02
03