Rapid diversification of coevolving marine Synechococcus and a virus (original) (raw)
Abstract
Marine viruses impose a heavy mortality on their host bacteria, whereas at the same time the degree of viral resistance in marine bacteria appears to be high. Antagonistic coevolution—the reciprocal evolutionary change of interacting species—might reconcile these observations, if it leads to rapid and dynamic levels of viral resistance. Here we demonstrate the potential for extensive antagonistic coevolution between the ecologically important marine cyanobacterium Synechococcus and a lytic virus. In a 6-mo-long replicated chemostat experiment, Synechococcus sp. WH7803 and the virus (RIM8) underwent multiple coevolutionary cycles, leading to the rapid diversification of both host and virus. Over the course of the experiment, we detected between 4 and 13 newly evolved viral phenotypes (differing in host range) and between 4 and 11 newly evolved Synechococcus phenotypes (differing in viral resistance) in each chemostat. Genomic analysis of isolates identified several candidate genes in both the host and virus that might influence their interactions. Notably, none of the viral candidates were tail fiber genes, thought to be the primary determinants of host range in tailed bacteriophages, highlighting the difficulty in generalizing results from bacteriophage infecting γ-Proteobacteria. Finally, we show that pairwise virus–host coevolution may have broader community consequences; coevolution in the chemostat altered the sensitivity of Synechoccocus to a diverse suite of viruses, as well as the virus’ ability to infect additional Synechococcus strains. Our results indicate that rapid coevolution may contribute to the generation and maintenance of Synechococcus and virus diversity and thereby influence viral-mediated mortality of these key marine bacteria.
Keywords: cyanophage, bacterial diversity, arms race
Viruses play a central role in marine biogeochemical cycles by killing as much as 20–40% of marine prokaryotic cells each day (1). At the same time, bacteria isolated from the marine environment are often resistant to many viral strains (2–4). These two sets of observations about marine virus–host interactions—high virus-induced host mortality on the one hand and a high degree of resistance on the other—have yet to be entirely reconciled (1, 5, 6). As a result, the controls on viral-mediated nutrient turnover in the oceans remain largely unknown.
In addition to their effects on host mortality, marine viruses affect host diversity in several ways. First, viruses contribute genetic material to their hosts via horizontal gene transfer (7). For instance, photosynthesis genes encoded by both cyanobacteria and their infecting viruses appear to have undergone repeated recombination events (8, 9). Second, viruses, like other pathogens, are thought to maintain host diversity by frequency dependence such that host strains (genotypes or taxa) that become relatively abundant are more susceptible to attack (the “kill the winner” hypothesis) (10–12). Finally, viruses might affect marine host diversity by promoting the emergence of viral resistance, leading to antagonistic coevolution (the reciprocal evolution of host resistance and viral infectivity) (13).
Experimental microcosms, focusing primarily on heterotrophic γ-Proteobacteria, such as Escherichia coli and Pseudomonas fluorescens, have demonstrated that viral-bacterial coevolution can quickly generate genetic and phenotypic diversity (14–17). At the same time, however, a classic microcosm study revealed constraints on the potential for coevolutionary “arms races” between viruses and heterotrophic bacteria (16). Using E. coli and various T-phages, that study found that after one or two “cycles” of coevolution (in which the host evolves resistance and the virus then evolves to overcome that resistance), a bacterial strain evolves resistance that the virus cannot overcome. The inability of the phage to continue to evolve host range mutants suggests that antagonistic coevolution is unlikely to be important in natural systems, at least over ecological time scales (16). Although extensive coevolution has recently been observed in P. fluorescens (15), possibly in E. coli (17), and indirectly in Archaea (18), the potential for such coevolution has never been tested in a marine system.
In the present study we tested the potential for antagonistic coevolution to generate diversity in an ecologically important, nonheterotrophic marine bacterium. Marine Synechococcus is a widespread unicellular cyanobacterium that may account for as much as 20% of primary productivity in coastal and upwelling regions (19, 20). Viruses that infect and lyse cyanobacteria (cyanophages) are also abundant and ubiquitous in the ocean (4, 21) and recycle as much as one-quarter of photosynthetically fixed carbon back to dissolved organic material pools (22). We conducted a 6-mo replicated chemostat experiment to address three main questions. First, what is the potential for coevolution and diversification between Synechoccocus and a lytic virus? In particular, we tested whether virus–host coevolution is limited to one or two coevolutionary cycles, with a cycle defined as the host evolving resistance to the virus and the virus evolving to overcome that resistance (in contrast to a predator–prey cycle defined by abundances). Second, can we identify candidate genes that underlie the phenotypic diversification observed in the chemostats? Currently, the genetic mechanisms underlying _Synechococcus_–virus interactions (or that of any marine viruses and its host) are largely unknown and based primarily on inferences from viruses infecting γ-Proteobacteria. Finally, do mutations that arise during pairwise coevolution have consequences for interactions with other Synechococcus and cyanophage strains? In natural settings, evolution occurs in the context of a diverse community; thus, we aimed to test whether antagonistic coevolution might alter these broader community interactions.
Results and Discussion
We established four chemostats with Synechococcus spp. WH7803 and RIM8 (family Myoviridae) and one control chemostat with Synechococcus only. Both Synechococcus and RIM8 persisted for the duration of the 6-mo experiment (∼170 generations). Host abundance was dramatically reduced after the addition of the virus, but recovered after 50–80 d. Thereafter, cell abundance remained similar to (chemostats C and D) or somewhat lower than (chemostats A and B) the abundance of Synechococcus in the control (no virus) chemostat (top third of the panels in Fig. 1).
Fig. 1.

Synechococcus sp. WH7803 and virus (RIM8) dynamics in the four replicate chemostats A_–_D. The top third of each panel plots the abundance of Synechococcus cells (red solid line) and infectious viral particles (blue dashed line) over time. For reference, the gray line is cell abundance in the control (no virus) chemostat. The middle of the panel indicates the host phenotypes detected at six time points, and the bottom of the panel, the viral phenotypes detected at the same six time points. Each chemostat was inoculated with ancestral virus (φ0) on day 0. Host range mutants are numbered in their order of infectivity (e.g., φ1–φ12), with higher numbers indicating the ability to infect a greater number of host phenotypes. Host phenotypes are labeled by their ability to resist infection by each host range mutant from the same chemostat. For example, S (sensitive to RIM8) is the ancestral host, and R0–2 is resistant to φ0, φ1, and φ2. We cannot determine whether a particular phenotype evolved directly from another phenotype, because some of the same phenotypes might have evolved more than once. Therefore, the dashed lines connecting the phenotypes are for ease of reading and make only the most parsimonious assumptions. The fully sequenced host and viral isolates are circled in A.
To test for the potential of antagonistic coevolution to lead to the diversification of marine Synechococcus and a lytic virus, we isolated single cells and viruses from each of the chemostats by colony isolation and plaque purification at numerous time points. Using these isolates, we conducted infection assays to determine the ability of the viral isolates to infect various Synechococcus isolates from the same chemostat. We then defined the phenotypes of the host and viral isolates based on these patterns of susceptibility and infectivity.
In the virus chemostats, Synechococcus spp. WH7803 and RIM8 underwent at least four cycles of coevolution, and we found no evidence indicating an imminent “end” to coevolution (bottom two-thirds of the panels in Fig. 1). We detected Synechococcus isolates that evolved resistance to the ancestral virus, viral isolates that infected this resistant strain, Synechococcus isolates that were resistant to the evolved virus, and so forth with continuing cycles of resistance and host range evolution. In each virus chemostat, we detected at least 4 newly evolved viral phenotypes over the course of the experiment, and in one chemostat, we found 13 viral phenotypes (Fig. 1_A_). At the same time, between 4 and 11 distinct Synechococcus phenotypes evolved as well. In contrast, in the control chemostat with no virus, we detected only the ancestral Synechococcus phenotype (sensitive to the ancestral virus and host range mutants from the other chemostats). Thus, Synechococcus and RIM8 underwent extensive antagonistic coevolution, resulting in the rapid (within fewer than 170 generations) appearance of considerable phenotypic diversity in both the host and virus. These results resemble the dynamics observed for P. fluorescens and the virus Phi 2 over ∼400 generations (15). They also lend support to recent evolutionary ecology models that predict the diversification and coexistence of multiple phenotypes (“quasispecies”) of bacteria and virus in chemostats (23).
Both the evolution of viral host range and the evolution of viral resistance in Synechococcus were directional. Viral infectivity (i.e., the ability to infect various Synechococcus isolates) of both isolates and whole populations increased over time, such that on average, viruses from later time points had a broader host range than those from earlier time points (Fig. 1 and Fig. S1). Similarly, Synechococcus cells isolated from later time points were on average more resistant than cells isolated from earlier time points in terms of both resistance to viral isolates and viral populations (Fig. 1 and Fig. S2). This directional arms race is typical of that observed in other laboratory studies of antagonistic coevolution (e.g., refs. 13, 17, but see ref. 24).
Although Synechococcus and virus evolution was generally directional, the phenotypic pattern was more variable in Synechococcus than in the virus. Viral phenotypes appeared to replace one another over time, such that we usually detected only one or two host range phenotypes in any one sample. At the same time, infectivity increased steadily (Fig. 1). In contrast, many Synechococcus phenotypes were observed at multiple time points, and up to three host phenotypes co-occurred in a sample. This greater phenotypic diversity among the hosts versus the viruses is particularly notable because at each time point we examined only two to four host isolates versus 10 viral isolates; thus, we likely greatly underestimated actual host diversity. Further, co-occurring host phenotypes often differed greatly in their degree of resistance (Fig. 1); for instance, in chemostat C, a sensitive (S) host phenotype and a R0–3 phenotype (resistant to four viral phenotypes) were present on the same day. The high degree of coexistence among host phenotypes suggests that resistance to RIM8 and its host range mutants is associated with a fitness cost (25, 26). Indeed, previous experiments have reported a fitness cost for viral resistance in Synechococcus (27) and Prochlorococcus (28).
To identify genes that might be involved in the interactions between Synechococcus and the virus, we sequenced the complete genomes of three viral isolates [the ancestral (φ0) phenotype from day 56, the φ9 phenotype from day 112, and the φ12 phenotype from day 167] and two Synechococcus isolates (an S phenotype from day 0 and an R0–13 phenotype from day 167) from chemostat A (circled in Fig. 1_A_). Among the viral genomes (∼171 kb each), we identified eight nonsynonymous nucleotide substitutions in genic regions, three indels in intergenic regions, and one large deletion of several genes; one of the nucleotide substitutions was in the φ9 isolate, and all of the other mutations were in the φ12 isolate (Table S1). Notably, none of these changes occurred in homologs to known tail fiber genes, which are thought to be involved in determining the specificity of myoviruses (T4-like phages) (29) and have been observed to evolve in other coevolution experiments (30). Half of the nucleotide substitutions were in a 130-bp region of a 972-bp gene of unknown function (RIM8.A.HR1_096), suggesting that the region is involved in viral–host interactions. To further explore this possibility, we sequenced the region in viral isolates obtained at each sampling time in the four chemostats.
Extensive parallel evolution in RIM8.A.HR1_096 was observed among the four chemostats, similar to that found in other viral evolution studies (31, 32). Of the 87 viral isolates genoytped, 71 had one or more substitutions in RIM8.A.HR1_096 compared with the ancestral RIM8 isolate. Nucleotide substitutions were observed at 11 positions in the gene (Table S2). All substitutions were nonsynonymous and led to changes in 10 amino acid sites, although not all changes were to the same amino acid (Table 1). Most (>60%) of the amino acid changes were observed in more than one chemostat.
Table 1.
Comparison of viral phenotypes and RIM8.A.HR1_096 genotypes in the four chemostats
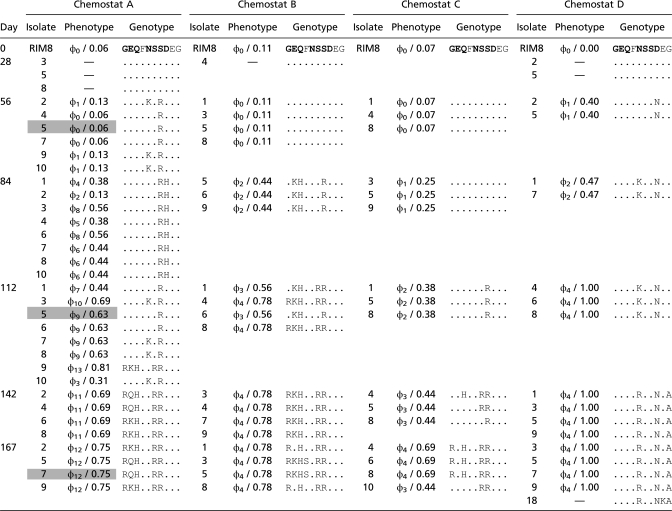
This large number of parallel, nonsynonymous substitutions suggests that the region was under strong positive selection. Such a pattern could be caused by host range adaptation or, alternatively, general adaptation to the chemostat environment. However, the number of amino acid changes was also highly correlated with the isolate's infectivity (_R_2 = 0.50–0.96, P < 0.0001 for all chemostats) (Fig. S3), strongly suggesting that RIM8.A.HR1_096 is involved in determining viral host range. At the same time, eight viruses isolated from chemostat A on day 84 had the same RIM8.A.HR1_096 amino acid sequence, but represented five phenotypes (Table 1), indicating the role of additional mutations in other parts of the genome. Thus, host range determination in this virus appears to involve multiple genes (in addition to this candidate gene), similar to that observed in heterotrophic bacteriophage (30).
Viral resistance in Synechococcus also appears to be a complex trait encoded by multiple loci. Sequencing of the two host genomes revealed four changes (in four different coding regions) between the ancestral Synechococcus and the derived isolate that was resistant to 13 viral phenotypes: three nucleotide substitutions and a single nucleotide deletion, leading to an early stop codon (Table S3). As in the viral genomes, the mutations observed were nonsynonymous, suggesting that they are adaptive. Only one mutation—a point mutation in the glucose-1-phosphate thymidylyltransferase gene (SynWH7803_0102)—was found in isolates from additional chemostats (using PCR amplification and sequencing), further supporting the idea that it is an adaptive mutation (Table S4). This core gene (all sequenced Synechoccocus genomes contain homologs) is located in a highly variable region, identified as genomic island 1 (ISL1) in Synechococcus sp. WH7803 (33). The gene encodes the first enzyme in the dTDP-l-rhamnose biosynthesis pathway. l-rhamnose is one of the important residues of the O antigen of LPS in Gram-negative organisms (34) and has been detected in the LPS of marine Synechococcus (35). LPS is also involved in bacteriophage attachment in WH7803 (36). The two additional mutations were found in genes encoding a glycosyltransferase, which could be involved in LPS biosynthesis (37), and a histidine kinase, which could regulate proteins exposed outside the cell wall to which viruses attach (38) (Table S3).
Two of the mutations were not observed in other chemostat A Synechococcus isolates (SynWH7803_0140 and SynWH7803_1555). The lack of shared mutations among the chemostat A Synechococcus isolates is notable, because the isolates are also resistant to subsets of the same viruses (Fig. 1_A_). Thus, Synechococcus appears to evolve resistance to some viruses in more than one way, such that mutations in entirely different genes can confer resistance to the same virus (39). This finding, in contrast to that seen in the virus, also supports the hypothesis that parallel evolution within a gene occurs more often in bacteriophages than in their hosts (40).
In seawater, viral–host coevolution occurs in the context of a diverse community of Synechococcus and virus strains. To test whether coevolution in this experimental setting has potential consequences in this broader community, we challenged the evolved Synechococcus populations with 31 genetically distinct myovirus strains isolated from Rhode Island (RI) waters. Over the course of the experiment, the number of RIM strains to which the Synechococcus populations were resistant increased (_F_1,18 = 90.83, P < 0.0001, ANCOVA) (Table S5). Similar trends were observed with Synechococcus isolates, suggesting that this increased resistance is related to the increased resistance of individual cells (Table S5). Furthermore, resistance to similar RI strains evolved in parallel among the four chemostats; the phenotypic profile of Synechococcus populations differed significantly by sampling day (_R_2 = 0.742, P = 0.006, ANOSIM), but not by chemostat (_R_2 = 0.33, P = 0.186) (Fig. 2). Such pleiotropic effects—with resistance to one virus also conferring resistance to other viruses—have recently been reported in other marine bacteria, including Prochlorococcus (28) and Flavobacterium (41). Notably, in both Synechococcus and Prochlorococcus, there are examples where gaining resistance to one virus simultaneously increases sensitivity to other viruses (28, 39); for instance, in the present study, some of the Synechococcus cells coevolving with RIM8 lost their initial resistance to RIM26 (Table S5).
Fig. 2.

Dendrogram of the phenotype of the chemostat Synechococcus populations, where phenotype is defined as the sensitivity or resistance to 31 virus strains from Rhode Island waters (Table S5). The populations are indicated by chemostat (A, B, C, D, and X for the no-virus control) and sampling day. The number of virus strains to which the populations are resistant is noted. Of note, the Synechococcus population from the control chemostat at the end of the experiment (X-167) was similar to populations from day 0 of the experiment, indicating that resistance did not increase in the absence of virus.
Coevolution with Synechococcus sp. WH7803 also altered the interactions of the virus with other Synechococcus strains. We challenged cyanophage isolates from each chemostat with Synechococccus sp. WH8018 and five strains derived either from WH7803 or WH8108. These derived strains were previously selected for resistance to other RIM viruses (39), but were resistant to the ancestral RIM8 virus as well. Coevolution altered the ability of RIM8 to infect three of the five previously resistant strains, including those derived from the genetically distinct Synechococcus sp. WH8018 (Table 2).
Table 2.
Susceptibility of Synechococcus spp. WH7803 and WH8018 and five derived RIM8-resistant strains to viral isolates from the chemostats
| Chemostat | Day | Isolate | Phenotype | WH7803 | WH7803R17 | WH8018 | WH8018R16 | WH8018R19 | WH8018R20 | WH8018R34 |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | RIM8 | S | S | R | S | R | R | R | R | |
| A | 28 | 5 | — | S | R | S | R | R | R | R |
| 28 | 8 | — | S | R | S | R | R | R | R | |
| 56 | 5 | Φ0 | S | R | S | R | R | R | S | |
| 112 | 5 | Φ9 | S | S | S | R | R | R | S | |
| 167 | 7 | Φ12 | S | S | S | S | R | R | S | |
| B | 56 | 1 | Φ0 | S | R | S | R | R | R | R |
| 112 | 4 | Φ4 | S | S | S | S | R | R | S | |
| 167 | 3 | Φ4 | S | S | S | S | R | R | S | |
| C | 56 | 4 | Φ0 | S | R | S | R | R | R | R |
| 112 | 8 | Φ2 | S | R | S | R | R | R | S | |
| 167 | 10 | Φ3 | S | R | S | R | R | R | S | |
| D | 28 | 2 | — | S | R | S | R | R | R | R |
| 28 | 5 | — | S | R | S | R | R | R | R | |
| 56 | 5 | Φ1 | S | R | S | R | R | R | S | |
| 167 | 9 | Φ4 | S | S | S | R | R | R | S |
Conclusions
The potential for rapid and extensive antagonistic coevolution between marine Synechococcus and their viruses has various implications. In particular, we have shown that persistent, coevolutionary dynamics quickly generate and maintain diversity in this ecologically important aquatic bacterium. Furthermore, although Synechococcus readily evolved viral resistance, the cells are often susceptible to co-occurring viral genotypes. Such dynamics offer a possible explanation for the paradoxical observations of simultaneously high host mortality and high viral resistance, given that resistance may be highly temporally dynamic.
Coevolution within a broader community context might further maintain Synechococcus diversity. Within the chemostat, _Synechococcus_–virus coevolution led to a generally directional arms race. However, this pairwise coevolution also altered interactions with host and virus strains not in the chemostat. Coevolution conferred sensitivity in Synechococcus sp. WH7803 to at least one other RIM virus, whereas RIM8 developed the ability to infect resistant strains of a different Synechococcus species. This suggests that in a diverse natural community, pairwise coevolution may be interrupted when changes in resistance and/or host range of the coevolving pair alter their interactions with other members of the community. These new interactions might then aid in the coexistence of Synechococcus and infecting viruses by preventing a particular host strain from “winning” a directional arms race. New theoretical models that consider virus–host coevolution in a community context, as well as the complex genetic mechanisms observed here, are needed to provide insight into marine virus–host dynamics.
The phenotypic diversification in Synechococcus and RIM8 was driven by small genetic changes across a variety of genes. Although genetic manipulations are needed to pinpoint the molecular mechanisms involved in virus–host interactions, we identified several candidate genes that may provide alternative markers for host–phage interactions. This study identifies host range genes in cyanophage, and notably none of these candidates are homologous to tail fiber genes in known bacteriophages. Indeed, the gene with the most nucleotide substitutions in the present study (RIM8.A.HR1_096) is not even conserved among cyanophages and apparently exists in only two of the other fully sequenced cyanophage genomes (Syn1 and P-HM1). Thus, the genetic mechanisms underlying virus–host interactions may be quite different from those previously characterized for terrestrial γ-Proteobacteria systems.
Our findings also confirm previous studies indicating that neither Synechococcus susceptibility nor cyanophage infectivity is correlated with the genotype of highly conserved genes (39, 42). Thus, coevolutionary and/or frequency-dependent (i.e., kill the winner) dynamics are unlikely to be observed using conserved genetic markers such as bacterial _rpo_C and viral _g_20. Instead, taxa defined by these markers may be composed of multiple co-occurring genotypes, differing in their sensitivity (for the host) or infectivity (for the virus) (3, 28, 43, 44). Such fine-scale dynamics may explain why in natural communities, particular bacterial taxa remain dominant for extended periods despite high viral abundances (45).
In conclusion, viral coevolution led to rapid diversification and a highly dynamic degree of viral resistance in Synechococcus. Selection for viral resistance is known to alter nutrient acquisition rates in marine bacteria (27, 41), and in laboratory experiments, the fraction of Synechococcus cells that are resistant to co-occurring cyanophages influences nutrient turnover rates (46). Taken together, this evidence suggests that rapid coevolutionary processes may influence how viruses mediate nutrient cycling in the ocean.
Materials and Methods
Strains and Chemostats.
Synechococcus sp. WH7803 was obtained from the Woods Hole Collection of Cyanobacteria (Woods Hole Oceanographic Institution). RIM8 was isolated from Narragansett Bay, Rhode Island in 2000 (47). Cells derived from a single colony of WH7803 were used to inoculate five chemostats; this Synechococcus culture is nonaxenic, containing heterotrophic bacteria (46). An artificial seawater medium (48) was supplied to each 35-mL chemostat at a dilution of ∼1.02/d. The contents were stirred with magnetic stir bars and maintained at 23.0 ºC on a 14:10 light:dark cycle at 10 μE m−2 s−1. The cells were allowed to equilibrate for 2 wk before RIM8 (derived from a single plaque) was added to four of the chemostats. The fifth chemostat with no virus was maintained as a control.
Sampling.
After the addition of viruses (day 0), cell and virus abundance was estimated every day for the first week, then approximately once a week thereafter until day 167. Cells were counted by epifluorescence microscopy and viruses were counted by counting plaques on dilution plates (47). Single cells and viral particles were isolated from each of the chemostats by colony isolation and plaque purification (39) at eight time points. At each time point, 6–10 Synechococcus colonies and ∼10 viral plaques were chosen and grown in liquid media (with ancestral host for the virus). To examine whether the isolate results were biased by selecting for plaques on the ancestral host or colony formation on a solid media, host and virus populations were collected as well. Synechococcus populations (containing virus) were collected by transferring an aliquot directly from the chemostats into the AN medium. Viral populations were obtained by filtering 1–2 mL of the chemostat sample through a 0.22-μm filter. For long-term storage, lysates were stored in cryogenic tubes at 4 ºC, and Synechococcus cultures were stored in 7.5% (vol/vol) DMSO at −80 ºC.
Phenotype Assays.
Resistance and infectivity were assayed by challenging the Synechococcus isolates and populations with the viral isolates and/or populations. All assays were carried out in duplicate in 24-well microtiter plates as described previously (39). Two control wells containing cells but no virus were also included on each plate. Plates were incubated under constant illumination at room temperature and scored once a week for 4 wk. Synechococcus cultures were scored as susceptible (and the virus as infective) if growth was visibly inhibited compared with the control wells. Five sets of these resistance/infectivity assays were performed: (i) host isolates vs. viral isolates, (ii) host isolates vs. viral populations, (iii) hosts from the no-virus chemostat vs. ancestral virus, (iv) host populations and isolates vs. other RI viruses, and (v) viral isolates vs. other Synechococcus species. See the SI Materials and Methods for details of these assays.
Genetic Analysis.
The genomes of three viral isolates from chemostat A were fully sequenced. Genomic DNA was purified, sequenced by 454 FLX technology, assembled, and annotated as described previously (49, 50). Coverage ranged from 44% to 60% for the three genomes. The sequences—JF974288, the ancestral (φ0) phenotype from day 56; JF974289, the φ9 phenotype from day 112; and HQ317385, the φ12 phenotype from day 167—are available from GenBank. To further determine genetic variability among the viral populations in the chemostats, the variable portion of RIM8.A.HR1_096 was PCR-amplified and sequenced from the ancestral RIM8 isolate and 87 viral isolates with representatives from each time point in each chemostat (Table 1 and Table S6).
Two Synechococcus isolates from chemostat A also were fully sequenced, including the ancestral Synechococcus inoculated into the chemostats (S in Fig. 1_A_) and a derived isolate (R0–13 in Fig. 1_A_). The regions surrounding four identified mutations were PCR-amplified (Table S6) and sequenced in 13 isolates from chemostat A (and confirmed in the fully sequenced isolates). If the mutation was found to be variable in chemostat A, then 19 additional isolates were sequenced from the other chemostats. Further details of the bacterial genome sequencing and analysis are provided in the SI Materials and Methods.
Supplementary Material
Supporting Information
Acknowledgments
We thank the Broad Institute's Genome Sequencing, Assembly, Annotation, and Finishing teams for their efforts in generating the genomic data reported here. We also thank Lisa Crummett, Brandon Gaut, China Hanson, Adam Martiny, and Olivier Tenaillon for constructive suggestions on the manuscript. This research was supported by National Science Foundation Grants OCE-0314523 and OCE-1029684 (to M.F.M.) and OCE-0315645 and OCE-1031783 (to J.B.H.M.), the Gordon and Betty Moore Foundation (awards to J.B.H.M., M.R.H., and S.C.S.), Rhode Island Experimental Program to Stimulate Competitive Research Grant 0554548 (to M.F.M.), and the Roger Williams University Foundation to Promote Scholarship (to M.F.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Database deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. JF974288, JF974289, and HQ317385).
References
- 1.Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–548. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- 2.Waterbury JB, Valois FW. Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl Environ Microbiol. 1993;59:3393–3399. doi: 10.1128/aem.59.10.3393-3399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmfeldt K, Middelboe M, Nybroe O, Riemann L. Large variabilities in host strain susceptibility and phage host range govern interactions between lytic marine phages and their Flavobacterium hosts. Appl Environ Microbiol. 2007;73:6730–6739. doi: 10.1128/AEM.01399-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan MB, Waterbury JB, Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–1051. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- 5.Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 6.Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Brüssow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeidner G, et al. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ Microbiol. 2005;7:1505–1513. doi: 10.1111/j.1462-2920.2005.00833.x. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan MB, et al. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4:e234. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thingstad TF. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr. 2000;45:1320–1328. [Google Scholar]
- 11.Janzen DH. Herbivores and the number of tree species in tropical forests. Am Nat. 1970;104:501–527. [Google Scholar]
- 12.Hamilton WD. Pathogens as causes of genetic diversity in their host populations. In: Anderson RM, May RM, editors. Population Biology of Infections Diseases. New York: Springer; 1982. pp. 269–296. [Google Scholar]
- 13.Buckling A, Rainey PB. Antagonistic coevolution between a bacterium and a bacteriophage. Proc Biol Sci. 2002;269:931–936. doi: 10.1098/rspb.2001.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paterson S, et al. Antagonistic coevolution accelerates molecular evolution. Nature. 2010;464:275–278. doi: 10.1038/nature08798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockhurst MA, Morgan AD, Fenton A, Buckling A. Experimental coevolution with bacteria and phage: The Pseudomonas fluorescens–Phi2 model system. Infect Genet Evol. 2007;7:547–552. doi: 10.1016/j.meegid.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Lenski RE, Levin BR. Constraints on the coevolution of bacteria and virulent phage: A model, some experiments, and predictions for natural communities. Am Nat. 1985;125:585–602. [Google Scholar]
- 17.Mizoguchi K, et al. Coevolution of bacteriophage PP01 and Escherichia coli O157:H7 in continuous culture. Appl Environ Microbiol. 2003;69:170–176. doi: 10.1128/AEM.69.1.170-176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Held NL, Whitaker RJ. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ Microbiol. 2009;11:457–466. doi: 10.1111/j.1462-2920.2008.01784.x. [DOI] [PubMed] [Google Scholar]
- 19.Li WKW. Primary production of prochlorophytes, cyanobacteria, and eukaryotic ultraphytoplankton: Measurements from flow cytometric sorting. Limnol Oceanogr. 1994;39:169–175. [Google Scholar]
- 20.Jardillier L, Zubkov MV, Pearman J, Scanlan DJ. Significant CO2 fixation by small prymnesiophytes in the subtropical and tropical northeast Atlantic Ocean. ISME J. 2010;4:1180–1192. doi: 10.1038/ismej.2010.36. [DOI] [PubMed] [Google Scholar]
- 21.Williamson SJ, et al. The Sorcerer II Global Ocean Sampling Expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE. 2008;3:e1456. doi: 10.1371/journal.pone.0001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilhelm SW, Suttle CA. Viruses and nutrient cycles in the sea. Bioscience. 1999;49:781–788. [Google Scholar]
- 23.Weitz JS, Hartman H, Levin SA. Coevolutionary arms races between bacteria and bacteriophage. Proc Natl Acad Sci USA. 2005;102:9535–9540. doi: 10.1073/pnas.0504062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gómez P, Buckling A. Bacteria–phage antagonistic coevolution in soil. Science. 2011;332:106–109. doi: 10.1126/science.1198767. [DOI] [PubMed] [Google Scholar]
- 25.Chao L, Levin BR, Stewart FM. A complex community in a simple habitat: An experimental study with bacteria and phage. Ecology. 1977;58:369–378. [Google Scholar]
- 26.Frank SA. Models of plant–pathogen coevolution. Trends Genet. 1992;8:213–219. doi: 10.1016/0168-9525(92)90236-w. [DOI] [PubMed] [Google Scholar]
- 27.Lennon JT, Khatana SAM, Marston MF, Martiny JBH. Is there a cost of virus resistance in marine cyanobacteria? ISME J. 2007;1:300–312. doi: 10.1038/ismej.2007.37. [DOI] [PubMed] [Google Scholar]
- 28.Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. Genomic island variability facilitates Prochlorococcus–virus coexistence. Nature. 2011;474:604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
- 29.Tétart F, Desplats C, Krisch HM. Genome plasticity in the distal tail fiber locus of the T-even bacteriophage: Recombination between conserved motifs swaps adhesin specificity. J Mol Biol. 1998;282:543–556. doi: 10.1006/jmbi.1998.2047. [DOI] [PubMed] [Google Scholar]
- 30.Scanlan PD, Hall AR, Lopez-Pascua LDC, Buckling A. Genetic basis of infectivity evolution in a bacteriophage. Mol Ecol. 2011;20:981–989. doi: 10.1111/j.1365-294X.2010.04903.x. [DOI] [PubMed] [Google Scholar]
- 31.Bollback JP, Huelsenbeck JP. Parallel genetic evolution within and between bacteriophage species of varying degrees of divergence. Genetics. 2009;181:225–234. doi: 10.1534/genetics.107.085225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. Different trajectories of parallel evolution during viral adaptation. Science. 1999;285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]
- 33.Dufresne A, et al. Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 2008;9:R90. doi: 10.1186/gb-2008-9-5-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blankenfeldt W, Asuncion M, Lam JS, Naismith JH. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA) EMBO J. 2000;19:6652–6663. doi: 10.1093/emboj/19.24.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder DS, Brahamsha B, Azadi P, Palenik B. Structure of compositionally simple lipopolysaccharide from marine synechococcus. J Bacteriol. 2009;191:5499–5509. doi: 10.1128/JB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zwirglmaier K, Spence E, Zubkov MV, Scanlan DJ, Mann NH. Differential grazing of two heterotrophic nanoflagellates on marine Synechococcus strains. Environ Microbiol. 2009;11:1767–1776. doi: 10.1111/j.1462-2920.2009.01902.x. [DOI] [PubMed] [Google Scholar]
- 37.Osborn MJ, Gander JE, Parisi E. Mechanism of assembly of the outer membrane of Salmonella typhimurium: Site of synthesis of lipopolysaccharide. J Biol Chem. 1972;247:3973–3986. [PubMed] [Google Scholar]
- 38.Wang SP, Sharma PL, Schoenlein PV, Ely B. National Academy of Sciences A histidine protein kinase is involved in polar organelle development in Caulobacter crescentus. Proc Natl Acad Sci USA. 1993;90:630–634. doi: 10.1073/pnas.90.2.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoddard LI, Martiny JBH, Marston MF. Selection and characterization of cyanophage resistance in marine Synechococcus strains. Appl Environ Microbiol. 2007;73:5516–5522. doi: 10.1128/AEM.00356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woods R, Schneider D, Winkworth CL, Riley MA, Lenski RE. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci USA. 2006;103:9107–9112. doi: 10.1073/pnas.0602917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middelboe M, Holmfeldt K, Riemann L, Nybroe O, Haaber J. Bacteriophages drive strain diversification in a marine Flavobacterium: Implications for phage resistance and physiological properties. Environ Microbiol. 2009;11:1971–1982. doi: 10.1111/j.1462-2920.2009.01920.x. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan MB, et al. Portal protein diversity and phage ecology. Environ Microbiol. 2008;10:2810–2823. doi: 10.1111/j.1462-2920.2008.01702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marston MF, Amrich CG. Recombination and microdiversity in coastal marine cyanophages. Environ Microbiol. 2009;11:2893–2903. doi: 10.1111/j.1462-2920.2009.02037.x. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Brito B, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4:739–751. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- 45.Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459:193–199. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 46.Lennon JT, Martiny JBH. Rapid evolution buffers ecosystem impacts of viruses in a microbial food web. Ecol Lett. 2008;11:1178–1188. doi: 10.1111/j.1461-0248.2008.01225.x. [DOI] [PubMed] [Google Scholar]
- 47.Marston MF, Sallee JL. Genetic diversity and temporal variation in the cyanophage community infecting marine Synechococcus species in Rhode Island's coastal waters. Appl Environ Microbiol. 2003;69:4639–4647. doi: 10.1128/AEM.69.8.4639-4647.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waterbury J, Willey J. Isolation and growth of marine planktonic cyanobacteria. Methods Enzymol. 1988;167:100–105. [Google Scholar]
- 49.Sullivan MB, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henn MR, et al. Analysis of high-throughput sequencing and annotation strategies for phage genomes. PLoS ONE. 2010;5:e9083. doi: 10.1371/journal.pone.0009083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information