Analyzing Endodontic Infections by Deep Coverage Pyrosequencing (original) (raw)
Abstract
Bacterial diversity in endodontic infections has not been sufficiently studied. The use of modern pyrosequencing technology should allow for more comprehensive analysis than traditional Sanger sequencing. This study investigated bacterial diversity in endodontic infections through taxonomic classification based on 16S rRNA gene sequences generated by 454 GS-FLX pyrosequencing and conventional Sanger capillary sequencing technologies. Sequencings were performed on 7 specimens from endodontic infections. On average, 47 vs. 28,590 sequences were obtained per sample for Sanger sequencing vs. pyrosequencing, representing a 600-fold difference in “depth-of-coverage”. Based on Ribosomal Database Project (RDP II) Classifier analysis, pyrosequencing identified 179 bacterial genera in 13 phyla, which was significantly more than Sanger sequencing. The phylum Bacteroidetes was the most prevalent bacterial phylum. These results indicate that bacterial communities in endodontic infections are more diverse than previously demonstrated. In addition, deep-coverage pyrosequencing of the 16S rRNA gene revealed low-abundance micro-organisms with potential clinical implications.
Keywords: endodontic infection, pyrosequencing, 16S rRNA gene, bacterial diversity, taxonomy
Introduction
Microbial diversity associated with the human body is much greater than previously thought (Turnbaugh et al., 2007; Hsiao and Fraser-Liggett, 2009). Clonal analysis of the 16S ribosomal RNA gene has identified over 700 species in the human oral cavity (Paster et al., 2006). The application of this approach to endodontic infections has revealed previously unrecognized bacterial diversity (Rolph et al., 2001; Munson et al., 2002; Saito et al., 2006; Sakamoto et al., 2006; Vickerman et al., 2007). Thus far, more than 400 species-level bacterial taxa belonging to 9 phyla have been reported in various forms of endodontic infections, namely, Firmicutes, Bacteroidetes, Spirochaetes, Actinobacteria, Fusobacteria, Proteobacteria, Synergistes, TM7, and SR1 (Rocas and Siqueira, 2008; Siqueira and Rocas, 2009).
16S rRNA gene-based clonal analysis utilizes the conventional Sanger capillary sequencing technique (Weisburg et al., 1991), which is limited in “depth of coverage” because it is expensive and labor-intensive. As a result, the less-abundant micro-organisms that constitute the majority of the biodiversity cannot be selected for sequencing (Petrosino et al., 2009). Pyrosequencing is a next-generation deep-coverage sequencing technique (Ronaghi et al., 1998). The 454 pyrosequencing technology facilitates metagenomic investigation through a high-throughput, deep-coverage, massively parallel and multiplex barcoded approach (Margulies et al., 2005). As a result, an in-depth study of endodontic microbial diversity can be carried out with amplified bacterial 16S rRNA sequences without cloning bias. Recently, a pyrosequencing study in the human oral cavity revealed bacterial diversity at least one order of magnitude higher than previously described (Keijser et al., 2008).
The purpose of this study was to investigate bacterial diversity in endodontic infections through taxonomic classification of 16S rRNA tag sequences by the current generation GS FLX pyrosequencing as well as by conventional Sanger capillary sequencing. Taxonomic information is critical in relating 16S rRNA gene sequences in a community to their hosting micro-organisms, with potential physiological and pathogenic functions (Wang et al., 2007). At this time, we are not aware of any other work that has applied this generation of pyrosequencing technology to the investigation of endodontic infections.
Materials & Methods
Participant Recruitment, Sample Collection, and DNA Extraction
All clinical procedures were approved by the institutional review board at the University of Maryland, Baltimore. Seven teeth in different individuals who had endodontic infection were included. All teeth had periapical lesions that were 3-10 mm in diameter, but did not have active periodontal disease. Two individuals had lesions that were deemed symptomatic, as determined by a score of 30% or more on a visual analogue scale, whereas the rest were asymptomatic. One tooth had a pre-operative sinus tract, and another one had received previous endodontic treatment and was receiving a re-treatment due to persistent asymptomatic disease. Two teeth were mandibular molars, 4 were maxillary premolars, and 1 was a maxillary anterior tooth. Following participants’ informed consent, microbial specimens were collected from the root canal space under strict aseptic conditions according to an established protocol (Fouad et al., 2002). The total genomic DNA was extracted with the use of a QIAamp DNA mini kit (Qiagen, Valencia, CA, USA).
16S rRNA Gene Amplification, Clone Library Construction, and Sanger Sequencing
The 16S rRNA genes were amplified under standardized conditions with a universal bacterial 16S forward primer 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and reverse primer 1512R (5′-ACG GCT ACC TTG TTA CGA CTT-3′) (Weisburg et al., 1991). PCR-amplified 16S rRNA genes were cloned with the TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). Transformation was done with competent E. coli TOP10 cells. The size of inserts (~1500 bp) was determined by PCR with flanking vector primers followed by electrophoresis on 1% agarose gel. The numbers of clones in each library that were successfully sequenced, which were affected by the DNA quality and yield in each sample, ranged from 20 to 59, with an average of 48 clones per library. Purified products were sequenced by the Sanger capillary technique with a 519R primer (5′-TTA CCG CGG CTG CTG-3′) (Paster et al., 2001). Sequencing reactions were run on ABI model 3100 DNA Sequencer; reads of ~500 bp in length were obtained for each Sanger sequence. In total, 25 full 16S Sanger sequences of ~1500 bp were obtained for the potential novel sequences.
16S rRNA Gene Amplification and Pyrosequencing
The 16S rRNA gene was amplified with the composite forward primer 5′GCCTTGCCAGCCCGCTCAG **TCAGAGTTTGATCC TGGCTCAG**-3′ and reverse primer 5′GCCTCCCTCGCGCCATCAGNNNNNNNN _CATGCTGCCTCCCGTAGGA-GT_-3′, where the underlined sequence is 454 Life Sciences® Adaptor primer A and B; the bold sequence is the broadly conserved bacterial primer 27F and 338R; NNNNNNNN designates a unique eight-base barcode used to tag PCR products from each DNA sample. The amplified PCR products from different samples were pooled and cleaned by means of a MinElute PCR purification kit (Qiagen, Valencia, CA, USA) before they were subjected to sequencing by the Genome Sequencer FLX System (Roche, Mannheim, Germany) according to the manufacturer’s protocol, at the Institute for Genome Sciences, University of Maryland. All pyrosequences were passed through the built-in quality filter, and sequences of questionable qualities or shorter than 200 bp were discarded. The average length of the sequencing products is about 250 nucleotides.
Data Analysis
Chimeric Sanger sequences were checked by the Chimera Check program in the Ribosomal Database Project (RDP II). Sanger sequences were compared with over 35,000 16S rRNA gene sequences in the Forsyth Institute Human Oral Microbiome Database (HOMD) (http://www.homd.org) and with GenBank by the BLAST algorithm with 99% similarity value (http://www.ncbi.nih.nlm.gov). Phylogenetic tree construction was based only on Sanger sequence BLAST matches at the species level according to the method described previously (Paster et al., 2001). Both pyrosequences and Sanger sequences were compared with over 250,000 rRNA sequences in the Ribosomal Database Project (RDP II) (http://www.rdp.cmc.msu.edu). The taxonomic affiliations were determined to the genus level by the Classifier tool in RDP II, with a cut-off of 0.8 (Wang et al., 2007).
Results
Total Sequence Information
In total, 337 Sanger sequences were obtained from 7 specimens. Three chimeric Sanger sequences were identified and discarded. A total of 200,129 pyrosequences passed the quality control process, representing ~85% of the total number of sequences obtained. Most of the sequences that were discarded had insufficient length rather than poor sequence qualities. The number of high-quality sequences from pyrosequencing represented a 600-fold increase in depth-of-coverage than from Sanger sequencing. A comparison of the total numbers of bacterial taxa revealed by RDPII Classifier analysis at different taxonomic ranks showed that Sanger sequencing and pyrosequencing yielded 8 vs. 13 phyla, 10 vs. 22 classes, 11 vs. 43 orders, 20 vs. 97 families, and 25 vs. 179 genera, respectively (Fig. 1).
Figure 1.
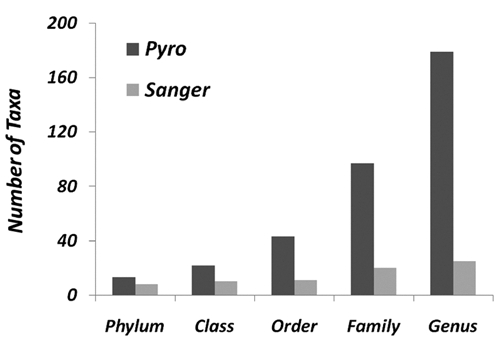
Difference in taxonomic coverage by Sanger sequencing and pyrosequencing. Sanger sequences and pyrosequences in the same specimens from 7 cases of endodontic infections were analyzed by the RDP Classifier program in the Ribosomal Database Project (RDP II). The numbers of taxa at different taxonomic levels were compared between Sanger sequencing and pyrosequencing approaches.
Pyrosequencing Data
In total, 13 bacterial phyla and 179 genera were assigned by RDP II Classifier to pyrosequences (Fig. 2a and Appendix Table). In total, 5.89% and 46% of the sequences could not be assigned to phylum or genus levels, respectively, and were listed as “unclassified bacteria” or were classified at the next higher taxonomic rank possible. The overall phylum abundance was Bacteroidetes (59.44%), Firmicutes (19.92%), Actinobacteria (4.79%), Fusob ac teria (3.49%), Proteobacteria (3.18%), Spirochaetes (2.28%), and TM7 (0.10%). An additional 6 phyla were not previously reported in endodontic infections: Tenericutes (0.68%), Deinococcus-Thermus (0.16%), Chloro-flexi (0.10%), Cyanobacteria (0.01%), OD1 (0.003%), and Acidobacteria (0.001%) (Fig. 2a). The most abundant bacterial phylum in six out of seven individuals by both sequencing approaches was the phylum Bacteroidetes. The top 10 most abundant genera and the top 25 most prevalent genera revealed by pyrosequencing were summarized (Figs. 2b, 2c). Only 6 genera appeared in all 7 specimens (Fig. 2c). Most of the genera were of relatively low abundance (Appendix Table).
Figure 2.
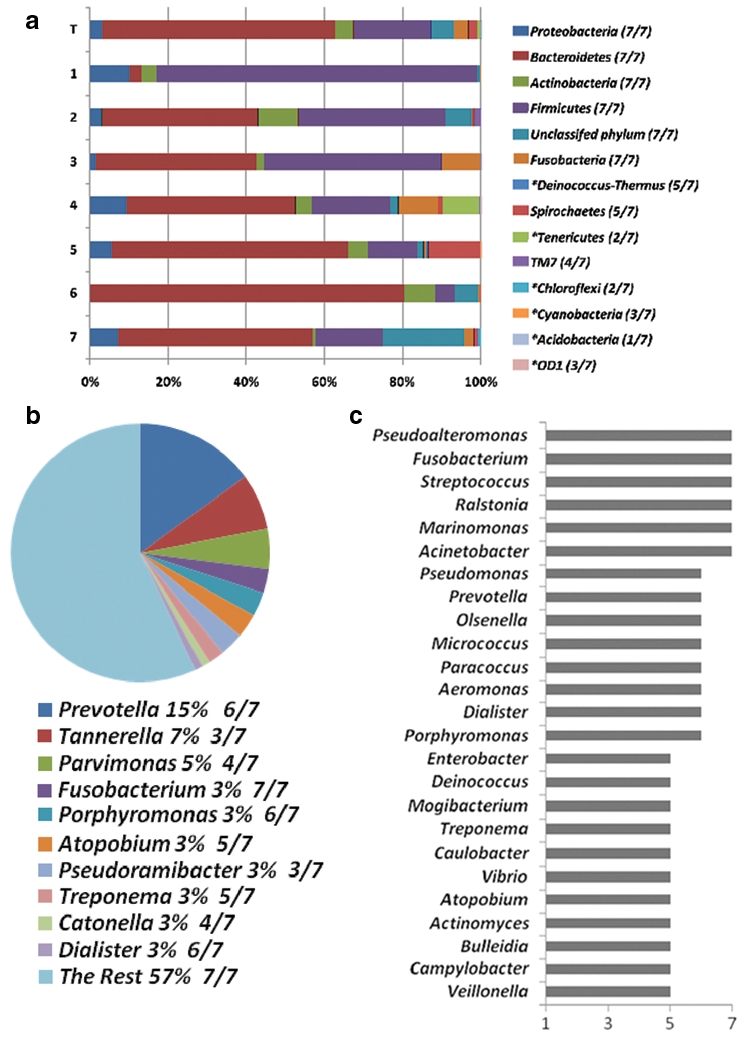
Bacterial abundance and prevalence distribution at phylum and genus levels by pyrosequencing in 7 endodontic infection samples. “T” represents the combined total. (a) Phylum distribution in seven individuals. In total, 13 phyla were identified; 6 *phyla were newly detected in endodontic infection. Prevalence for each phylum is shown inside the brackets. (b) The top 10 most abundant genera with their abundance and prevalence values. (c) The top 25 most prevalent genera listed according to their frequency of occurrence.
Sanger Sequencing Data
Fifty-nine species-level designations were matched to the total 334 quality Sanger sequences through the BLAST algorithm at 99% similarity level against the GenBank and the HOMD databases. The taxonomic designations, phylogenetic lineage, and frequency of detection were shown on a phylogenetic tree (Appendix Fig.). Two novel phylotypes, Bacteroidetes sp. clone YT053 and Lachnospiraceae sp. clone YT026, were identified. 1500-base full 16S sequences were obtained and deposited to GenBank under the accession numbers GQ495892 and GQ 495893, respectively (Appendix Fig.).
Discussion
Recent metagenomic studies in the human digestive tract have revealed an overall diversity orders of magnitude higher than previously described (Gill et al., 2006; Andersson et al., 2008; Dethlefsen et al., 2008). The biggest limitation of pyrosequencing has been its short reading lengths: ~100 bases for the first-generation GS 20 system, ~250 bases for the current GS FLX platform, and ~400 bases for the emerging next-generation GS XLR Titanium instrument. A recent computer simulation study demonstrated that accurate taxonomy assignments can be obtained from 16S rRNA tag sequences by pyrosequencing (Liu et al., 2008). The previous pyrosequencing study in the oral cavity utilized the first-generation GS 20 system (Keijser et al., 2008). The second-generation GS FLX system used in the present study generated informative sequences (after removal of the primer sequences) nearly 3 times longer than those from the GS 20 system, therefore enabling more accurate taxonomic assignments to be made. The present study also generated, on average, 25 times more sequences per sample compared with the study by Keijser et al. (2008); however, analysis of our data revealed a less diverse microflora in endodontic infection than their study in saliva and supragingival plaque, confirming that endodontic microflora are a restricted community, supposedly a subset derived from the total oral microflora.
The clinical parameters of the endodontic infections included in this study were relatively diverse. The emphasis was on the “depth of coverage” that results from the use of pyrosequencing compared with traditional methods in analyzing these infections. The effect of increase in “depth of coverage” was demonstrated in a taxonomic context in the present study. In total, 13 bacterial phyla and 179 genera were detected by pyrosequencing, and only 8 phyla and 25 genera in the same samples with Sanger sequencing. Consistent with previous studies, the top 5 abundant bacterial phyla in endodontic infection were Bacteroidetes, Firmicutes, Actinobacteria, Fusobacteria, and Proteobacteria (Munson et al., 2002; Vickerman et al., 2007). They occupied 90.2% of all pyrosequences. A large degree of inter-subject variability was consistently observed at different taxonomic ranks by both sequencing approaches, indicating an underlying heterogeneneous microbial etiology for the development of endodontic infections in different individuals with different clinical presentations. In several earlier studies, Firmicutes has been recognized as the most abundant phylum in endodontic infection (Munson et al., 2002; Saito et al., 2006; Vickerman et al., 2007). In the present study, both sequencing strategies showed a clear dominance of the members of the phylum Bacteroidetes. Different bacterial dominances might contribute to different clinical expressions, favor different clinical interventions, or result in different treatment outcomes. While pyrosequencing may tend to overestimate diversity if the unit used was an Operational Taxonomic Unit (OTU), here the RDP Classifier was used to classify 16S genes, a method that is less sensitive to sequencing errors.
Among the 6 previously undetected bacterial phyla in endodontic infection, Cyanobacteria, Acidobacteria, and Chloroflexi are known bacterial phyla in water, soil, and wastewater plants, respectively. Phylum OD1 is a newly recognized bacterial phylum known only by its 16S sequences (Harris et al., 2004). Deinococcus spp. within phylum Deinococcus-Thermus was previously known to exist only in extreme environments and animal feces until it was recently identified in the human stomach (Bik et al., 2006). Mycoplasma spp., belonging to phylum Tenericutes, was detected by pyrosequencing in two individuals in the present study. Sanger sequences in the same sample identified Mycoplasma salivarium. _Mycoplasma_-like species was first isolated from an inflamed human dental pulp nearly 5 decades ago (Barile and Sheingorn, 1960). The Synergistes are a group of Gram-negative anaerobic organisms that have been frequently found in the oral cavity (Vartoukian et al., 2007) as well as in endodontic infections (Siqueira and Rocas, 2007). Although they are phylogenetically distinct, the current Bergey’s Taxonomic Outline of the Prokaryotes does not list it as a separate phylum (Garrity and Castenholtz, 2004). As a result, these sequences in the present study were classified by RDP II Classifier as “unclassified bacteria”. Nevertheless, when the BLAST algorithm was used on Sanger sequences, abundant Synergistes spp. were identified, indicating that they are an important member of the endodontic microflora. So far, all known oral and endodontic Spirochetes have been classified under the genus Treponema (Chan and McLaughlin, 2000; Sakamoto et al., 2009). In the present study, pyrosequencing detected genus Spirochaeta, non-Treponema spirochetes, in one individual. The existence of non-Treponema spirochetes in endodontic infection has been speculated before, based on the findings from a nested PCR-DGGE (Siqueira et al., 2005). This was the first time a non-Treponema spirochete was detected in the oral cavity. Spirochaeta spp. are free-living, facultative, or obligate anaerobes, often found in ponds and marshes (Paster et al., 1991).
Endodontic microflora is established within previously sterile pulp spaces. Unlike other parts of the oral cavity, there is no supposedly indigenous endodontic microflora. All bacteria inside the infected root canal are opportunistic pathogens; they can either be the commensal oral bacteria associated with a healthy oral cavity, or the pathogenic bacteria associated with a diseased oral cavity, such as dental caries and periodontal disease. Nevertheless, it is essential to determine the composition of the endodontic microflora, because this may relate to the various clinical presentations or stages of development of an endodontic infection as well as its responses to different treatments. The present study demonstrated that deep-coverage pyrosequencing technology facilitated access to low-abundance bacteria in infected root canals, and revealed bacterial diversity in previously inaccessible endodontic microflora. This, in turn, will result in more accurate estimation of species abundance and prevalence in the endodontic microbial community. Low-abundance members may occupy critical niches within a complex microbial community, and therefore are potentially important in maintaining the stability and virulence of a microbial community (Siqueira and Rocas, 2009). The application of this technology can broaden our understanding of the pathogenesis of endodontic infections and has the potential to improve treatment outcomes.
Supplementary Material
Data Supplement
Acknowledgments
We thank Dr. Benli Chai from the Ribosomal Database Project, Center for Microbial Ecology, Michigan State University, for his excellent technical support on RDP-related applications.
Footnotes
This work was funded by a research grant from the NIDCR (No. DE015320-01-A1).
References
- Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile MF, Sheingorn A. (1960). The isolation and cultivation of a filterable form resembling pleuropneumonia-like organisms from the human dental pulp: report of a case. Oral Surg Oral Med Oral Pathol 13:756-760 [DOI] [PubMed] [Google Scholar]
- Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. (2006). Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA 103:732-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EC, McLaughlin R. (2000). Taxonomy and virulence of oral spirochetes. Oral Microbiol Immunol 15:1-9 [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6:e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad AF, Barry J, Caimano M, Clawson M, Zhu Q, Carver R, et al. (2002). PCR-based identification of bacteria associated with endodontic infections. J Clin Microbiol 40:3223-3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity GM, Castenholtz RW, editors (2004). Taxonomic outline of the prokaryotes. Bergey’s manual of systematic bacteriology: Vol. 1. New York, NY: Springer-Verlag [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312:1355-1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JK, Kelley ST, Pace NR. (2004). New perspective on uncultured bacterial phylogenetic division OP11. Appl Environ Microbiol 70:845-849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao WW, Fraser-Liggett CM. (2009). Human Microbiome Project—paving the way to a better understanding of ourselves and our microbes. Drug Discov Today 14:331-333 [DOI] [PubMed] [Google Scholar]
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al. (2008). Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res 87:1016-1020 [DOI] [PubMed] [Google Scholar]
- Liu Z, DeSantis TZ, Andersen GL, Knight R. (2008). Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 36:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson MA, Pitt-Ford T, Chong B, Weightman A, Wade WG. (2002). Molecular and cultural analysis of the microflora associated with endodontic infections. J Dent Res 81:761-766 [DOI] [PubMed] [Google Scholar]
- Paster BJ, Dewhirst FE, Weisburg WG, Tordoff LA, Fraser GJ, Hespell RB, et al. (1991). Phylogenetic analysis of the spirochetes. J Bacteriol 173:6101-6109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. (2001). Bacterial diversity in human subgingival plaque. J Bacteriol 183:3770-3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Olsen I, Aas JA, Dewhirst FE. (2006). The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000 42:80-87 [DOI] [PubMed] [Google Scholar]
- Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. (2009). Metagenomic pyrosequencing and microbial identification. Clin Chem 55:856-866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocas IN, Siqueira JF., Jr (2008). Root canal microbiota of teeth with chronic apical periodontitis. J Clin Microbiol 46:3599-3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolph HJ, Lennon A, Riggio MP, Saunders WP, MacKenzie D, Coldero L, et al. (2001). Molecular identification of microorganisms from endodontic infections. J Clin Microbiol 39:3282-3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaghi M, Uhlen M, Nyren P. (1998). A sequencing method based on real-time pyrophosphate. Science 281:363, 365 [DOI] [PubMed] [Google Scholar]
- Saito D, Leonardo Rde T, Rodrigues JL, Tsai SM, Hofling JF, Goncalves RB. (2006). Identification of bacteria in endodontic infections by sequence analysis of 16S rDNA clone libraries. J Med Microbiol 55(Pt 1):101-107 [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Rocas IN, Siqueira JF, Jr, Benno Y. (2006). Molecular analysis of bacteria in asymptomatic and symptomatic endodontic infections. Oral Microbiol Immunol 21:112-122 [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Siqueira JF, Jr, Rocas IN, Benno Y. (2009). Diversity of spirochetes in endodontic infections. J Clin Microbiol 47:1352-1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira JF, Jr, Rocas IN. (2007). Molecular detection and identification of Synergistes phylotypes in primary endodontic infections. Oral Dis 13:398-401 [DOI] [PubMed] [Google Scholar]
- Siqueira JF, Jr, Rocas IN. (2009). Community as the unit of pathogenicity: an emerging concept as to the microbial pathogenesis of apical periodontitis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 107:870-878 [DOI] [PubMed] [Google Scholar]
- Siqueira JF, Jr, Rocas IN, Cunha CD, Rosado AS. (2005). Novel bacterial phylotypes in endodontic infections. J Dent Res 84:565-569 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. (2007). The human microbiome project. Nature 449:804-810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartoukian SR, Palmer RM, Wade WG. (2007). The division “Synergistes”. Anaerobe 13:99-106 [DOI] [PubMed] [Google Scholar]
- Vickerman MM, Brossard KA, Funk DB, Jesionowski AM, Gill SR. (2007). Phylogenetic analysis of bacterial and archaeal species in symptomatic and asymptomatic endodontic infections. J Med Microbiol 56(Pt 1):110-118 [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261-5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ. (1991). 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement