MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity (original) (raw)
Abstract
MCScan is an algorithm able to scan multiple genomes or subgenomes in order to identify putative homologous chromosomal regions, and align these regions using genes as anchors. The MCScanX toolkit implements an adjusted MCScan algorithm for detection of synteny and collinearity that extends the original software by incorporating 14 utility programs for visualization of results and additional downstream analyses. Applications of MCScanX to several sequenced plant genomes and gene families are shown as examples. MCScanX can be used to effectively analyze chromosome structural changes, and reveal the history of gene family expansions that might contribute to the adaptation of lineages and taxa. An integrated view of various modes of gene duplication can supplement the traditional gene tree analysis in specific families. The source code and documentation of MCScanX are freely available at http://chibba.pgml.uga.edu/mcscan2/.
INTRODUCTION
Comparative genomic studies often rely on the accurate identification of homology (genes that share a common evolutionary origin) within or across genomes. Homologous genes are further classified as either orthologous, if they were separated by a speciation event, or paralogous, if they were separated by a gene duplication event. Recently, comparisons between related eukaryotic genomes reveal various degrees to which homologous genes remain on corresponding chromosomes (synteny) and in conserved orders (collinearity) during evolution (1). Over evolutionary time, genomes have been shaped and dynamically restructured by several forces such as whole-genome duplication (WGD), segmental duplication, inversions and translocations (2–5). These forces have acted in various combinations and to differing degrees to result in taxonomic groups with different modes of genome structure modification and gene family expansion. For example, angiosperm (flowering plants) genomes appear more volatile than mammalian genomes (1). Angiosperm genomes show remarkable fluctuations in size and organization, even among close relatives, and all examined angiosperms have undergone one or more ancient WGD (6). In contrast, karyotype evolution among major vertebrate lineages appears to have been slower, with a single whole-genome duplication event ∼500 million years ago (4,7). However, hundreds of invertebrates are paleopolyploids (8) and their rates of chromosomal rearrangement have been suggested to be almost twice that of vertebrates (1,9,10). Further, there is also a remarkable lack of synteny and high rate of rearrangement in the parasitic and pathogenic protistan phylum Apicomplxa compared to what is seen in vertebrates (11).
Traditionally, synteny was identified via the clustering of neighboring matching gene pairs, as implemented in various programs including ADHoRe (12), TEAM (13), LineUp (14), the Max-gap Clusters by Multiple Sequence Comparison (MCMuSeC) (15) and OrthoCluster (16). However, detection of synteny is often complicated by gene loss, tandem duplications, gene transpositions and chromosomal rearrangements, any of which may produce artifacts. Collinearity, a more specific form of synteny, requires conserved gene order. More recent methods apply dynamic programming to chains of pair-wise collinear genes, and often specify a certain scoring scheme that rewards the adjacent collinear gene pairs (or ‘anchor genes’) and penalizes the distance between anchor genes. This class of methods has been implemented in software tools such as DAGchainer (17), ColinearScan (18), MCScan (19), SyMAP (20), FISH (21) and CYNTENATOR (22). In addition to algorithmic differences, synteny and collinearity detection tools often differ in application ranges, inputs, presentation of results and/or computational costs.
Although pair-wise collinear relationships among chromosomal regions have been widely studied, the multi-alignment (alignment of three or more regions) of collinear chromosomal regions (referred to as collinear blocks) is more important as it can reveal ancient WGD events (19,23) and complex chromosomal duplication/rearrangement relationships (24). Collinear blocks are comprised of anchor genes which are located at collinear positions and non-anchor genes which are assumed to have experienced gene gains, losses or transposition. Further, anchor genes are more likely to be homologs (25) and tend to be under stronger purifying selection than non-anchor genes (26). Patterns of synteny and collinearity can provide insight into the evolutionary history of a genome, and inform on potentially useful downstream analyses. However, although graphic interfaces for visualizing synteny and collinearity may be incorporated, many available software packages for synteny and collinearity detection do not directly provide downstream analysis tools. Further, genes may be duplicated by mechanisms other than whole-genome duplication, such as tandem, proximal and/or dispersed duplications, each of which may make different contributions to evolution (11,27). In addition, analysis of gene family evolution may require that it be placed in the context of genome evolution. To analyze the evolution of a genome, it may be helpful to correlate gene family analysis with different duplication modes for a more integrated view. To our knowledge, only the MicroSyn package (28) provides analysis of collinearity within gene families, but it cannot superimpose such analysis on a context of whole-genome collinearity.
MCScan is able to identify collinear blocks in genomes or subgenomes and then conduct multi-alignments of collinear blocks using collinear genes as anchors (19,23). MCScan is also customizable for genomes of different sizes and with different average intergenic distances. Using MCScan, a Plant Genome Duplication Database (PGDD) has been constructed and is publicly available at http://chibba.pgml.uga.edu/duplication/. The MCScan software package and PGDD database have been applied to a variety of research areas such as genome duplication and evolution (11,29–36), annotation of newly sequenced genomes (37) and the evolution of gene families (38–48).
Building on the MCScan algorithm, here we describe a software package named MCScanX for synteny and collinearity detection, visualization and diverse downstream analyses. Compared with MCScan, the usage of MCScanX has been greatly simplified. To more clearly show how frequently chromosomal regions are duplicated, multi-alignments of collinear blocks against reference chromosomes can be viewed through a web browser with various highlighted features (e.g. tandem arrays, coverage statistics). The overall pattern of synteny and collinearity between or among genomes can be visualized by up to four types of plots. Compared with existing synteny and collinearity detection tools, a distinct feature of MCScanX is that diverse tools for evolutionary analyses of synteny and collinearity are incorporated, aiding efforts to construct gene families using collinearity information, infer gene duplication modes and enrichments, characterize collinear genes with nucleotide substitution rates, detect collinear tandem arrays, perform statistical analyses of duplication depths and collinear orthologs, and analyze collinearity within gene families. MCScanX enables rapid and convenient conversion of synteny and collinearity information into evolutionary insights.
MATERIALS AND METHODS
Gene set and homology search
Whole-genome protein sequences and gene positions for Arabidopsis thaliana, Populus trichocarpa, Vitis vinifera, Glycine max, Oryza sativa and Brachypodium distachyon were retrieved from Phytozome v7.0 (http://www.phytozome.net/). Whole-genome protein sequences and gene positions for Sorghum bicolor and Zea mays were retrieved from EnsemblPlants (http://plants.ensembl.org/index.html) and MaizeSequence Release 5b.60 (http://www.maizesequence.org/index.html) respectively. If a gene had more than one transcript, only the first transcript in the annotation was used. To search for homology, the protein-coding genes from each genome was compared against itself and other genomes using BLASTP (49). For a protein sequence, the best five non-self hits in each target genome that met an _E_-value threshold of 10−5 were reported.
MCScanX algorithm
The MCScanX algorithm is a modified version of MCScan (19). Whole-genome BLASTP results are used to compute collinear blocks for all possible pairs of chromosomes and scaffolds. First, BLASTP matches are sorted according to gene positions. To avoid high numbers of local collinear gene pairs due to tandem arrays, if consecutive BLASTP matches have a common gene and its paired genes are separated by fewer than five genes, these matches are collapsed using a representative pair with the smallest BLASTP _E_-value. Then, dynamic programming is employed to find the highest scoring paths (i.e. chains of collinear gene pairs) using the following scoring schema, assuming that two gene pairs, u and v, are on the path where u precedes v,
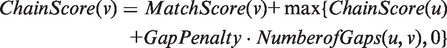
where by default MatchScore(v) = 50 for one gene pair, GapPenalty = −1, and NumberofGaps (u,v), the maximum number of intervening genes between u and v, should be fewer than 25. Non-overlapping chains with scores over 250 (i.e. involving at least 5 collinear gene pairs) are reported. In a pair of collinear blocks, there are two distinct genomic locations with aligned collinear genes as anchors.
The expected number of occurrences (_E_-value) of a pair of collinear blocks is estimated using the formula introduced by Wang et al. (18),
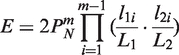
where N is the number of matching gene pairs between the two chromosomal regions defined by the pair of collinear blocks, m is the number of anchors in the pair of collinear blocks, _L_1 and _L_2 are respective lengths of the two chromosomal regions, and l_1_i and l_2_i are distances (in terms of nucleotide numbers) between two adjacent anchors in the pair of collinear blocks. The default _E-_value cutoff of MCScanX is 10−5.
Multiple chromosomal regions threaded by consecutive ancestral loci are progressively aligned against reference chromosomes, where each genome being tested is used as a reference successively, according to the following procedure: (i) any reference chromosome is scanned from start to end, and empty tracks are placed alongside the reference chromosome to hold potential aligned collinear blocks; (ii) collinear blocks are progressively aligned against reference chromosomes pinpointed by anchors and assigned to the nearest empty tracks (once a track region is filled, it cannot be assigned collinear blocks again). In aligned collinear blocks, only symbols of anchor genes are shown while un-matched positions (gaps) between anchors (regardless of numbers of intervening genes) are denoted by ‘||’; (iii) at each locus of reference chromosomes, the number of tracks occupied by collinear blocks is recorded to reflect the duplication depth.
Classification of duplicate gene origins
Genes within a single genome can be classified as singletons, dispersed duplicates, proximal duplicates, tandem duplicates and segmental/WGD duplicates depending on their copy number and genomic distribution. The following procedure is used to assign gene classes: (i) All genes are initially classified as ‘singletons’ and assigned gene ranks according to their order of appearance along chromosomes; (ii) BLASTP results are evaluated and the genes with BLASTP hits to other genes are re-labeled as ‘dispersed duplicates’; (iii) In any BLASTP hit, the two genes are re-labeled as ‘proximal duplicates’ if they have a difference of gene rank<20 (configurable); (iv) In any BLASTP hit, the two genes are re-labeled as ‘tandem duplicates’ if they have a difference of gene rank = 1; (v) _MCScanX_ is executed. The anchor genes in collinear blocks are re-labeled as ‘WGD/segmental’. So, if a gene appears in multiple BLASTP hits, it will be assigned a unique class according to the order of priority: WGD/segmental>tandem>proximal>dispersed.
Detection of orthologous gene pairs using OrthoMCL
Whole-genome protein sequences from Arabidopsis, Populus, Vitis, Glycine, Oryza, Brachypodium, Sorghum and Zea were merged and searched against themselves for homology using BLASTP with an _E_-value cutoff of 10−5. Default parameters of OrthoMCL (50) were used. The combination of OrthoMCL intermediate files ‘orthologs.txt’ and ‘coorthologs.txt’ (generated by orthomclDumpPairsFiles) was used as the whole set of ortholog pairs.
Enrichment analysis
Enrichment analysis is performed using Fisher's exact test. The _P_-value was calculated for the null hypothesis that there is no association between the members of a gene family and a particular gene duplication mode and is corrected with the total number of duplication modes for multiple comparisons (i.e. Bonferroni correction). The _P_-value cutoff of 0.05 is used to suggest putative enrichment of certain gene duplication modes.
Computing _K_a and _K_s
Non-synonymous (Ka) and synonymous (Ks) substitution rates are estimated by Nei-Gojobori statistics (51), available through the ‘Bio::Align::DNAStatistics’ module of the BioPerl package (http://www.bioperl.org/wiki/Module:Bio::Align::DNAStatistics). Note that the ‘Bio::Align::DNAStatistics’ module may generate invalid Ka or Ks (i.e. non-digital output) for some homologous gene pairs due to mis-alignments.
Gene family examples
Lists of published Arabidopsis gene families were obtained from TAIR (http://www.arabidopsis.org/browse/genefamily/index.jsp). Only families with more than nine genes were considered in order to have enough statistical power to detect enrichment of duplication modes. Arabidopsis disease resistance gene homologs were downloaded from the NIBLRRS Project website (http://niblrrs.ucdavis.edu).
Execution of the MCScanX package
MCScanX is freely available at http://chibba.pgml.uga.edu/mcscan2. All programs in the MCScanX package should be executed using command line arguments on Mac OS or Linux systems. On Mac OS, Xcode (http://developer.apple.com/xcode/) should be installed prior to the installation of MCScanX package. On Linux systems, the Java SE Development Kit (JDK) and ‘libpng’ should be installed before the installation of MCScanX package. To list available command line options, the user can simply type the name of a program without any options.
RESULTS
Structure of the MCScanX package
The MCScanX package consists of two main components: (i) three core programs that implement an adjusted MCScan algorithm to generate pairwise and multiple alignments of collinear blocks and (ii) 12 downstream analysis programs for displaying and analyzing identified synteny and collinearity output by the core programs. The structure of the MCScanX package is shown in Figure 1. Compared with the previous version (0.8) of MCScan, there are numerous improvements in MCScanX. First, preprocessing of BLASTP input has been pipelined into the execution of core programs. Next, in MCScan, each gene was assigned a family ID to identify tandem genes, where the family ID has to be pre-computed using the Markov Clustering Algorithm (MCL) software (52). In MCScanX, tandem genes are assessed by gene rank according to chromosomal positions and thus, execution of MCL is no longer required. The aforementioned two improvements have made the installation and execution of MCScanX easier and more efficient. Furthermore, multi-alignments of collinear blocks, which are output as HTML files in MCScanX, can be easily and clearly viewed. In addition, numerous visualization and downstream analysis tools are incorporated into the MCScanX package, greatly enhancing the biological applications of the MCScan algorithm. In the following, we describe in detail each program in the MCScanX package.
Figure 1.

The structure of the MCScanX package illustrating major components and their dependencies.
The first core program, named MCScanX, can generate both pair-wise and multiple alignments of collinear blocks, similar to the previous MCScan version (0.8). However, MCScanX takes only a simplified GFF format file and a BLASTP tabular file as inputs. The simplified GFF file should contain the gene locations (which include chromosome, gene symbol, start and end) for the genomes to be compared. The BLASTP input file is one BLASTP output or combined multiple BLASTP outputs in tabular format (option ‘-m8’ in BLAST and ‘−outfmt 6’ in BLAST+) for all protein sequences in the species of interest. Note that when MCScanX is applied to multiple species, it may be useful to guard against over-enrichment of gene pairs from closely related species and we recommend that the BLASTP input file include the combined BLASTP outputs of pairwise genome comparisons and self-genome comparisons with a cutoff of best hits instead of a single BLASTP output of pooled protein sequences from different species. Alternatively, the BLASTP input can be replaced by a tab-delimited file containing pair-wise homologous relationships detected by third party software. In this case, the user needs to implement MCScanX_h (the second core program). In addition, MCScanX_h can generate statistics on numbers of collinear homolog pairs and their percentages (relative to the numbers of input homolog pairs).
We also adopted an adjusted MCScan algorithm. Matches among genes are first sorted according to chromosomal positions for all possible pairs of chromosomes and scaffolds, and in both transcriptional directions. Adjacent collinear genes are chained using dynamic programming (see ‘Materials and Methods’ section), outputting pairwise collinear blocks and tandem gene pairs to ‘.collinearity’ and ‘.tandem’ files respectively. Note that during the chaining of collinear genes, distances between genes are calculated in terms of differences in gene ranks. Use of differences in gene ranks provides relative gene distances, which can mitigate the effects of different gene densities (per unit physical DNA) among species on collinearity detection. Next, multiple chromosomal regions threaded by consecutive anchor loci are progressively aligned against ‘reference’ genomes. Because there could be many intervening/non-anchor genes between consecutive anchor genes, especially for divergent genomes, the alignment of non-anchor genes is highly flexible and could clutter the view of results. Thus, in MCScanX, the alignment among non-anchor genes is discarded in the output and non-anchor genes (mismatches) are simply denoted by ‘||’ in the multi-alignment of gene orders. As a result, the layout of multiple alignments is less affected by alignment parameters and anchor genes and duplication depths can be easily discerned in the resulting multiple alignments.
The results of MCScanX multiple alignments are presented in HTML format with variously colored features that can be displayed using a web browser. An example is shown in Figure 2. In a reference chromosome, both anchor and non-anchor genes are shown, while in aligned collinear blocks only anchor genes are shown. Along the reference chromosome, duplication depth (i.e. number of aligned collinear blocks) is shown at each locus to indicate how frequently chromosomal regions are duplicated, and tandem genes are highlighted in red. In principle, all aligned collinear blocks can be also references. Note that in certain cases, in a specific alignment (e.g. A–B–C), an anchor locus is lost in the reference chromosome (A) and in turn cannot be shown in aligned collinear blocks (B and C) due to the non-reciprocity of the employed algorithm. To study differential gene loss, the user is suggested to analyze the results using the gene or genome of interest as the reference (i.e. the alignments B–A–C and C–A–B can show that the anchor locus exists between B and C but is lost in A) to ensure that complete chromosomal neighborhoods and matching segments are observed.
Figure 2.

Sample HTML output displaying multiple alignments of collinear blocks by MCScanX. The first and second columns show duplication depth and gene symbol at each locus of reference chromosomes, where tandems are marked in red. The remaining columns show aligned collinear blocks, where only the symbols of anchor genes are shown.
The third core program, named duplicate gene classifier, can classify the duplicate genes of a single species into WGD/segmental, tandem, proximal and dispersed duplicates. WGD/segmental duplicates are inferred by the anchor genes in collinear blocks. Tandem duplicates are defined as paralogs that are adjacent to each other on chromosomes, which are suggested to arise from illegitimate chromosomal recombination (27). Proximal duplicates are paralogs near each other, but interrupted by several other genes (e.g. separated by fewer than 20 genes, configurable). Proximal duplicates are inferred to result from localized transposon activities (53), or ancient tandem arrays interrupted by more recent gene insertions. Dispersed duplicates are paralogs that are neither near each other on chromosomes, nor do they show conserved synteny (54). Distant single gene translocations mediated by transposons may explain the wide spread of dispersed duplicates (27), often via pack-MULEs (55), helitrons (56), or CACTA elements (37) in plant genomes, or through ‘retropositions’ (57). Inferences about the mechanism(s) responsible for duplication of genes may reveal unusual evolutionary characteristics for particular lineages. Duplicate gene classifier, incorporating the MCScanX procedure, takes in the same input files as MCScanX, and returns statistics of duplicate gene origins and a file showing the likely origin of each gene.
Once the outputs of the core programs are generated, various visualization and downstream analysis tools can be applied. To display synteny and collinearity, four types of plots can be generated: dual synteny plot (Figure 3A), circle plot (Figure 3B), dot plot (Figure 3C) and bar plot (Figure 3D) using the Java programs: dual synteny plotter, circle plotter, dot plotter and bar plotter, respectively. The ‘.collinearity’ file generated by MCScanX can be annotated with non-synonymous (Ka) and synonymous (Ks) substitution rates using the Perl program add ka and ks to collinearity.pl. Gene families constructed based on collinear relationships (instead of BLAST hits) can be generated based on the ‘.collinearity’ file using the Perl program group collinear genes. It may be interesting to see how frequently chromosomal regions are duplicated within or across species for understanding species-specific or shared evolutionary events, and the program dissect multiple alignment can compute the number of intra- and inter-species collinear blocks at each locus of reference genomes and show statistics on gene numbers at different duplication depths. To avoid high numbers of local collinear gene pairs generated by MCScanX due to tandem arrays, tandem matches are collapsed using a representative pair with the smallest BLASTP _E_-value during MCScanX execution. However, a tandem array at an ancestral locus may imply positional gene family expansion (58). Thus, a tool named detect collinear tandem arrays is provided for detection of collinear tandem arrays.
Figure 3.

Different types of plots showing patterns of synteny and collinearity: (A) dual synteny plot, (B) circle plot, (C) dot plot and (D) bar plot, generated by ‘_dual synteny plotter, circle plotter, dot plotter and bar plotter_’, respectively. Chromosomes are labeled in the format ‘species abbreviation’ + ‘chromosome ID’. os, Oryza sativa; sb, Sorghum bicolor.
The MCScanX package provides a variety of tools for analyzing gene family evolution based on the synteny and collinearity identified by MCScanX. Origin enrichment analysis can detect potential enrichment of duplicate gene origins for gene families, based on the classification of whole-genome duplicate genes (the output of duplicate gene classifier). Detect collinearity within gene families outputs all collinear gene pairs among gene family members. Family circle plotter can detect all collinear gene pairs within a gene family and plot them using a genomic circle Family tree plotter, with a Newick-format tree (direct results from most phylogenetic software) and ‘.collinearity’ and ‘.tandem’ files (generated by MCScanX) as inputs, can graphically annotate a phylogenetic tree with collinear and tandem relationships.
Application examples
Estimation of the number of WGD events
MCScan version 0.8 was implemented to estimate the number of WGD events of Arabidopsis, Carica, Populus and Vitis, through analysis of the duplication depths of their collinear blocks using Vitis as the reference genome (19,23). To facilitate this analysis using the output of MCScanX, the tool dissect multiple alignment is provided. When the user applies the MCScanX package, the BLASTP and GFF inputs should be restricted to a single genome for self-genome comparison or between two genomes for cross-genome comparison. Alternatively, a BLASTP of self-genome comparison and cross-genome comparison may be merged for both comparisons. However, self-genome comparison may not be as sensitive as cross-genome comparison due to the differential loss of functionally redundant genes, sometimes in a complementary fashion (19). Although the determination of an exact number of WGD events may be heuristic, the output of ‘_dissect multiple alignment_’ can give a reasonable estimate. Note that a duplication depth x indicates that there are x and x+1 aligned collinear blocks in the target genome using cross-genome and self-genome comparisons respectively. For example, ‘_dissect multiple alignment_’ was applied to both self-genome and cross-genome comparisons between Arabidopsis and Vitis. Using Arabidopsis and Vitis as references, the maximum duplication depths of Arabidopsis collinear blocks are 7 (self-genome comparison, so the maximum number of aligned Arabidopsis collinear blocks is 8) and 11 (cross-genome comparison, so the maximum number of aligned Arabidopsis collinear blocks is 11), respectively, suggesting that the lineage experienced at least three WGD events to achieve this duplication depth, i.e. a triplication WGD event γ × two duplication WGD events α and β (6,19,23). By applying dissect multiple alignment to self-genome comparison of Vitis, the maximum duplication depth of Vitis collinear blocks is 4. However, the gene numbers at levels 3 and 4 (297 and 6, respectively) are much smaller than at level 2 (6993). A whole-genome triplication (WGT) plus small scale chromosomal duplications is the simplest explanation for this duplication pattern (19,23). Note that analysis of duplication depths of collinear blocks can generate good estimates on relatively recent WGD events. Very ancient WGD events often do not result in discernable collinear blocks in extant species due to extensive chromosome rearrangement, loss or gain of chromosomal segments, loss or transposition of duplicate genes, horizontal gene transfers, etc. A recent study, through analyzing the phylogenetic trees of cross-species gene families, reported two ancestral WGD events for seed plants and angiosperms respectively (59).
Detection of collinear orthologs
Detection of collinear orthologs is important for understanding gene evolution. The comparison between collinear orthologs and all orthologs can reveal how gene orders are conserved (or inversely, how frequently chromosomes are rearranged) between species. Limited only by the state of a genome's annotation and the assumption that sufficient sequence similarity is present for detection, a complete set of orthologs for a set of species can be generated by third-party software such as OrthoMCL (50). We implemented OrthoMCL to find ortholog pairs among Arabidopsis, Populus, Vitis, Glycine, Rice, Brachypodium, Sorghum and Zea. The ortholog pairs identified by OrthoMCL were regarded as the whole set of orthologs, and were then used as the input of MCScanX_h. Besides standard MCScanX output, MCScanX_h generated statistics on the numbers of collinear ortholog pairs and all ortholog pairs, and percentages of collinear ortholog pairs between any two of the selected angiosperm genomes (Table 1). As expected, gene order is better conserved within monocots and within eudicots than between monocots and eudicots. Within eudicots, Vitis shows the highest level of collinearity with the other 3 species, suggesting that Vitis most closely resemble the gene order of the eudicot ancestral genome, due in part to the lack of recent WGDs (60).
Table 1.
Numbers of collinear ortholog pairs and total ortholog pairs and percentage of collinear ortholog pairs in selected angiosperm genomes
| Species | No. of collinear ortholog pairs, No. of total ortholog pairs and percentage of collinear ortholog pairs | ||||||
|---|---|---|---|---|---|---|---|
| Pt | Gm | Vv | Os | Bd | Sb | Zm | |
| At | 14 278, 46 944, 30.4% | 17 498, 58 038, 30.1% | 7378, 24 086, 30.6% | 319, 24 992, 1.3% | 202, 22 719, 0.9% | 350, 24 120, 1.5% | 142, 24 689, 0.6% |
| Pt | – | 34 545, 92 901, 37.2% | 15 734, 38 727, 40.6% | 2121, 37 575, 5.6% | 1632, 32 790, 5.0% | 1523, 36 059, 4.2% | 687, 35 596, 1.9% |
| Gm | – | – | 18 310, 47 652, 38.4% | 1437, 46 916, 3.1% | 1308, 43 130, 3.0% | 1263, 46 631, 2.7% | 501, 47 326, 1.1% |
| Vv | – | – | – | 1315, 19 678, 6.7% | 981, 18 080, 5.4% | 1194, 19 137, 6.2% | 293, 19 501, 1.5% |
| Os | – | – | – | – | 15 492, 34 413, 45.0% | 15 664, 39 695, 39.5% | 14 112, 35 206, 40.1% |
| Bd | – | – | – | – | – | 14 070, 32 701, 43.0% | 13 111, 30 841, 42.5% |
| Sb | – | – | – | – | – | – | 18084, 36826, 49.1% |
Differences in duplicate gene origins among angiosperms
Using self-genome BLASTP outputs and the tool duplicate gene classifier, we classified the origins of duplicate genes for Arabidopsis, Populus, Vitis, Glycine, Oryza, Brachypodium, Sorghum and Zea respectively_._ The results are shown in Table 2. The collinear blocks in the self-genome comparisons result from segmental or whole-genome duplications. Most collinear blocks within these flowering plant genomes were derived from WGDs because of their high coverage throughout the genome as well as supporting Ks evidence (19).
Table 2.
Numbers of genes from different origins as classified by duplicate gene classifier in eight angiosperm genomes
| Species | No. of genes | No. of genes from different origins (percentage) | ||||
|---|---|---|---|---|---|---|
| Singletons | WGD | Tandem | Proximal | Dispersed | ||
| Arabidopsis | 27 105 | 5272 (19.5) | 7321 (27.0) | 769 (2.8) | 892 (3.3) | 12 851 (47.4) |
| Populus | 40 650 | 5014 (12.3) | 20 989 (51.6) | 713 (1.8) | 999 (2.5) | 12 935 (31.8) |
| Glycine | 46 360 | 1459 (3.1) | 35 233 (76.0) | 582 (1.3) | 670 (1.4) | 8416 (18.2) |
| Vitis | 23 647 | 6275 (26.5) | 3539 (15.0) | 688 (2.9) | 1590 (6.7) | 11 555 (48.9) |
| Oryza | 40 634 | 12 720 (31.3) | 5896 (14.5) | 960 (2.4) | 2184 (5.4) | 18 874 (46.4) |
| Brachypodium | 25 524 | 4842 (19.0) | 4575 (17.9) | 697 (2.7) | 827 (3.2) | 14 583 (57.1) |
| Sorghum | 34 564 | 5839 (16.9) | 5260 (15.2) | 895 (2.6) | 1283 (3.7) | 21 287 (61.6) |
| Zea | 39 365 | 8212 (20.9) | 11 506 (29.2) | 774 (2.0) | 1175 (3.0) | 17 698 (45.0) |
WGDs have had different impacts on the gene repertoires of the investigated taxa. Strikingly, ∼76.0% of Glycine genes were duplicated and retained from WGD events, versus only 14.5% of Oryza genes. The proportions of genes involved in WGD events may reflect the relative timing of the most recent WGD event, as well as the level of gene retention following the WGD. For example, Vitis, with only 15.0% of genes created by WGD (actually WGT), was inferred to have undergone the γ WGT event, which likely predated the divergence of most eudicots >100 million years ago (19,23). Other eudicot lineages have experienced lineage-specific WGDs in addition to the shared γ event. Twenty-seven percent of Arabidopsis appear to have been created through WGD, having experienced α and β WGD events since its divergence from other members of the Brassicales clade (6,23). Populus, with 51.6% of genes created by WGD, was inferred to have undergone an additional WGD event in the Salicoid lineage (23). Glycine, with the highest proportion of WGD genes, was reported to have experienced two additional WGD events, with the most recent occurring 13 million years ago (61). A total of 29.2% of Zea genes were created through WGD, which experienced a lineage-specific WGD after its divergence from Sorghum (15.2% genes created by WGD) (62,63). Although tandem genes are volatile after gene duplication, those retained may indicate functional significance. We find that tandem genes account for about 1–3% of genes in each genome, smaller than ∼10% reported by Rizzon et al. (64). This difference is due to the algorithm of duplicate gene classifier, which treats the tandem duplicates located at ancestral loci as WGD duplicates. Proximal duplicates account for larger proportions of genes in the genomes with fewer WGD duplicates, e.g. there are 5.4% of Oryza genes and 6.7% of Vitis genes created by proximal duplications, while in other genomes, the numbers of proximal duplicates are comparable to those of tandem duplicates.
Detection of collinear tandem arrays
In the MCScanX package, tandem arrays are defined as clusters of consecutive tandem duplicates. Via ‘_detect collinear tandem arrays_’, tandem arrays are first determined according to successive gene ranks in all chromosomes. Collinear gene pairs are then searched against these tandem arrays. If any gene of a collinear pair is located within a tandem array, the gene is replaced by the tandem array and then reported. If a tandem array is located at an anchor locus of a collinear block, it is termed a collinear tandem array. Collinear tandem arrays can indicate positional gene family expansions (58), which could be important for forming large gene families, or adopted as an alternative path to increasing gene copy number in the genomes that experienced fewer WGD events. For example, we applied the tool ‘_detect collinear tandem arrays_’ to a comparison of the Arabidopsis and Vitis genomes. A total of 1160 pairs of collinear tandem arrays were detected between Arabidopsis and Vitis, of which only 68 (5.9%) pairs have equal numbers of tandem duplicates in each species, while 54.3% of pairs have more tandem duplicates in Vitis than Arabidopsis. In conjunction with the finding above that Vitis has more proximal duplicates than other species, we suggest that tandem and proximal duplications contribute relatively more to the expansion of the Vitis genome than to other eudicots that experienced more WGDs in their evolutionary histories.
Analysis of gene family evolution
While MCScanX can detect synteny and collinearity using whole-genome homology and gene positional information, it is also of interest to analyze collinearity within a gene family, toward clarifying gene family evolution (65). We used the Arabidopsis MADS-box gene family as an example to illustrate the usefulness of MCScanX for analyzing the history of gene family expansion. Using the tool ‘_detect collinearity with gene families_’, we detected 14 collinear gene pairs from the members of the MADS box gene family. The inferred collinear relationships of the MADS box gene family members can be displayed and placed within the context of whole-genome collinearity using a genomic circle generated by ‘_family circle plotter_’ (Figure 4). Next, a phylogenetic tree was constructed for the MADS box gene family using PhyML package (66). The Newick tree was then used as the input of ‘_family tree plotter_’. A plot that showed the phylogenetic tree, collinear and tandem relationships for the MADS box gene family was generated (Figure 5). The overlay of positional history over the gene clades reveals interesting characteristics of the MADS-box gene family. We note that the clade with many collinear relationships (WGD or segmentally duplicated) appears to be the MIKCc-type (67). In contrast, the remaining clades of MADS-box genes appear to favor dispersed duplications (27,68).
Figure 4.

Circle plot showing collinearity in the MADS box gene family over the gray background of collinearity in Arabidopsis (the collinear blocks in Arabidopsis). The circle plot can be generated by ‘_family tree plotter_’. Chromosomes are labeled in the format ‘species abbreviation’ + ‘chromosome ID’. at, Arabidopsis thaliana.
Figure 5.

Phylogenetic tree of the MADS box gene family in Arabidopsis annotated with collinear and tandem relationships. Curves connecting pairs of gene names suggest either the collinear relationship (red) or tandem relationship (blue). This annotated tree is output from ‘_family tree plotter_’.
The tool ‘_origin enrichment analysis_’, which is able to detect potential enrichments of duplicate gene origins, was applied to 126 published Arabidopsis gene families of 10 or more genes, available at TAIR (http://www.arabidopsis.org/). We found that 46 (36.5%) gene families were enriched for at least one of the four types of origins at α = 0.05. For example, disease resistance gene homologs and the cytochrome P450 gene family are enriched for dispersed and proximal duplicates, while the cytoplasmic ribosomal protein gene family and C2H2 zinc finger proteins are enriched for WGD duplicates, as previously noted (68).
Comparison with other synteny and collinearity tools
Existing tools for synteny and collinearity detection mainly include ADHoRe (12), TEAM (13), LineUp (14), MCMuSeC (15), OrthoCluster (16), DiagHunter (69), DAGChainer (17), ColinearScan (18), MCScan (19), SyMAP (20), FISH (21), Cyntenator (22), MicroSyn (28) and Cinteny (70), of which OrthoCluster, ADHoRe and SyMAP are currently upgraded to OrthoClusterDB (71), i-ADHoRe 3 (72) and SyMAP 3.4 (73), respectively. We summarized the functions of synteny and collinearity detection tools regarding five elements: graphic visualization, operation on multiple (>2) genomes, multi-alignments, evolutionary analyses of synteny and collinearity (e.g. estimating WGD events, gene-order conservation and duplicate gene origins, constructing collinear gene groups/families, etc.) and analyses of gene families. Functional comparison of different synteny and collinearity detection tools is shown in Table 3. If there were multiple versions for a tool, we used the latest one for comparison. Seven tools output synteny or collinearity information as plain texts, while the other tools provide graphic visualization options, though types and numbers of plots vary among different tools. As for the data scale, most tools published in the past 4 years can operate on multiple genomes. Five tools can perform multi-alignments of collinear blocks. MicroSyn is focused on collinearity analysis within gene families. i-ADHoRe 3 has provided several post-processing programs for dissecting multi-alignments of collinear blocks, in addition to detecting and visualizing synteny and collinearity. Among these synteny and collinearity detection tools, 11 tools cover no more than two functions, and OrthoclusterDB, MicroSyn and i-ADHoRe 3 cover three functions. MCScanX, with all five functions, can perform more biological analyses than any other synteny or collinearity detection tool.
Table 3.
Functional comparison of different synteny and collinearity detection tools (‘+’ and ‘−’ represent ‘yes’ and ‘no’, respectively)
| Tool | Year published | Graphic visualization | Multiple genomes | Multi-alignments | Evolutionary analyses of synteny and collinearity | Analyses of gene families |
|---|---|---|---|---|---|---|
| i-ADHoRe 3 | 2011 | + | + | + | − | − |
| LineUp | 2003 | − | − | − | − | − |
| TEAM | 2003 | − | + | − | − | − |
| MCMuSeC | 2009 | − | + | − | − | − |
| OrthoClusterDB | 2009 | + | + | + | − | − |
| DiagHunter | 2003 | + | − | − | − | − |
| DAGChainer | 2004 | + | − | − | − | − |
| ColinearScan | 2006 | − | − | − | − | − |
| MCScan | 2008 | − | + | + | − | − |
| SyMAP 3.4 | 2011 | + | + | − | − | − |
| FISH | 2003 | − | − | − | − | − |
| Cyntenator | 2010 | − | + | + | − | − |
| MicroSyn | 2011 | + | + | − | − | + |
| Cinteny | 2007 | + | + | − | − | − |
| MCScanX | + | + | + | + | + |
MCScanX is unique in providing multiple programs for evolutionary analysis of synteny and collinearity, which are a necessary step towards biological discovery. Further, MCScanX has connected collinearity analyses between whole-genome and gene family scales. To our knowledge, the following biological analyses implemented in MCScanX are not yet available in other synteny and collinearity detection tools: constructing gene families using collinearity information, inferring gene duplication modes and enrichments, detecting collinear tandem arrays, performing statistical analyses of duplication depths and collinear orthologs and annotating phylogenetic trees with collinearity and tandems.
For synteny and collinearity detection tools, effective identification of collinear gene pairs is the basis for collinear block construction and downstream analyses. It is informative to perform a quantitative evaluation of MCScanX on the identification of collinear gene pairs. Two widely implemented tools, MCScan and i-ADHoRe 3 were chosen as competitors. Since a benchmark for assessing synteny and collinearity tools has not been established (72), we compared their performances by applying them to the Arabidopsis thaliana genome. Note that a higher number of detected collinear gene pairs does not simply indicate better performance, as true and false positives must be simultaneously considered and well balanced (69). A total of 5794 collinear gene pairs (i.e. WGD duplicate gene pairs) in the Arabidopsis genome including 3822 α, 1451 β and 521 γ pairs profiled using an integrated phylogenomic approach in the study from Bowers et al. (6), were regarded as the whole set of collinear gene pairs. The performances of MCScan, MCScanX and i-ADHoRe 3 were evaluated by power (i.e. sensitivity), defined as the ratio between numbers of true positives and all collinear gene pairs; and precision, defined as the ratio between numbers of true positives and all positives (i.e. true positives + false positives). When MCScan and MCScanX were compared, the same parameters were used. Based on the default parameters of MCScanX (match size = 5, max gaps = 25), MCScan and MCScanX identified 4134 and 4225 collinear gene pairs, of which 3375 and 3407 were true positives, respectively. Power was 0.58 and 0.59, and precision was 0.82 and 0.81 for MCScan and MCScanX, respectively. The above statistics suggest that MCScan and MCScanX are generally comparable in detecting collinear gene pairs, while MCScanX has a slightly higher power and a slightly lower precision. Based on its default parameters, i-ADHoRe 3 identified 6233 non-overlapping collinear gene pairs, of which 3459 were true positives. Its power and precision was 0.60 and 0.55. However, direct comparison between MCScanX and i-ADHoRe 3 using their respective default parameters was not reasonable because i-ADHoRe 3 output many more positives. To this end, we executed MCScan and MCScanX using a more relaxed set of parameters (match size = 3, max gaps = 50), which output 5554 and 6110 positives, respectively. Based on the new parameters, power was 0.65 and 0.67, and precision was 0.68 and 0.64 for MCScan and MCScanX, respectively. The new statistics suggest that in terms of identification of collinear gene pairs, MCScan and MCScanX each perform better than i-ADHoRe 3 and remain comparable to one another, with MCScan having higher precision and MCScanX having higher power. The small difference between MCScan and MCScanX is because in order to make MCScanX more easily and efficiently implemented, pre-processing of BLASTP input was pipelined into the execution of the main programs and the dependency of MCL was dropped. In MCScan, cross-family BLASTP hits are removed based on MCL output, while in MCScanX, all non-self BLASTP hits are considered, leading to an enlarged pool of BLASTP hits. MCL may generate 5–20% incorrect families and its performance is affected by inflation value (a parameter of the MCL algorithm used to control the granularity/tightness of protein clusters) (52). So the cross-family BLASTP hits based on MCL gene families indeed contain some collinear gene pairs, though the proportion of collinear gene pairs is smaller in cross-family BLASTP hits than in within-family BLASTP hits. This results in marginally higher power and lower precision for MCScanX than MCScan, though their performances on identifying collinear gene pairs are very similar. Since MCScan was successfully applied to the distantly related apicomplexans (11), we believe that MCScanX is also applicable over a wide range of organisms besides angiosperms.
DISCUSSION
Synteny and collinearity information is important for elucidating the evolutionary histories of both genomes and gene families. Although many synteny and collinearity tools are available, their output files are often difficult to read and downstream evolutionary analysis programs are rarely provided. For this reason, users often have to write additional programs or reformat the synteny and collinearity output files in order to use third-party evolutionary analysis tools. This incompleteness of functionality has reduced the usefulness of existing synteny and collinearity detection tools. A distinguishing feature of MCScanX is that diverse tools for evolutionary analyses of synteny and collinearity are incorporated, which enables rapid and convenient conversion of synteny and collinearity information into evolutionary insights. In addition, many biological analyses implemented in MCScanX are unique. MCScanX can be used to effectively analyze chromosome structural changes and evolution, annotate new genomes and reveal the history of gene family expansions.
In conclusion, MCScanX is a toolkit that implements an adjusted MCScan algorithm for detection of synteny and collinearity and incorporates 14 computer programs for visualizing and analyzing identified synteny and collinearity. The usefulness of the MCScanX toolkit has been demonstrated through a series of real data applications and comparison with other synteny and collinearity detection tools. MCScanX is freely available at http://chibba.pgml.uga.edu/mcscan2/.
FUNDING
National Science Foundation (NSF: DBI 0849896, MCB 0821096, MCB 1021718 to A.H.P.); National Institutes of Health (R01 AI068908to J.C.K.) in part; resources and technical expertise from the University of Georgia Georgia Advanced Computing Resource Center in part. Funding for open access charge: NSF (DBI 0849896).
Conflict of interest statement. None declared.
REFERENCES
- 1.Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L. Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 2005;21:673–682. doi: 10.1016/j.tig.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pohlmann R, Luedi P, Choi SD, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 2004;304:304–307. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- 3.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuveglise C, Talla E, et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 4.Nakatani Y, Takeda H, Kohara Y, Morishita S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007;17:1254–1265. doi: 10.1101/gr.6316407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salse J, Abrouk M, Bolot S, Guilhot N, Courcelle E, Faraut T, Waugh R, Close TJ, Messing J, Feuillet C. Reconstruction of monocotelydoneous proto-chromosomes reveals faster evolution in plants than in animals. Proc. Natl Acad. Sci. USA. 2009;106:14908–14913. doi: 10.1073/pnas.0902350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–438. doi: 10.1038/nature01521. [DOI] [PubMed] [Google Scholar]
- 7.Hufton AL, Groth D, Vingron M, Lehrach H, Poustka AJ, Panopoulou G. Early vertebrate whole genome duplications were predated by a period of intense genome rearrangement. Genome Res. 2008;18:1582–1591. doi: 10.1101/gr.080119.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otto SP, Whitton J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000;34:401–437. doi: 10.1146/annurev.genet.34.1.401. [DOI] [PubMed] [Google Scholar]
- 9.Ranz JM, Casals F, Ruiz A. How malleable is the eukaryotic genome? Extreme rate of chromosomal rearrangement in the genus Drosophila. Genome Res. 2001;11:230–239. doi: 10.1101/gr.162901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourque G, Pevzner PA, Tesler G. Reconstructing the genomic architecture of ancestral mammals: lessons from human, mouse, and rat genomes. Genome Res. 2004;14:507–516. doi: 10.1101/gr.1975204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debarry JD, Kissinger JC. Jumbled genomes: missing Apicomplexan synteny. Mol. Biol. Evol. 2011;28:2855–2871. doi: 10.1093/molbev/msr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandepoele K, Saeys Y, Simillion C, Raes J, Van De Peer Y. The automatic detection of homologous regions (ADHoRe) and its application to microcolinearity between Arabidopsis and rice. Genome Res. 2002;12:1792–1801. doi: 10.1101/gr.400202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luc N, Risler JL, Bergeron A, Raffinot M. Gene teams: a new formalization of gene clusters for comparative genomics. Comput. Biol. Chem. 2003;27:59–67. doi: 10.1016/s1476-9271(02)00097-x. [DOI] [PubMed] [Google Scholar]
- 14.Hampson S, McLysaght A, Gaut B, Baldi P. LineUp: statistical detection of chromosomal homology with application to plant comparative genomics. Genome Res. 2003;13:999–1010. doi: 10.1101/gr.814403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ling X, He X, Xin D. Detecting gene clusters under evolutionary constraint in a large number of genomes. Bioinformatics. 2009;25:571–577. doi: 10.1093/bioinformatics/btp027. [DOI] [PubMed] [Google Scholar]
- 16.Vergara IA, Chen N. Using OrthoCluster for the detection of synteny blocks among multiple genomes. Curr. Protoc. Bioinform. 2009 doi: 10.1002/0471250953.bi0610s27. 27, 6.10.1–6.10.18. [DOI] [PubMed] [Google Scholar]
- 17.Haas BJ, Delcher AL, Wortman JR, Salzberg SL. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics. 2004;20:3643–3646. doi: 10.1093/bioinformatics/bth397. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Shi X, Li Z, Zhu Q, Kong L, Tang W, Ge S, Luo J. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinformatics. 2006;7:447. doi: 10.1186/1471-2105-7-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18:1944–1954. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soderlund C, Nelson W, Shoemaker A, Paterson A. SyMAP: a system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006;16:1159–1168. doi: 10.1101/gr.5396706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calabrese PP, Chakravarty S, Vision TJ. Fast identification and statistical evaluation of segmental homologies in comparative maps. Bioinformatics. 2003;19(Suppl. 1):i74–i80. doi: 10.1093/bioinformatics/btg1008. [DOI] [PubMed] [Google Scholar]
- 22.Rodelsperger C, Dieterich C. CYNTENATOR: progressive gene order alignment of 17 vertebrate genomes. PLoS One. 2010;5:e8861. doi: 10.1371/journal.pone.0008861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. Synteny and collinearity in plant genomes. Science. 2008;320:486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 24.Abrouk M, Murat F, Pont C, Messing J, Jackson S, Faraut T, Tannier E, Plomion C, Cooke R, Feuillet C, et al. Palaeogenomics of plants: synteny-based modelling of extinct ancestors. Trends Plant Sci. 2010;15:479–487. doi: 10.1016/j.tplants.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Jun J, Mandoiu II, Nelson CE. Identification of mammalian orthologs using local synteny. BMC Genomics. 2009;10:630. doi: 10.1186/1471-2164-10-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casneuf T, De Bodt S, Raes J, Maere S, Van de Peer Y. Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 2006;7:R13. doi: 10.1186/gb-2006-7-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freeling M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009;60:433–453. doi: 10.1146/annurev.arplant.043008.092122. [DOI] [PubMed] [Google Scholar]
- 28.Cai B, Yang X, Tuskan GA, Cheng ZM. MicroSyn: a user friendly tool for detection of microsynteny in a gene family. BMC Bioinformatics. 2011;12:79. doi: 10.1186/1471-2105-12-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Tang H, Paterson AH. Seventy million years of concerted evolution of a homoeologous chromosome pair, in parallel, in major Poaceae lineages. Plant Cell. 2011;23:27–37. doi: 10.1105/tpc.110.080622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Tang H, Bowers JE, Paterson AH. Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 2009;19:1026–1032. doi: 10.1101/gr.087288.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch D, et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 2008;148:1772–1781. doi: 10.1104/pp.108.124867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charles M, Tang HB, Belcram H, Paterson A, Gornicki P, Chalhoub B. Sixty million years in evolution of soft grain trait in grasses: emergence of the softness locus in the common ancestor of Pooideae and Ehrhartoideae, after their divergence from Panicoideae. Mol. Biol. Evol. 2009;26:1651–1661. doi: 10.1093/molbev/msp076. [DOI] [PubMed] [Google Scholar]
- 33.Lin L, Pierce GJ, Bowers JE, Estill JC, Compton RO, Rainville LK, Kim C, Lemke C, Rong J, Tang H, et al. A draft physical map of a D-genome cotton species (Gossypium raimondii) BMC Genomics. 2010;11:395. doi: 10.1186/1471-2164-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin L, Tang H, Compton RO, Lemke C, Rainville LK, Wang X, Rong J, Rana MK, Paterson AH. Comparative analysis of Gossypium and Vitis genomes indicates genome duplication specific to the Gossypium lineage. Genomics. 2011;97:313–320. doi: 10.1016/j.ygeno.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Tang H, Bowers JE, Wang X, Paterson AH. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl Acad. Sci. USA. 2010;107:472–477. doi: 10.1073/pnas.0908007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Wang X, Tang H, Tan X, Ficklin SP, Feltus FA, Paterson AH. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One. 2011;6:e28150. doi: 10.1371/journal.pone.0028150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 38.Causier B, Castillo R, Xue YB, Schwarz-Sommer Z, Davies B. Tracing the evolution of the floral homeotic B- and C-function genes through genome synteny. Mol. Biol. Evol. 2010;27:2651–2664. doi: 10.1093/molbev/msq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knoller AS, Blakeslee JJ, Richards EL, Peer WA, Murphy AS. Brachytic2/ZmABCB1 functions in IAA export from intercalary meristems. J. Exp. Bot. 2010;61:3689–3696. doi: 10.1093/jxb/erq180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe M, Mochida K, Kato T, Tabata S, Yoshimoto N, Noji M, Saito K. Comparative genomics and reverse genetics analysis reveal indispensable functions of the serine acetyltransferase gene family in Arabidopsis. Plant Cell. 2008;20:2484–2496. doi: 10.1105/tpc.108.060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okazaki Y, Shimojima M, Sawada Y, Toyooka K, Narisawa T, Mochida K, Tanaka H, Matsuda F, Hirai A, Hirai MY, et al. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell. 2009;21:892–909. doi: 10.1105/tpc.108.063925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyun TK, Kim JS, Kwon SY, Kim SH. Comparative genomic analysis of mitogen activated protein kinase gene family in grapevine. Genes Genom. 2010;32:275–281. [Google Scholar]
- 43.Li C, Zhang YM. Molecular evolution of glycinin and beta-conglycinin gene families in soybean (Glycine max L. Merr.) Heredity. 2011;106:633–641. doi: 10.1038/hdy.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Liu B, Yu L, Feng D, Wang H, Wang J. Phylogenetic analysis, structural evolution and functional divergence of the 12-oxo-phytodienoate acid reductase gene family in plants. BMC Evol. Biol. 2009;9:90. doi: 10.1186/1471-2148-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kopriva S, Mugford SG, Matthewman C, Koprivova A. Plant sulfate assimilation genes: redundancy versus specialization. Plant Cell Rep. 2009;28:1769–1780. doi: 10.1007/s00299-009-0793-0. [DOI] [PubMed] [Google Scholar]
- 46.Palmieri F, Pierri CL, De Grassi A, Nunes-Nesi A, Fernie AR. Evolution, structure and function of mitochondrial carriers: a review with new insights. Plant J. 2011;66:161–181. doi: 10.1111/j.1365-313X.2011.04516.x. [DOI] [PubMed] [Google Scholar]
- 47.Higgins JA, Bailey PC, Laurie DA. Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS One. 2010;5:e10065. doi: 10.1371/journal.pone.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Gowik U, Tang H, Bowers JE, Westhoff P, Paterson AH. Comparative genomic analysis of C4 photosynthetic pathway evolution in grasses. Genome Biol. 2009;10:R68. doi: 10.1186/gb-2009-10-6-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 50.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 52.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao XP, Si Y, Hanson RE, Crane CF, Price HJ, Stelly DM, Wendel JF, Paterson AH. Dispersed repetitive DNA has spread to new genomes since polyploid formation in cotton. Genome Res. 1998;8:479–492. doi: 10.1101/gr.8.5.479. [DOI] [PubMed] [Google Scholar]
- 54.Ganko EW, Meyers BC, Vision TJ. Divergence in expression between duplicated genes in Arabidopsis. Mol. Biol. Evol. 2007;24:2298–2309. doi: 10.1093/molbev/msm158. [DOI] [PubMed] [Google Scholar]
- 55.Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–573. doi: 10.1038/nature02953. [DOI] [PubMed] [Google Scholar]
- 56.Yang L, Bennetzen JL. Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc. Natl Acad. Sci. USA. 2009;106:19922–19927. doi: 10.1073/pnas.0908008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, Zheng H, Fan C, Li J, Shi J, Cai Z, Zhang G, Liu D, Zhang J, Vang S, et al. High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell. 2006;18:1791–1802. doi: 10.1105/tpc.106.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vergara IA, Chen N. Large synteny blocks revealed between Caenorhabditis elegans and Caenorhabditis briggsae genomes using OrthoCluster. BMC Genomics. 2010;11:516. doi: 10.1186/1471-2164-11-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- 60.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 61.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 62.Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet. 2007;3:e123. doi: 10.1371/journal.pgen.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Quraishi UM, Calcagno T, Cooke R, Delseny M, Feuillet C. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell. 2008;20:11–24. doi: 10.1105/tpc.107.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rizzon C, Ponger L, Gaut BS. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput. Biol. 2006;2:e115. doi: 10.1371/journal.pcbi.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sampedro J, Lee Y, Carey RE, dePamphilis C, Cosgrove DJ. Use of genomic history to improve phylogeny and understanding of births and deaths in a gene family. Plant J. 2005;44:409–419. doi: 10.1111/j.1365-313X.2005.02540.x. [DOI] [PubMed] [Google Scholar]
- 66.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 67.Becker A, Theissen G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003;29:464–489. doi: 10.1016/s1055-7903(03)00207-0. [DOI] [PubMed] [Google Scholar]
- 68.Freeling M, Lyons E, Pedersen B, Alam M, Ming R, Lisch D. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 2008;18:1924–1937. doi: 10.1101/gr.081026.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cannon SB, Kozik A, Chan B, Michelmore R, Young ND. DiagHunter and GenoPix2D: programs for genomic comparisons, large-scale homology discovery and visualization. Genome Biol. 2003;4:R68. doi: 10.1186/gb-2003-4-10-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinha AU, Meller J. Cinteny: flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC Bioinformatics. 2007;8:82. doi: 10.1186/1471-2105-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng MP, Vergara IA, Frech C, Chen Q, Zeng X, Pei J, Chen N. OrthoClusterDB: an online platform for synteny blocks. BMC Bioinformatics. 2009;10:192. doi: 10.1186/1471-2105-10-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fostier J, Proost S, Dhoedt B, Saeys Y, Demeester P, Van de Peer Y, Vandepoele K. A greedy, graph-based algorithm for the alignment of multiple homologous gene lists. Bioinformatics. 2011;27:749–756. doi: 10.1093/bioinformatics/btr008. [DOI] [PubMed] [Google Scholar]
- 73.Soderlund C, Bomhoff M, Nelson WM. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011;39:e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]