Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line (original) (raw)
Abstract
Both stem cells and cancer cells are thought to be capable of unlimited proliferation. Paradoxically, however, some cancers seem to contain stem-like cells (cancer stem cells). To help resolve this paradox, we investigated whether established malignant cell lines, which have been maintained for years in culture, contain a subpopulation of stem cells. In this article, we show that many cancer cell lines contain a small side population (SP), which, in many normal tissues, is thought to contain the stem cells of the tissue. We demonstrate that in the absence of serum the combination of basic fibroblast growth factor and platelet-derived growth factor maintains SP cells in the C6 glioma cell line. Moreover, we show that C6 SP cells, but not non-SP cells, can generate both SP and non-SP cells in culture and are largely responsible for the in vivo malignancy of this cell line. Finally, we provide evidence that C6 SP cells can produce both neurons and glial cells in vitro and in vivo. We propose that many cancer cell lines contain a minor subpopulation of stem cells that is enriched in an SP, can be maintained indefinitely in culture, and is crucial for their malignancy.
Like stem cells, cancer cells are widely thought to be able to proliferate indefinitely, and yet it is notoriously difficult to establish immortal cell lines in culture from primary cancers. When such cell lines have been established, however, they retain the ability to reform the original cancer in vivo. One possibility is that cancers arise in cells with the characteristics of stem cells, which, by definition, can self-renew indefinitely. Another possibility is that cancer cells acquire the property of unlimited proliferation by mutation, which might be essential for malignancy, as well as for primary human cells in culture to pass through the Hayflick limit (1).
There is increasing evidence that cancers might contain their own stem cells. Many cancers, like normal organs, seem to be maintained by a hierarchical organization that includes slowly dividing stem cells, rapidly dividing transit amplifying cells (precursor cells), and differentiated cells (2–6). Malignant gliomas, for example, often contain both undifferentiated and differentiated cells and sometimes contain cells that express neuronal markers as well as cells that express glial markers, suggesting that they may contain multipotent neural stem cell (NSC)-like cells (7–9). Such mixed gliomas may develop from NSCs or from more restricted glial lineage cells that acquire multipotential stem-cell-like properties by mutation (10, 11). Yet, even normal oligodendrocyte precursor cells can be induced by extracellular signals in vitro to acquire stem-cell-like properties, and some NSCs in vivo express the astrocyte marker glial fibrillary acidic protein (GFAP) (12–17).
The presence of a small subpopulation of slowly dividing cancer stem cells might explain why so many cancers recur after treatment with irradiation or cytotoxic drugs, even when most of the cancer cells seem to be killed by the therapy. Usually, some cancer cells survive the treatment, and these surviving cells may be cancer stem cells, which may be not only resistant to the therapy but also essential for the malignancy of the cancer. It has been shown that various types of ATP-binding cassette (ABC) transporters, including those encoded by the multidrug-resistant (MDR) gene 1, the MDR protein (MRP), and the breast cancer-resistant protein 1 (BCRP1), contribute to drug resistance in many cancers by pumping the drugs out of the cell (18). Interestingly, some of these transporters are expressed also by many kinds of stem cells. BCRP1, for example, pumps out the fluorescent dye Hoechst 33342, identifying an unlabeled side population (SP), which is enriched for stem cells (19–22). Taken together, these findings suggest that cancers might contain an SP with the characteristics of stem cells.
Here we show that many established cancer cell lines contain SP cells, which are apparently maintained in normal serum-containing cultures over decades. We demonstrate that platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF) can maintain the SP cells in the C6 glioma cell line in the absence of serum and that the SP cells can produce both neuronal and glial cells. Finally, we show that FACS-sorted C6 SP cells, but not non-SP C6 cells, can produce both SP and non-SP cells in culture and form tumors in multiple tissues in nude mice, which contain both neurons and glia, indicating that these cells have the characteristics of multipotent cancer stem cells. Our findings suggest that the SP may be a general source of cancer stem cells, which need to be targeted for effective cancer therapy.
Materials and Methods
Chemicals. Chemicals were purchased from Sigma unless indicated otherwise. Recombinant cytokines were purchased from PeproTech (Rocky Hill, NJ) unless indicated otherwise.
Cell Culture. Various cancer cell lines were studied, including the rat glioma line C6, the human breast cancer line MCF-7, the human osteosarcoma lines U-20S and SaOS-2, the rat neuroblastoma line B104, and the human adenocarcinoma line HeLa. The cells were cultured in DMEM, supplemented with 10% FCS, 100 units/ml penicillin G, and 100 μg/ml streptomycin (GIBCO). In some experiments, C6 cells were cultured in serum-free DMEM containing 10 μg/ml bovine insulin, 100 μg/ml human transferrin, 100 μg/ml BSA, 60 ng/ml progesterone, 16 μg/ml putrescine, 40 ng/ml sodium selenite, 63 μg/ml _N_-acetylcysteine, 5 μM forskolin, 50 units/ml penicillin, and 50 μg/ml streptomycin (GIBCO), as well as 10 ng/ml bFGF, 10 ng/ml PDGF, or both. In all experiments, cells were maintained in 100-mm culture dishes (Nunc) at 37°C in a humidified 5% CO2/95% air atmosphere.
Flow Cytometry. To identify and isolate the SP cells in the cancer cell lines, we cultured the lines, as described above, in either FCS or serum-free culture medium with bFGF, PDGF, or both. The cells were removed from the culture dish with trypsin and EDTA (GIBCO BRL), washed, suspended at 106 cells per ml in DMEM containing 2% FCS (staining medium), and preincubated in a 1.5-ml Eppendorf tube at 37°C for 10 min. The cells were then labeled in the same medium at 37°C for 90 min with 2.5 μg/ml Hoechst 33342 dye (Molecular Probes), either alone or in combination with 50 μM verapamil (Sigma), which is an inhibitor of some (verapamil-sensitive) ABC transporters (19). Finally, the cells were counterstained with 1 μg/ml propidium iodide to label dead cells.
Then, 3–5 × 104 cells were analyzed in a FACSVantage fluorescence-activated cell sorter (Becton Dickinson) by using a dual-wavelength analysis (blue, 424–444 nm; red, 675 nm) after excitation with 350-nm UV light. Propidium iodide-positive dead cells (<15%) were excluded from the analysis. In the case of C6, the SP cells or non-SP cells were sorted and cultured in serum-free culture medium with bFGF and PDGF.
RNA Extraction and RT-PCR Assay. Cells were harvested, and poly(A)+ RNA was prepared by using a QuickPrep Micro mRNA purification kit (Amersham Biosciences) and reverse transcribed by using a First-Strand cDNA synthesis kit (Amersham Biosciences), as described (13). The RT-PCR was carried out in a 20-μl reaction mixture that contained 1 μl of cDNA as template, 1 μM specific oligonucleotide primer pair, and 0.5 unit of Taq DNA polymerase (Takara Shuzo, Kyoto). Cycle parameters for bcrp1, mdr1, or g3pdh cDNAs were 30 sec at 94°C, 30 sec at 60°C, and 60 sec at 72°C for 33, 32, and 25 cycles, respectively. The identity of the amplified products was checked by digestion with appropriate restriction enzymes. Oligonucleotide DNA primers were synthesized as follows. For rat bcrp1, we used sequences conserved between human and mouse: 5′, 5′-CCAGTTCCATGGCACTGGCCATA-3′ and 3′, 5′-CAGGGCCACATGATTCTTCCACA-3′. For rat mdr1, we used sequences conserved between human and mouse: 5′, 5′-GCAAAGCTGGAGAGATCCTCACCA-3′ and 3′, 5′-CAACATTTTCATTTCAACAACTCCTGC-3′. For rat g3pdh, the following sequences were used: 5′, 5′-ACCACAGTCCATGCCATCAC-3′ and 3′, 5′-TCCACCACCCTGTTGCTGTA-3′.
Immunostaining of Cultured Cells. To examine the expression of neuronal and glial markers in C6 SP cells cultured for 1 or 10 d, the cells were cultured overnight in chamber slides (Nunc) precoated with 1 μg/ml fibronectin (Invitrogen) and 15 μg/ml ornithine (Sigma). The cells were fixed with 2% paraformaldehyde for 10 min at room temperature, treated with 20% FCS, and then stained with the following mouse monoclonal antibodies: anti-GFAP (1:200; Sigma), anti-β-III tubulin (1:200; Sigma), anti-microtubule-associated protein 2 (MAP2; 1:500; Sigma), and anti-nestin (1:200; Pharmingen). The primary antibodies were detected with Texas red-conjugated goat anti-mouse IgM or IgG (1:100; Jackson ImmunoResearch) as described (13). The cells were counterstained with Hoechst 33342 to identify all nuclei.
Transplantation into Nude Mice. KSL/slc nude mice were purchased from SLC (Shizuoka, Japan). FACS-sorted C6 SP and non-SP cells were cultured for 7 d in bFGF plus PDGF, and 105 cells of each type were injected i.p. into three 4-wk-old female nude mice. The same experiment was repeated twice with similar results. The mice were killed 18 d after injection and examined for tumors, as described below. Mice were treated according to the guidelines of the Kumamoto University Animal Committee.
Hematocrit Analysis. Blood was collected in EDTA at a final concentration of 1 mM. Glass microcapillary tubes (Hirschmann) were filled with blood, capped with Parafilm, and centrifuged at 3,000 × g for 1 min, and the hematocrit was calculated as the proportion of the tube containing erythrocytes.
Immunostaining of Tissue Sections. Tumor-bearing tissues were fixed in 4% paraformaldehyde, embedded in Tissue-Tek OCT (optimal cutting temperature) compound, and then frozen at –20°C. Cryostat sections (12 μm) were cut, mounted on poly-l-lysine-coated slides, and air-dried for 24 h. To characterize the cells in tumors, the sections were treated with 10% normal goat serum (DAKO) for 30 min at room temperature and then stained with the following mouse monoclonal antibodies: anti-nestin antibody, anti-GFAP antibody, and anti-low molecular weight neurofilament antibody (1:200; Sigma). The primary antibodies were detected with Alexa 594-conjugated goat anti-mouse IgG (1:200; Molecular Probes), as described (13). The cells were counterstained with Hoechst 33342 to identify all nuclei. The stained sections were examined and photographed in an AX70 fluorescence microscope (Olympus, Orangeburg, NY).
Results
Many Cancer Cell Lines Contain a Small SP. To determine whether any of the six established cancer cell lines in our collection (see Materials and Methods) contained SP cells, we removed the cells from the culture dishes with trypsin and EDTA and stained them with the fluorescent dye Hoechst 33342, which has been shown to be extruded actively by the SP cells in various tissues by means of verapamil-sensitive ABC transporters. We then analyzed them by flow cytometry. As shown in Fig. 1, the C6 glioma cells contained 0.4% SP cells (A), the MCF7 breast cancer cells contained 2.0% SP cells (B), the B104 neuroblastoma cells contained 0.4% SP cells (C), and the HeLa carcinoma cells contained 1.2% SP cells (D). In each case, the SP population was decreased greatly by treatment with verapamil (Fig. 1), indicating that the populations were bona fide SPs. Thus, some cancer cell lines contain a small SP, despite having been maintained in culture for many years. We could not detect SP cells in the two human osteosarcoma lines (U-2OS and SaOS-2) that we tested (data not shown), suggesting that not all cancer cell lines have an SP. The rest of our experiments were focused on C6 cells.
Fig. 1.

Existence of SP cells in established cancer cell lines. Cells of the rat C6 glioma (A), human MCF7 breast carcinoma (B), rat B104 neuroblastoma (C), and human HeLa carcinoma (D) cell lines were labeled with the Hoechst 33342 and then analyzed by flow cytometry. (Lower) Results when the cells were treated with 50 μM verapamil during the labeling procedure. The SP, which disappears in the presence of verapamil, is outlined and shown as a percentage of the total cell population. These experiments were repeated at least three times with similar results.
C6 SP Cells Can Be Expanded in bFGF Plus PDGF. We then investigated which factor(s) can maintain C6 SP cells in serum-free culture media. We tested PDGF and bFGF (FGF-2) as possible candidates. PDGF is the main mitogen for oligodendrocyte precursor cells (23–25), whereas bFGF is a major mitogen for NSCs (26, 27). We first cultured unfractionated C6 cells on uncoated dishes in 10% FCS or in serum-free culture medium with PDGF, bFGF, or both. Surprisingly, the morphology of the cells was very different in the different culture conditions. In FCS or bFGF alone, the cells had a flat, fibroblast-like shape (Fig. 2 A and B). In PDGF, they had a round body but were still attached to the dish (Fig. 2_C_). In the presence of both bFGF and PDGF, by contrast, they formed floating, neurosphere-like cell aggregates after 10 d (Fig. 2_D_), just as CNS NSCs do under similar conditions (28). We then stained the cells with Hoechst 33342 and analyzed them by flow cytometry. When cultured in serum-free medium with both PDGF and bFGF, SP cells were maintained, and their proportion increased with time (Fig. 2 E, H, and I). By contrast, when cultured in either bFGF or PDGF alone, the C6 cells survived and proliferated, but by 3 wk few SP cells could be detected (Fig. 2 F, G, and I). These findings suggest that C6 SP cells can expand in a combination of bFGF and PDGF but cannot be maintained in either growth factor alone.
Fig. 2.

The roles of PDGF and bFGF on C6 SP cells. C6 cells were cultured in FCS (A) or serum-free medium with bFGF (B), PDGF (C), or bFGF plus PDGF (D) for 3 wk and were then photographed while alive in an inverted phase-contrast microscope. C6 SP cells were cultured for 3 wk in FCS (E) or serum-free medium with bFGF (F), PDGF (G), or both (H) and were analyzed by flow cytometry as shown in Fig. 1. (I) C6 cells were cultured in FCS (○), bFGF (▴), PDGF (▪), or bFGF plus PDGF (•) for the indicated times, and the proportion of SP cells was analyzed by flow cytometry. (J) C6 cells were cultured as in A_–_D for 3 wk, and then the expression of bcrp1, mdr1, or g3pdh mRNAs was analyzed by RT-PCR. All experiments were repeated at least three times with similar results. (Scale bar in D, 100 μm.)
It was shown previously that BCRP1, which is a verapamil-sensitive ABC transporter, exports the Hoechst 33342 from some types of SP cells (21, 22, 29). We, therefore, examined the expression of bcrp1 mRNA, as well as mdr1 mRNA, which encodes another ABC transporter, in C6 cells cultured in the four conditions described above. As shown in Fig. 2 J, bcrp1 mRNA expression was detected in the presence of both PDGF and bFGF but not in FCS or in bFGF or PDGF alone. In contrast, mdr1 mRNA was expressed in FCS, bFGF, or bFGF plus PDGF, although it was not expressed in PDGF alone. Together, these data suggest that BCRP1, but not MDR1, is responsible for the SP character of some C6 cells, as reported for some other types of SP cells (21, 22, 29).
To investigate the individual roles of PDGF and bFGF in expanding C6 SP cells, we cultured C6 cells in either bFGF or PDGF for 2 wk and then in bFGF plus PDGF for an additional 2 wk. We then stained the cells with Hoechst 33342 and analyzed them by flow cytometry. As shown in Fig. 3_B_, 1.8% of the cells cultured in bFGF and then in bFGF plus PDGF were SP cells. By contrast, although there seemed to be SP cells after culturing in PDGF and then in bFGF plus PDGF, these cells were still seen when stained in the presence of verapamil (Fig. 3_A_ Lower), indicating that they were not bona fide SP cells. We also examined the expression of both bcrp1 and mdr1 mRNA in C6 cells cultured under these two conditions. As shown in Fig. 3_D_, we detected bcrp1 mRNA in the cells that were cultured in bFGF and then in PDGF plus bFGF, but we could not do so in the cells cultured in PDGF and then in PDGF plus bFGF; by contrast, we could detect mdr1 mRNA in both conditions. It is possible, therefore, that bFGF on its own maintains C6 SP cells at an undetectable low level and that PDGF stimulates the proliferation of these cells.
Fig. 3.

Different roles of bFGF and PDGF on C6 SP cells. C6 cells were cultured in PDGF (A), bFGF (B), or both (C) for 2 wk and then expanded in bFGF plus PDGF for an additional 2 wk. They were then analyzed by flow cytometry as described for Fig. 1. (D) C6 cells were cultured as shown in A_–_C, and the expression of bcrp1, mdr1, or g3pdh mRNAs was analyzed by RT-PCR. All experiments were repeated at least three times with similar results.
C6 SP Cells Can Repopulate both SP and Non-SP C6 Cells. To compare the ability of C6 SP cells with the ability of non-SP cells to produce SP cells, we cultured C6 cells without FCS and in PDGF plus bFGF for 2 wk, stained them with Hoechst 33342, and sorted them into SP and non-SP fractions by flow cytometry, and then we expanded the SP and non-SP cells separately in the same medium for an additional 2 wk. As they proliferated, the cells in the two populations had different morphologies. The cells in the SP cultures formed floating spheres (Fig. 4_A_), whereas the cells in the non-SP cultures remained attached to the culture dishes and had a fibroblast-like morphology (Fig. 4_B_). When we restained the cells with Hoechst 33342 and reanalyzed them by flow cytometry, we found that the cultures initiated with SP cells contained both SP and non-SP cells, whereas the cultures initiated with non-SP cells contained only non-SP cells (Fig. 4_C_). These data are consistent with previous findings that only the SP cells in primary neurospheres can produce both SP and non-SP cells in culture (30). Furthermore, when single FACS-sorted SP cells were cultured alone in the same medium in a well of a 98-well culture plate, ≈70% of the cells proliferated and reformed floating spheres; in contrast, single non-SP cells cultured in the same way proliferated much more slowly, and almost all of the cells died by 3 wk (data not shown). Thus, apparently C6 SP cells, but not non-SP cells, can form floating spheres, proliferate extensively, and produce C6 SP cells in culture.
Fig. 4.

SP cells in the C6 glioma cell line can generate both SP and non-SP C6 cells. C6 cells were cultured in bFGF plus PDGF for 2 wk and then sorted by flow cytometry as described for Fig. 1. The SP (A) and non-SP (B) cells were then cultured separately in the same conditions for an additional 2 wk. The cells were then photographed while alive and analyzed by flow cytometry (C) as shown in Fig. 1. (Scale bar in B, 100 μm.)
C6SP Cells Can Produce both Neurons and Glia in Culture. To explore the developmental potential of C6 SP cells, we sorted SP cells, cultured them for 1 or 10 d in PDGF plus bFGF on ornithine/fibronectin-coated chamber slides, and then immunolabeled them for neuronal and glial markers. After 1 d, >90% of the cells were labeled for the NSC marker nestin (92 ± 2%), but none were labeled for the neuronal markers MAP2 or β-III tubulin or the astrocyte marker GFAP (Fig. 5_A_), suggesting that C6 SP cells might be undifferentiated NSC-like cells. After 10 d, however, ≈70% were immunolabeled for β-III tubulin, ≈5% for MAP2, and ≈7% for GFAP (Fig. 5_B_). Thus, C6 SP cells can apparently generate both neurons and glial cells in culture.
Fig. 5.
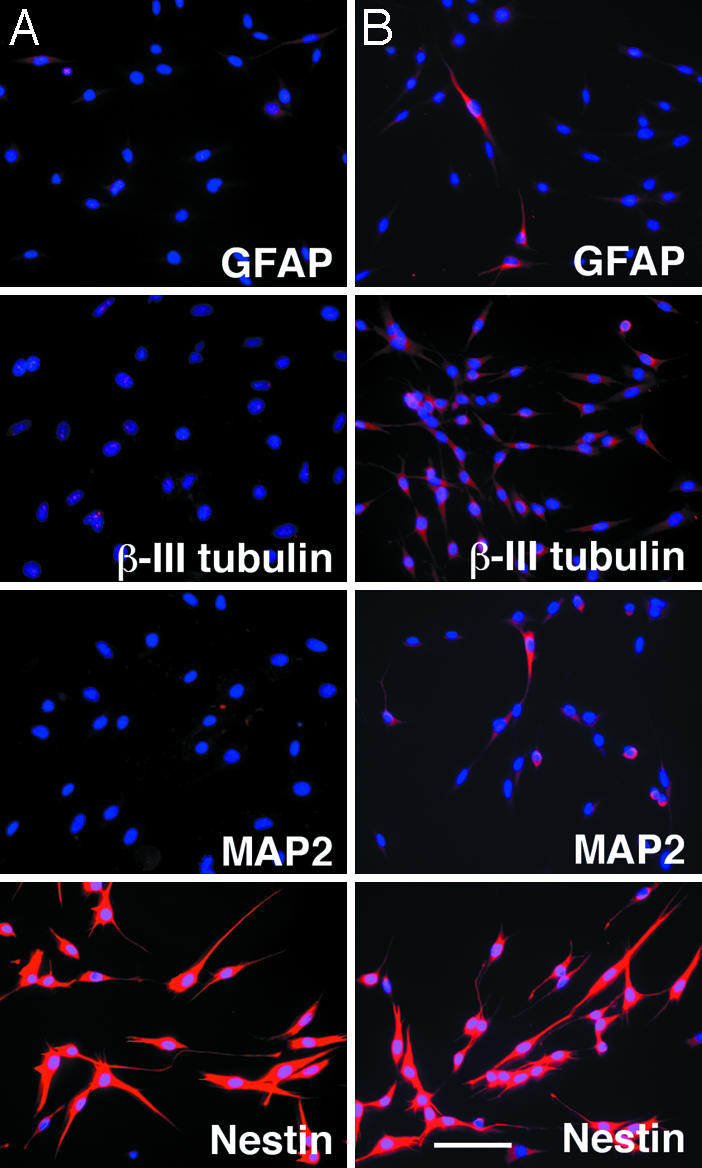
C6 SP cells can differentiate into both neurons and glia. C6 SP cells were prepared as described for Fig. 4_A_ and then cultured in serum-free medium with bFGF plus PDGF on ornithine/fibronectin-coated chamber slides for 1 (A) or 10 (B) d. Cells were then immunolabeled for neuronal and glial markers. (Scale bar in B, 30 μm.)
The Malignancy of C6 Cells in Vivo Is Largely Dependent on the SP Cells. To address whether SP and non-SP C6 cells differ in their malignancy, we sorted C6 cells growing in PDGF plus bFGF into SP and non-SP cells, expanded them in the same medium for 1 wk, and then injected 105 cells from either population i.p. into nude mice. After 18 d, all mice injected with cells from the SP cultures showed intraabdominal hemorrhages (Fig. 6 A and B) and tumor invasion into the mesentery (Fig. 6_C_, right), uterus (Fig. 6_D_, right) and lymph nodes. In four of six mice, there was also tumor invasion into the lungs (Fig. 6_E_, right). In contrast, after the same period, the cells from non-SP cultures had not formed tumors that invaded into these tissues, although we detected one s.c. tumor (Fig. 6_A_) and an occasional small metastasis in mesenteric lymph nodes (Fig. 6_C_, left, arrow). Thus, much of the malignancy of the C6 line apparently depends on SP cells.
Fig. 6.

Evidence of malignancy of C6 SP cells in vivo. SP or non-SP C6 cells (105) were injected i.p. into 4-wk-old female nude mice (A, right and left mice, respectively). The mice were killed 18 d later. (B_–_E) The hematocrit was measured (B), and tumor tissue in the mesentery (C), uterus (D), and lung (E) was photographed (right, SP-injected tissue; left, non-SP-injected tissue). Frozen sections of the tumors in the mesentery in SP-injected mice were immunolabeled for nestin (F), GFAP (G), or low molecular weight neurofilament (NF-L; H). The sections were labeled also with Hoechst 33342 (blue) to identify nuclei. (Scale bar in H, 50 μm.)
To determine whether C6 cells could produce neurons and glia in vivo, we fixed the tumor-bearing tissues in 4% paraformaldehyde, cut frozen sections, and immunolabeled them for neuronal and glial markers. As shown in Fig. 6, ≈35% of the cells in the tumors were immunolabeled for nestin (Fig. 6_F_), ≈30% for GFAP (Fig. 6_G_), and ≈15% for the neuronal marker NF-L (low molecular weight neurofilament) (Fig. 6_H_), suggesting that C6 SP cells can differentiate into both neurons and glia in vivo.
Discussion
Although the concept of cancer stem cells has been around for many years (31–33), there has been surprisingly little attention paid to them in recent years. Now, however, there is increasing evidence that at least some cancers contain stem cells that are largely responsible for the malignancy of the cancer. This possibility has been shown, for example, for acute myeloid leukemia (34) and breast carcinoma (35). Cancer stem cells are potentially extremely important because it is likely that they are often responsible for recurrences that occur after treatment. Thus, for cancer therapy to be curative, it probably must eliminate these cells, which is why it is important to identify and study cancer stem cells.
Our findings suggest that cancer stem cells may be present also in many cancer cell lines in culture, even when they have been maintained for many years. We can detect an SP in four of the six cancer cell lines that we have tested, and in most normal tissues, the stem cells are found in the SP. We find that the SP of the C6 glioma line, which is only 0.4% of the cells maintained in serum, has a number of characteristics that are expected of cancer stem cells. The SP cells in culture can self-renew and produce both SP and non-SP C6 cells, whereas the non-SP cells under the same culture conditions can produce non-SP cells only. Also, the C6 SP cells in culture can form neurospheres and produce neurons as well as glial cells, suggesting that they have NSC-like properties. Finally, they produce tumors in nude mice with high efficiency, whereas the non-SP C6 cells do not.
Besides providing further evidence for the existence of cancer stem cells, our finding that cancer stem cells can be enriched by isolating the SP in some cancer cell lines has a number of important implications. It may, for example, provide a simple and general strategy for isolating cancer stem cells. Because stem cells in any tissue are usually present in very small numbers, they are difficult to identify and even more difficult to isolate. There is a pressing need for methods to identify and isolate cancer stem cells so that their properties, including their sensitivity to various anti-cancer therapeutic agents, may be characterized.
Our findings that cancer SP cells can be maintained indefinitely in permanent cancer cell lines raise the possibility that these cell lines may provide attractive models for studying the molecular basis that defines stem cells. It seems likely that most of the stem cells and the other cells in a particular cancer cell line contain the same mutations, including the activation of specific protooncogenes and the loss of specific tumor-suppressor genes, but these changes are apparently not enough to make every cancer cell a stem cell. The difference between the stem cells and all of the other cells in a cancer is likely to be epigenetic, and cancer cell lines should be useful in trying to identify what these epigenetic differences are, which is a fundamental question in stem cell biology.
We find that the combination of PDGF and bFGF, but not either alone, allows C6 SP cells to expand in culture, as it has been shown previously that both mitogens can contribute to glial oncogenesis. For example, inhibition of bFGF signaling was shown not only to block cell proliferation but also to induce apoptosis in glioma cells (36, 37). Enforced expression of PDGF in either NSCs or GFAP-positive astrocytes was found to induce malignant gliomas (10, 11). We find that the two growth factors seem to have different roles in promoting the proliferation of C6 SP cells in culture. bFGF on its own seems to be able to maintain these cells but not to allow their expansion, whereas PDGF on its own apparently cannot maintain them, but in the presence of bFGF, helps to stimulate their proliferation.
It is of interest that C6 SP cells can generate neurons as well as glial cells; we and others (13, 16, 17) showed that oligodendrocyte precursor cells can be induced by extracellular signals to convert to an NSC-like phenotype and generate neurons and astrocytes, as well as oligodendrocytes. The molecular mechanisms involved in this conversion are unknown. C6 cells might provide an attractive model to study the molecular basis of this type of plasticity.
In summary, our studies underline the importance of cancer stem cells and suggest a simple strategy for isolating them. They also suggest that cancer cell lines may be important models for studying the basic biology of stem cells.
Acknowledgments
We thank Martin Raff for editing the manuscript and Yuko Saiki for technical assistance with flow cytometry. This work was supported in part by a grant-in-aid from the Japan Ministry of Education, Science, Sports, and Culture; the Nakajima Foundation; the Uehara Memorial Foundation; the Japan Brain Foundation; the Higo Foundation; the Yamada Science Foundation; and Merck Sharp and Dohme.
Abbreviations: SP, side population; PDGF, platelet-derived growth factor; bFGF, basic fibroblast growth factor; NSC, neural stem cell; GFAP, glial fibrillary acidic protein; ABC, ATP-binding cassette; MDR, multidrug-resistant; BCRP1, breast cancer-resistant protein 1; MAP2, microtubule-associated protein 2.
References
- 1.Hayflick, L. (1965) Exp. Cell Res. 37**,** 614–636. [DOI] [PubMed] [Google Scholar]
- 2.Poste, G. & Greig, R. (1982) Invasion Metastasis 2**,** 137–176. [PubMed] [Google Scholar]
- 3.Woodruff, M. F. (1983) Br. J. Cancer 47**,** 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sell, S. & Pierce, G. B. (1994) Lab. Invest 70**,** 6–22. [PubMed] [Google Scholar]
- 5.Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. (2001) Nature 414**,** 105–111. [DOI] [PubMed] [Google Scholar]
- 6.Tu, S. M., Lin, S. H. & Logothetis, C. J. (2002) Lancet Oncol. 3**,** 508–513. [DOI] [PubMed] [Google Scholar]
- 7.Wolf, H. K., Buslei, R., Blumcke, I., Wiestler, O. D. & Pietsch, T. (1997) Acta Neurophathol. 94**,** 436–443. [DOI] [PubMed] [Google Scholar]
- 8.Wharton, S. B., Chan, K. K., Hamilton, F. A. & Anderson, J. R. (1998) Neuropathol. Appl. Neurobiol. 24**,** 302–308. [DOI] [PubMed] [Google Scholar]
- 9.Katsetos, C. D., Del, V. L., Geddes, J. F., Aldape, K., Boyd, J. C., Legido, A., Khalili, K., Perentes, E. & Mork, S. J. (2002) J. Neuropathol. Exp. Neurol. 61**,** 307–320. [DOI] [PubMed] [Google Scholar]
- 10.Dai, C., Celestino, J. C., Okada, Y., Louis, D. N., Fuller, G. N. & Holland, E. C. (2001) Genes Dev. 15**,** 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holland, E. C. (2001) Curr. Opin. Neurol. 14**,** 683–688. [DOI] [PubMed] [Google Scholar]
- 12.Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. (1999) Cell 97**,** 703–716. [DOI] [PubMed] [Google Scholar]
- 13.Kondo, T. & Raff, M. (2000) Science 289**,** 1754–1757. [DOI] [PubMed] [Google Scholar]
- 14.Laywell, E. D., Rakic, P., Kukekov, V. G., Holland, E. C. & Steindler, D. A. (2000) Proc. Natl. Acad. Sci. USA 97**,** 13889–13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotz, M., Hartfuss, E. & Malatesta, P. (2002) Brain Res. Bull. 57**,** 777–788. [DOI] [PubMed] [Google Scholar]
- 16.Belachew, S., Chittajallu, R., Aguirre, A. A., Yuan, X., Kirby, M., Anderson, S. & Gallo, V. (2003) J. Cell Biol. 161**,** 169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nunes, M. C., Roy, N. S., Keyoung, H. M., Goodman, R. R., McKhann, G., Jiang, L., Kang, J., Nedergaard, M. & Goldman, S. A. (2003) Nat. Med. 9**,** 439–447. [DOI] [PubMed] [Google Scholar]
- 18.Gottesman, M. M., Fojo, T. & Bates, S. E. (2002) Nat. Rev. Cancer 2**,** 48–58. [DOI] [PubMed] [Google Scholar]
- 19.Goodell, M. A., Brose, K., Paradis, G., Conner, A. S. & Mulligan, R. C. (1996) J. Exp. Med. 183**,** 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doyle, L. A., Yang, W., Abruzzo, L. V., Krogmann, T., Gao, Y., Rishi, A. K. & Ross, D. D. (1998) Proc. Natl. Acad. Sci. USA 95**,** 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou, S., Schuetz, J. D., Bunting, K. D., Colapietro, A. M., Sampath, J., Morris, J. J., Lagutina, I., Grosveld, G. C., Osawa, M., Nakauchi, H., et al. (2001) Nat. Med. 7**,** 1028–1034. [DOI] [PubMed] [Google Scholar]
- 22.Zhou, S., Morris, J. J., Barnes, Y., Lan, L., Schuetz, J. D. & Sorrentino, B. P. (2002) Proc. Natl. Acad. Sci. USA 99**,** 12339–12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noble, M., Murray, K., Stroobant, P., Waterfield, M. D. & Riddle, P. (1988) Nature 333**,** 560–562. [DOI] [PubMed] [Google Scholar]
- 24.Raff, M. C., Lillien, L. E., Richardson, W. D., Burne, J. F. & Noble, M. D. (1988) Nature 333**,** 562–565. [DOI] [PubMed] [Google Scholar]
- 25.Richardson, W. D., Pringle, N., Mosley, M. J., Westermark, B. & Dubois-Dalcq, M. (1988) Cell 53**,** 309–319. [DOI] [PubMed] [Google Scholar]
- 26.Nurcombe, V., Ford, M. D., Wildschut, J. A. & Bartlett, P. F. (1993) Science 260**,** 103–106. [DOI] [PubMed] [Google Scholar]
- 27.Kitchens, D. L., Snyder, E. Y. & Gottlieb, D. I. (1994) J. Neurobiol. 25**,** 797–807. [DOI] [PubMed] [Google Scholar]
- 28.Chiasson, B. J., Tropepe, V., Morshead, C. M. & van der Kooy, D. (1999) J. Neurosci. 19**,** 4462–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunting, K. D. (2002) Stem Cells (Dayton) 20**,** 11–20. [DOI] [PubMed] [Google Scholar]
- 30.Hulspas, R. & Quesenberry, P. J. (2000) Cytometry 40**,** 245–250. [PubMed] [Google Scholar]
- 31.Makino, S. (1956) Ann. N.Y. Acad. Sci. 63**,** 813–883. [Google Scholar]
- 32.Kleinsmith, L. J. & Pierce, G. B. (1964) Cancer Res. 24**,** 1544–1552. [PubMed] [Google Scholar]
- 33.Gale, R. P. & Butturini, A. (1992) Leukemia 6**,** 80–85. [PubMed] [Google Scholar]
- 34.Wulf, G. G., Wang, R.-Y., Kuehnle, I., Weidner, D., Marini, F., Brenner, M. K., Andreeff, M. & Goodell, M. A. (2001) Blood 98**,** 1166–1173. [DOI] [PubMed] [Google Scholar]
- 35.Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J. & Clarke, M. F. (2003) Proc. Natl. Acad. Sci. USA 100**,** 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George, D. (2001) Semin. Oncol. 28**,** 27–33. [PubMed] [Google Scholar]
- 37.Aoki, T., Kato, S., Fox, J. C., Okamoto, K., Sakata, K., Morimatsu, M. & Shigemori, M. (2002) Int. J. Oncol. 21**,** 629–636. [PubMed] [Google Scholar]