Smith-Magenis Syndrome Results in Disruption of CLOCK Gene Transcription and Reveals an Integral Role for RAI1 in the Maintenance of Circadian Rhythmicity (original) (raw)
Abstract
Haploinsufficiency of RAI1 results in Smith-Magenis syndrome (SMS), a disorder characterized by intellectual disability, multiple congenital anomalies, obesity, neurobehavioral abnormalities, and a disrupted circadian sleep-wake pattern. An inverted melatonin rhythm (i.e., melatonin peaks during the day instead of at night) and associated sleep-phase disturbances in individuals with SMS, as well as a short-period circadian rhythm in mice with a chromosomal deletion of Rai1, support SMS as a circadian-rhythm-dysfunction disorder. However, the molecular cause of the circadian defect in SMS has not been described. The circadian oscillator temporally orchestrates metabolism, physiology, and behavior largely through transcriptional modulation. Data support RAI1 as a transcriptional regulator, but the genes it might regulate are largely unknown. Investigation into the role that RAI1 plays in the regulation of gene transcription and circadian maintenance revealed that RAI1 regulates the transcription of circadian locomotor output cycles kaput (CLOCK), a key component of the mammalian circadian oscillator that transcriptionally regulates many critical circadian genes. Data further show that haploinsufficiency of RAI1 and Rai1 in SMS fibroblasts and the mouse hypothalamus, respectively, results in the transcriptional dysregulation of the circadian clock and causes altered expression and regulation of multiple circadian genes, including PER2, PER3, CRY1, BMAL1, and others. These data suggest that heterozygous mutation of RAI1 and Rai1 leads to a disrupted circadian rhythm and thus results in an abnormal sleep-wake cycle, which can contribute to an abnormal feeding pattern and dependent cognitive performance. Finally, we conclude that RAI1 is a positive transcriptional regulator of CLOCK, pinpointing a novel and important role for this gene in the circadian oscillator.
Introduction
Smith-Magenis syndrome (SMS, MIM 182290) is a complex genomic disorder that results from an interstitial deletion of chromosomal region 17p11.2, including RAI1 (retinoic acid induced one [MIM 607642]), or heterozygous mutation of RAI1. Along with behavioral, skeletal, and neurological abnormalities, sleep disturbance is a hallmark of SMS.
Circadian rhythms are endogenous ∼24 hr cycles affecting and modulating physiological processes and behavior found in organisms that range from bacteria to mammals. The circadian oscillator enables organisms to anticipate predictable changes in the environment and temporally orchestrate their metabolism, physiology, and behavior largely through transcriptional modulation. Reports indicate that 75%–100% of all individuals with SMS present with sleep disturbance, and this is one of the early indicators that an individual might have SMS.1,2 Although infants typically present with hypersomnolence early in life, sleep disturbance, including difficulty falling asleep, inability to enter or maintain REM (rapid-eye movement) sleep, reduced night sleep, shortened and broken sleep cycles with frequent night-time and early-morning awakenings and excessive daytime sleepiness, begin in early toddlerhood and last well into adulthood.3,4 Studies in older adults are limited; however, sleep disturbance continues to be a common complaint among families.
Several studies have implicated an inverted rhythm of melatonin secretion as the root cause of the sleep disturbance, providing evidence that these circadian-rhythm difficulties can complicate behavior and learning.5–8 Furthermore, individuals with RAI1 mutation also have altered melatonin secretion, implicating RAI1 directly in circadian function.9 However, complicating this analysis is a report of a 17p11.2-deletion individual who has normal melatonin secretion but who still has sleep disturbance, indicating a possible molecular role for RAI1 in the sleep cycle and in the core molecular pathway of circadian rhythm.4 Additionally, mice in which Rai1 has been deleted have abnormal circadian behavior, including a shortened period length, which was observed after _Rai1_-deletion mice and wild-type (WT) littermates were entrained to the 12 hr light/12 hr dark cycle and then tested in constant darkness.10
Given the phenotypic consequences of RAI1 mutation or deletion, RAI1 must be involved in pathways associated with developmental, behavioral, and neurological function and circadian rhythm,11 and identification of the specific molecular changes and subsequent affected pathways that result from RAI1 haploinsufficiency holds the potential for pharmaceutical-, behavioral-, or nutritional-based interventions. Thus, we set out to investigate the biological role of RAI1 and Rai1 in circadian rhythm. Data show that RAI1 is a critical player in the maintenance of the molecular clock, wherein haploinsufficiency of RAI1 results in the dysregulation of critical genes involved in circadian biology.
Material and Methods
Cell Culture
Unless otherwise noted, all cell lines used in this study were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM [Invitrogen]) with 10% (v/v) FBS, 2 mM L-glutamine, and 1X antibiotic-antimycotic (Invitrogen) and were maintained at 37°C in a 5% CO2 incubator. Fibroblasts utilized in this study included GM0063712 (Coriell Cell Repository), BAB23913 (common SMS 17p11.2 deletion), SMS18214 (atypical SMS 17p11.2 deletion), and SMS17515 (RAI1 mutation [p.1562Gln>Arg]). Fibroblasts were collected from skin biopsies obtained from consenting participants in the National Human Genome Research Institute of the National Institutes of Health under protocol number 01-HG-0109.
siRNA Knockdown of RAI1 in HEK 293T Cells
RAI1 in human embryonic kidney (HEK) 293T cells was knocked down as previously described.16 Cells were reverse transfected with Trans-it LT1 (Mirus Bio) to a final concentration of 45 nM siRNA specific to RAI1 (Ambion) or with a 45 nM scrambled-sequence control. After 24 hr, RNA was isolated with the RNeasy kit according to the standard manufacturer (Invitrogen) protocols. Next, RNA was reverse transcribed with qScript (Quanta Biosciences). Transcription levels were measured with Syber Green Mastermix (Applied Biosystems [ABI]), and GAPDH was used as the internal control for performing the standard ΔΔCT relative quantification. Sybr Green primers are as follows: forward 5′-CCGACAAGGGTCGCAAACA-3′ and reverse 5′-CTTTGTGGCAGCACCCAATG-3′ for RAI1; forward 5′-ATGGGGAAGGTGAAGGTCG-3′ and reverse 5′-GGGGTCATTGATGGCAACAATA-3′ for GAPDH; forward 5′-TAAGGGGCAACAGTGGATTTG-3′ and reverse 5′-CAGCCCTAACTTCTGCATAACTT-3′ for CLOCK (circadian locomotor output cycles kaput [MIM 601851]); forward 5′-TTTGTGGCGATAAGTCCTCTGG-3′ and reverse 5′-CTTGGGCAGGAATAAGAAGCAT-3′ for RORA (MIM 600825); forward 5′-ATGTTGGAGGCATTAGATGGC-3′ and reverse 5′-TCCATGACATCCGACGGTAAA-3′ for NPAS2 (MIM 603347); and forward 5′-GCATGGATGCTTACCCAACT-3′ and reverse 5′-AATCTTCCAATGGCCACAAG-3′ for BMAL2 (Entrez Gene 56938).
Mouse-Tissue Collection
Rai1+/− mice were obtained from the Jackson Laboratories (B6.129s7-_Rai1tm1jrt/_J; stock 005981)17 and were bred with WT C57BI/6J females for the generation of Rai1+/− and WT pups. Genotyping was performed by PCR with primers specific to the _Rai1_-targeted allele and the WT allele, as reported.17 Mice were maintained in a 12 hr light/12 hr dark cycle and were fed normal chow ad libitum. Equal numbers of healthy WT and Rai1+/− male and female mice were euthanized at 9–15 months of age either during the day at ∼8 hr Zeitgeber time (Zeitgeber time 0 = 6 am) or at night at ∼16 hr Zeitgeber time. WT C57BI/6J and Rai1+/− mice were euthanized by CO2 anesthesia, and tissues were collected in accordance with standard protocols and frozen at −80°C.
Quantitative PCR
Total RNA was isolated from mouse tissues, HEK 293T cells, and SMS fibroblast cell lines with Trizol according to the manufacturer's (Invitrogen's) instructions. All cell lines were cultured for the same period of time to the same cell density, and all samples were processed independently according to standard protocols. Fibroblasts were serum shocked 24 hr prior to RNA isolation for the synchronization of cells. Prior to reverse transcription, RNA quality and concentration were determined by standard NanoDrop. First-strand cDNA synthesis was carried out with either Invitrogen's Superscript kit (with 4 μg of total RNA) or Quanta Biosciences's Qscript (with 2 μg of RNA) according to the manufacturers' instructions. For quantitative real-time PCR, predesigned Taqman MGB probes and primers from the Assays-on-Demand Gene Expression Products (ABI) were used for all genes, both human and mouse. All samples were run in triplicate in 10 μl reaction volumes. PCR conditions were the default settings of the ABI Prism 7900 HT Sequence Detection System. The cycle threshold (Ct) was determined during the geometric phase of the PCR amplification plots as recommended by the manufacturer. Relative differences in transcript levels were quantified with the ΔΔCt method with GAPDH (MIM 138400) mRNA as an endogenous control. All expression values are calculated relative to control levels set at 1.0. Data were analyzed by 1 sample t tests with an experimentally defined control value of 1.0 and a p value < 0.05 as described before.18 Acquired data were analyzed with Excel, and graphs were plotted with GraphPad Prism 4.
RAI1 Knockdown in U2OS-B Cells and Evaluation of Live-Cell Circadian Phenotype by Bioluminescence Rhythms
U2OS-B cells19 (∼1.5 × 10−4 cells), which have a stably integrated _Bmal1_-luciferase gene, were reverse transfected with Trans-it LT1 (Mirus Bio) to a final concentration of 15 nM, 30 nM, or 45 nM siRNA specific to RAI1 (Ambion) or with a 15 nM scrambled-sequence control in a 96-well tissue-culture plate suitable for luciferase analysis. All conditions were plated six times for the assessment of experimental variability. Cells were cultured in DMEM (Invitrogen) with 10% (v/v) FBS, 2 mM glutamine, 1X Anti-Anti (Invitrogen), and 0.1 mM D-Luciferin at 37°C in a Tecan luminometer as previously described.19 Luciferase readings were taken every 15 min for 7 days. The first and last days of readings were eliminated from the analysis because they were not reliable. The data obtained for each well were detrended with a 24 hr moving window average and smoothed with a 2 hr moving average.19 Period length was determined by COSOPT analysis with raw data as previously described.19,20
Chromatin Immunoprecipitation with Microarray
Chromatin immunoprecipitation with microarray (ChIP-Chip) was described previously.21 In brief, HEK 293T cells were transfected with the RAI1Flag plasmid, ChIP-Chip was processed with mouse IgG Dynabeads (Invitrogen), and the monoclonal Flag IgG antibody (Sigma Aldrich) was produced in mice with Nimblegen “ChIP sample preparation protocol v.2” (Roche Nimblegen) according to the manufacturers' protocols. Transfected cells were treated with formaldehyde for the crosslinking of protein complexes with nuclear chromatin. Protein-chromatin products were isolated, and crosslinks were reversed with heat treatment. After reverse crosslink, chromatin products were whole-genome amplified with the WGA2-10RXN kit (Sigma-Aldrich) as per recommendation by Nimblegen. Arrays were processed by Nimblegen with the Nimblegen HG18 RefSeq promoter array, which assays 5′UTRs across the genome, according to standard manufacturer protocols. Data were preprocessed by Nimblegen according to standard protocols. RAI1Flag binding sites were generated with genomic data points, Peak_Start and Peak_End, from Nimblegen preprocessed data files.
Creation of Plasmids
For RAI1Flag, a RAI1 coding sequence was cloned into pEntr/D-TOPO and transformed into One Shot Competent E. coli according to the manufacturer's (Invitrogen's) instructions. Plasmid DNA was isolated with the Fermentus GeneJET plasmid mini kit according to standard manufacturer instructions. Inserts were confirmed with RAI1 cDNA primers and were sequenced according to standard Sanger techniques. Next, RAI1pEntr/D-TOPO was recombined with pDest26Flag22 for the creation of pDest26_RAI1Flag_ according to the standard gateway protocol provided by Invitrogen.
For CLOCKLuc, the first CLOCK intron element was PCR amplified according to standard techniques (forward primer 5′-GGACCTTTGCAAGAGCCCAAG-3′ and reverse primer 5′-GCAGAGCACAGAGGGCTTTTAGGCCGATGT-3′) and was cloned into a StrataClone PCR Cloning vector according to the standard protocol provide by the manufacturer (Agilent Technologies) for the creation of the CLOCKStrata plasmid. The CLOCKStrata plasmid and the pGL3pro vector (Promega Corporation) were digested with KpnI and SacI (New England Biolabs) and were gel extracted with the QIAquick Gel Extraction Kit according to standard manufacturer (QIAGEN) protocols. The insert from CLOCKStrata was directionally ligated into the pGL3pro vector for the creation of CLOCKLuc according to standard T4 ligation protocols provided by the manufacturer (Promega Corporation). The constructs shown in Figure 5 were created as described above and in the supplemental data (see Supplemental Methods and Table S1, available online). All inserts were confirmed by standard Sanger sequencing.
Figure 5.
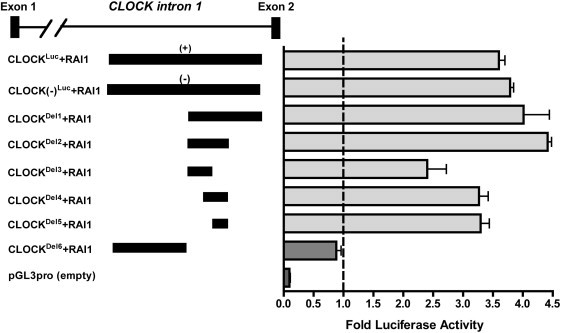
RAI1 Activation of CLOCK Gene Enhancer Element
HEK 293T cells were transfected in triplicate with three constructs (RAI1, β-GAL, and one of the test luciferase plasmids [CLOCKLuc, CLOCK(−)Luc, CLOCKDel1, CLOCKDel2, CLOCKDel3, CLOCKDel4, CLOCKDel5, or CLOCKDel6]) for a total of eight different combinations. The RAI1 plasmid represents the cDNA sequence of RAI1 in the pDEST26 vector (RAI1Flag). All assays were performed in triplicate. The x axis compares the luciferase activity of the luciferase constructs activated by RAI1 with that of the constructs alone, which are represented by the hatched line. Data shown are the average of three independent biological replicates for each construct. Error bars represent the standard deviation from the mean. Details for the creation of each construct are provided in the Material and Methods section and in the Supplemental Methods.
Transfections
HEK 293T cells were maintained in 6-well plates according to standard culturing practices described above. Transfections with pUC19, psvβ-Gal, CLOCKLuc constructs (see Figure 5), and RAI1Flag were performed with Lipofectamine 2000 according to the manufacturer's (Invitrogen's) instructions. In brief, ∼5 × 105 cells were plated in 2.0 ml of growth medium without antibiotics 24 hr prior to transfection. A total of 4 μg of total plasmid DNA, with the use of pUC19 plasmid as “filler” DNA so that transfection efficiencies were similar across all test groups, was diluted in 250 μl OptiMEM Reduced Serum Medium (Invitrogen). Similarly, 10 μl of Lipofectamine 2000 was diluted in 250 μl of OptiMEM Reduced Serum Medium, mixed well, and incubated for 5 min. After incubation, diluted plasmid DNA was mixed with diluted Lipofectamine 2000 to a total volume of 500 μl and was incubated for 20 min. Plasmid-Lipofectamine complexes (500 μl) were added to each well and were mixed by rocking. The cells were then incubated at 37°C in a 5% CO2 incubator for 24 hr.
Luciferase Assays
After the plasmid-DNA transfection and 24 hr incubation, cells were washed with 2 ml Dulbecco's phosphate-buffered saline (DPBS [Invitrogen]) and Tropix Glacto-Light (ABI); standard protocol was used. In brief, 500 μl of lysis solution was added to each well of the 6-well plate and scraped until all cells were detached. Lysates were collected and centrifuged at 12,000 RPM for 2 min so that the cell debris would pellet. Next, 50 μl of the supernatant was transferred to four wells of a 96-well white luminometer plate. Two wells were treated with 70 μl diluted Galacton substrate (1:100, reaction buffer diluent with Galacton) and incubated for 30 min; then, 100 μl of Accelerator(-II) was added. One hundred microliters of Steady-Glo Luciferase substrate (Promega Corporation) was added to the two wells that did not contain the diluted Galacton substrate. Each well was read with the Wallac 1420 VICTOR2 luminometer (PerkinElmer) on a maximum linear scale. We calculated the relative luciferase activity from each individual transfection by dividing the average number of light units from the wells containing the Steady-Glo Luciferase substrate (Promega Corp) by the average number of light units from the wells containing the Galacton substrate and Accelerator(-II). The equation (average luc/average β-gal = relative luciferase activity) was used. Wells containing pUC19, psvβ-Gal, and CLOCKLuc constructs were used as baseline luciferase activity. Each experiment was performed independently at least three times in triplicate wells for every assay. We generated p values by averaging relative luciferase activity from each independent study and performing a two-tailed Student's t test. Standard deviations were generated with GraphPad Prism 4.
Statistical Analyses
Figures 2B and 2C were generated with GraphPad Prism 4 software and then subjected to ANOVA followed by Dunnett's test. All other statistics were performed with a standard 1 sample Student's t test. For the analysis, individual values for each test group were uploaded to the St. John's University Department of Physics statistics website. Statistical significance was determined at a cutoff of p ≤ 0.05. All plots were generated with GraphPad Prism 4.
Figure 2.
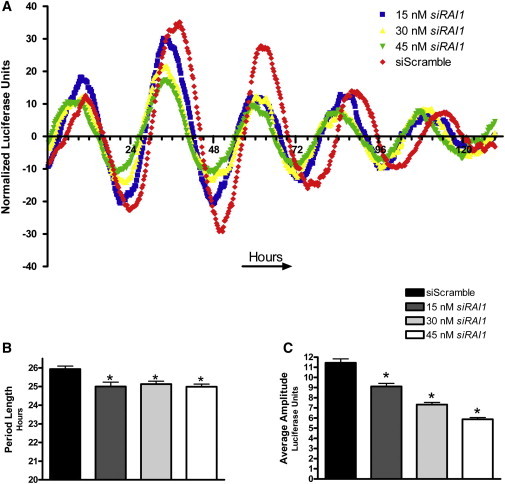
RAI1 siRNA Knockdown in U2OS-B Cells Results in a Shortened Period and Reduced Amplitude of Bmal1 Expression
Luciferase expression from U2OS-B cells treated with siRNA specific to RAI1 over a 5 day period and COSOPT analysis of period length are shown. Data reflect all luciferase measurements taken. The average for each siRNA concentration is compared to the control average, and asterisks represent p values < 0.001 (calculated with ANOVA and Dunnett's test). Data shown are representative of three independent experiments. Error bars represent the standard deviation from the mean.
(A) The x axis represents time, and ticks are shown in 4 hr increments. The y axis represents normalized and averaged luciferase activity of six wells for each experimental condition.
(B) Average period length of six wells after COSOPT analysis.
(C) Average amplitude of circadian wave.
Results
Knockdown of RAI1 Results in Altered Expression of the Key Circadian Components
Numerous studies have shown that when a major component of the circadian system is eliminated or modulated, significant downstream effects can occur.19,23 To examine our hypothesis that RAI1 is a component of the circadian feedback loop, we knocked down RAI1 in HEK 293T cells to ∼50% of normal expression (Figure 1). Consistent with the prediction that RAI1 plays a role in the maintenance of correct transcription of circadian components, the data show significant dysregulation of all tested circadian transcripts (Figure 1). When RAI1 expression is reduced by ∼50% in HEK 293T cells, expression of CLOCK, the “master regulator” of the central circadian clock, is reduced to ∼50%. Furthermore, in addition to reduced CLOCK expression, genes that CLOCK directly regulates and other essential components of the circadian feedback loop, including RORC (MIM 602943), RORA (MIM 600825), PER3 (MIM 603427), CRY1 (MIM 601933), CRY2 (MIM 603732), NR1D2 (MIM 602304), and NR1D1 (MIM 602408), also have significantly reduced or altered expression (Figure 1). These genes represent the major families of genes targeted by CLOCK and are all essential components of the feedback loop of transcription. These data show that haploinsufficiency of RAI1 results in significant disruption of circadian expression of core components in vitro, indicating a major role for RAI1 in maintaining transcription of these genes. Additionally, these data lend credence to the theory that RAI1 has a primary role in circadian-component regulation and are key to understanding the foundation of the SMS circadian phenotype.
Figure 1.
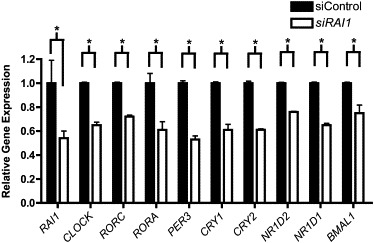
siRNA Knockdown of RAI1 in HEK 293T Cells Alters Circadian-Gene Expression
Circadian-gene expression in HEK 293T cells treated for knockdown of RAI1 by siRNA. Data shown are an average of three independent experiments performed in triplicate. Relative gene expression is shown (control siRNA is shown in black, and RAI1 siRNA is shown in white). Bars represent standard deviation. The quantification of the test gene is compared to that of the housekeeping gene GAPDH. All siRNA-treated samples are statistically different than controls, as shown by asterisks, which indicate p values < 0.05 (calculated with Student's t test). Error bars represent the standard deviation from the mean.
Knockdown of RAI1 in U2OS-B Cells Results in a Shortened Period Length and Dampened Amplitude of Bmal1 Expression
To further investigate the role of RAI1 in circadian rhythmicity, we ultimately knocked down RAI1 to ∼50% of endogenous levels in a dose-dependent fashion; these levels are consistent with theoretical expression in SMS subjects. To accurately assess the circadian cycle over a long period of time, we utilized a transgenic cell line that carries the firefly luciferase gene under the control of the Bmal1 promoter (Bmal1Luc).19 BMAL1 cycles with great accuracy and with robust expression, so its expression is a reliable measure of overall circadian stability. As seen in Figure 2, dose-dependent knockdown of RAI1 results in a significantly shortened period length of approximately 1 hr (Figures 2A and 2B) and decreased amplitude of Bmal1Luc expression in U20S-B cell lines (Figures 2B and 2C). Dampened Bmal1Luc expression was not surprising because it correlates with the reduced BMAL1 seen in Figure 1. However, it is worth noting that depending on the level of CLOCK knockdown in vitro, BMAL1 amplitude can be dampened significantly (this correlates with the data that we present), but the fact that it can also increase points to the level of fine tuning that occurs in the circadian feedback loop.24 These data show that reduction in RAI1 expression can affect the circadian system over time in vitro, further indicating that RAI1 is a key component in the maintenance of proper circadian rhythms. Importantly, these data are consistent with a report indicating that haploinsufficiency of Rai1 in mice results in a shortened-period phenotype as well.10
Dysregulation of Circadian Genes in SMS Fibroblasts
Human fibroblasts follow a rhythmicity of ∼24 hr in vitro just as they would in vivo. To assess the circadian cycling of a panel of core circadian genes, including RAI1, CLOCK, and NR1D1, we took RNA samples from unaffected (GM00637),18 common SMS 17p11.2 deletion (BAB239),19 atypical SMS 17p11.2 deletion (SMS182),20 and RAI1 mutation (SMS175 [p.1562Gln>Arg]21) cell lines. All cells were synchronized by serum shock 24 hr before measures were taken so that we could assess any desynchronization from a common starting point of transcription. Our data show that transcript levels of all assessed genes (RAI1, CLOCK, PER1, ARNTL2, NR1D1, and CRY1) are dysregulated when compared to those of normal fibroblasts (Figure 3). These data further support a role for RAI1 in the regulation of circadian genes and illustrate that when RAI1 is haploinsufficient as a result of deletion or mutation, transcriptional regulation of the molecular clock is disrupted. Although some aspects of the circadian feedback loop of expression have been analyzed in SMS subjects,25 these data show a true profile of the core circadian components in SMS tissue and that these circadian alterations are associated with both deletion and mutation of RAI1.
Figure 3.
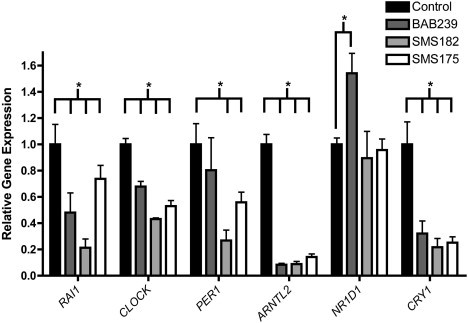
Altered Expression of Core Circadian Genes in SMS Fibroblasts
Gene expression in SMS fibroblasts was assessed 24 hr after cell synchronization by cell passage. Fibroblasts utilized included normal control fibroblasts BAB239 (common SMS deletion), SMS182 (small, unusual SMS deletion), and SMS175 (RAI1 missense mutation). Significant differences in gene expression, SMS versus control, are indicated by asterisks. All expression values are calculated relative to control levels set at 1.0. A p value < 0.05 was determined by 1 sample t tests. The quantification of the test gene is compared to that of the housekeeping gene GAPDH. Error bars represent the standard deviation from the mean.
Dysregulation of Circadian Genes in the Rai1+/− Mouse Hypothalamus
Rai1+/− mice, which harbor a targeted mutation of Rai1, are obese and hyperphagic, have decreased sensitivity to pain, and exhibit reduced muscle strength.17,21,26,27 In addition, a chromosomal deletion model of SMS, the Df(11)17 mouse, has the same overall phenotype as the Rai1+/− mouse but also has an altered circadian rhythm, which has not been assessed in the Rai1+/− mouse model.17 Expression analysis of Rai1 and Clock in the Rai1+/− mouse hypothalamus during the light phase (∼8 hr Zeitgeber) showed reduced expression of both genes (Figure 4A). The suprachiasmatic nucleus (SCN) lies within the hypothalamus and is the region of the brain that is responsible for controlling the central circadian rhythm. Furthermore, we analyzed the expression of downstream targets of Clock and other essential components of the circadian feedback loop, including Per2, Npas2, and Nr1d2, during the light phase, and all genes assessed had reduced expression when compared to those of the WT C57Bl/6J littermates (Figure 4A). These data illustrate dysregulation of diurnal gene expression in the Rai1+/− mouse hypothalamus.
Figure 4.
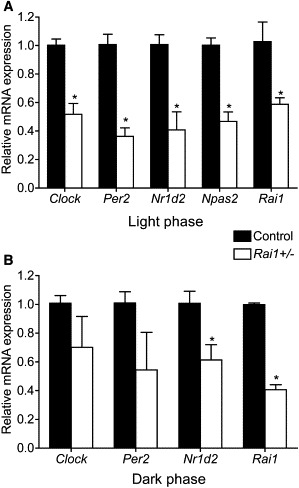
Altered Expression of Circadian Genes in Rai1+/− Mouse Hypothalamus during Light and Dark Phases
(A) Circadian-gene expression in mouse hypothalamus extracted from WT (black bars) or Rai1+/− mice (white bars) during the light phase at 8 hr Zeitgeber time.
(B) Circadian-gene expression in mouse hypothalamus extracted from WT (black bars) or Rai1+/− mice (white bars) during the dark phase at 16 hr Zeitgeber time. All values shown are the mean of at least three independent samples analyzed in triplicate. The quantification of the test gene is compared to that of the housekeeping gene Gapdh. Error bars represent the standard deviation from the mean. Asterisks indicate p < 0.05 by Student's t test.
We next evaluated diurnal gene expression in the mouse hypothalamus during the night (∼16 hr Zeitgeber) to ensure that expression differences remained and were not restored as a result of any external cues, such as the absence of light (Figure 4B). The data show that during the dark phase, the expression of core circadian genes is dysregulated; these genes include Per2, which results in a severe circadian phenotype when it is knocked out in mice,28 and Nr1d2, which was identified as one of the top genes with reduced expression in a study of human cells knocked down for RAI1 (Figure 4B).16 The variation in expression of these genes during the dark phase in Rai1+/− mice is illustrative of the dysregulation and malfunction of the circadian autoregulatory feedback loop.
RAI1 Transcriptionally Activates CLOCK via an Intron 1 Enhancer Element
The downstream targets of CLOCK are well characterized. However, the transcriptional regulators of CLOCK are largely unknown.29 Using peak data acquired from our chromatin immunoprecipitation for RAI1Flag and microarray analysis (ChIP-chip), we identified a ∼389 bp region in which RAI1Flag was bound. This element resides in CLOCK intron 1 (hg18, NCBI build 36, Chr4: 56,072,089–56,072,440). Initially, we cloned a 721 bp genomic region containing this element (CLOCKLuc) and performed luciferase assays to assess the ability of RAI1 to activate transcription via this potential enhancer element (Figure 5; see Material and Methods).
When the RAI1Flag construct was transfected with CLOCKLuc with either a (+) or a (−) strand, luciferase activity increased more than three times (Figure 5). We next designed deletion constructs to assess the smallest possible region that RAI1 could activate. When CLOCKDel1, CLOCKDel2, CLOCKDel3, and CLOCKDel4 were each activated by RAI1, luciferase activity was at least two times greater than that of each construct alone. When RAI1 was transfected with the smallest construct, CLOCKDel5, luciferase activity increased three times more than that of CLOCKDel5 alone, narrowing the enhancer binding region to 51 bp. This 51 bp region is the smallest tested region with the ability to enhance transcription via RAI1 (Figure 5). Additionally, when CLOCKDel6, which contains the 5′ end of the original CLOCKLuc, was tested, RAI1 did not have the ability to enhance transcription of this construct. These data indicate that CLOCKDel6 is not necessary for transcription control of this region, further validating that the 51 bp CLOCKDel5 is the most important location for RAI1 enhancer activity. Control experiments showed that RAI1 does not have the ability to enhance luciferase activity of the pGL3 promoter plasmid without an insert. All together, the ChIP-Chip and luciferase data suggest that RAI1 binds, directly or in a complex, to the first intron of CLOCK and enhances its transcriptional activity in vitro, supporting RAI1 as a positive regulator of CLOCK and an important part of the circadian loop of transcription.
Discussion
Mammalian circadian rhythm is an essential regulator of not only the sleep-wake cycle but also body temperature, feeding cycles, hormone secretion, drug and xenobiotic metabolism, glucose homeostasis, and cell-cycle progression.30 The master regulator of the circadian clock resides in the SCN located in the mammalian hypothalamus. The primary synchronizer of this clock is the 24 hr light/dark cycle, throughout which a feedback loop of gene transcription and the subsequent degradation of gene products occur and which signals the mentioned processes to react accordingly. Because of the complexity of this circadian loop, disturbances in the timing of gene expression can have a major impact on not only sleep pattern but also disease susceptibility and behavioral stability. Genes involved in fatty-acid metabolism, energy metabolism, and cholesterol biosynthesis are expressed in a circadian manner,30 and it has been shown that circadian disruption results in increased risk of not only sleep disturbance but also obesity,31 cancer,30 bipolar disorder, and schizophrenia.32 Interestingly, the ClockΔ19/Δ19 mice that harbor a homozygous mutation in Clock are obese and hyperphagic,33 a phenotype similar to that seen in RAI1+/− mice.21 Furthermore, altered circadian rhythm has been linked to bipolar disorder,34 and variants in CLOCK have been linked to sleep disorders in humans.35
The mammalian CLOCK is thought to be the master regulator of the central clock of circadian rhythm. The protein product of CLOCK is a basic-helix-loop-helix transcription factor and heterodimerizes with BMAL1 to activate transcription of a variety of downstream genes essential for the proper modulation of circadian rhythm. The period genes (PER1, PER2, and PER3), the cryptochrome genes (CRY1 and CRY2), the nuclear receptor subfamily 1 group D member 1 (NR1D1/Rev-Erbα), and RAR-related orphan receptor A genes (RORA, RORB, and RORC) are all regulated by the CLOCK/BMAL1 complex. Additionally, a recent report showed that JARID1a activates the CLOCK/BMAL1 complex and promotes trafficking to the PER2 promoter, where it inhibits histone methylation and thus allows transcription of PER2.36 Data presented here show that knockdown, deletion, or mutation of RAI1 results in altered expression of genes from all of these downstream gene families whether in mice, human SMS cell lines, or in vitro siRNA knockdown experiments. Furthermore, we were able to show that RAI1 is a direct regulator of CLOCK, adding an important element to the complex circadian feedback loop of transcription.
The circadian-rhythm defect seen in SMS is thought to contribute to a more severe behavioral phenotype.37 Data suggest that when sleep rhythmicity is improved, behavior and learning improve.5,7,32,38 Additionally, a survey of SMS caregivers indicated that partial alleviation of the sleep disturbance in individuals with SMS results in behavioral improvement.7,39 Thus, it is imperative to understand the molecular defects that result from RAI1 mutation or deletion because these defects signal global disruption of the central circadian clock in SMS.
We also show that RAI1 is a regulator of CLOCK, which might have broad implications in behavior and metabolism, pinpointing RAI1 as a new candidate for sleep disturbance and mental-health disorders in the general population. The current hypothesis with regard to the inverted circadian rhythm documented in SMS is that an inverted secretion of melatonin causes sleep disturbance.5,7,32,38 The data presented here suggest that this inverted circadian rhythm is probably due to a dysregulation of circadian-gene expression and that the inverted melatonin secretion observed in the majority of cases is probably a byproduct of this dysregulation. Further supporting our hypothesis are the facts that C57BL6 mice (which are the background strain for all mice in these experiments) do not secrete melatonin40 yet have altered circadian rhythm when Rai1 is haploinsufficient and that although not all individuals with SMS have inverted melatonin, they do have significant sleep disturbance similar to those who have melatonin inversion.4,8 However, these data do not exclude the possibility that RAI1 might be acting through multiple channels to influence circadian rhythm and not exclusively through CLOCK. Additional studies correlating hormonal regulation and external cues, such as feeding and light, in both humans and animals will be required for fully understanding the clinical implications of these findings and for identifying effective, targeted treatment.
RAI1 was recently shown to bind nucleosomes and to have low nuclear mobility, suggesting its ability to modify chromatin and regulate gene expression,41,42 but until now, true molecular evidence for RAI1 function has not been tractable. All together, these data suggest that RAI1 plays an important role in maintaining circadian rhythmicity in vitro and in vivo, and because circadian rhythms impact development, sleep, and behavior, these findings should help to further delineate the pathways involved in syndromes wherein these functions are disrupted.
Acknowledgments
We thank Ann C. M. Smith and the National Institutes of Health Smith-Magenis syndrome (SMS) Natural History Study for SMS fibroblasts, Kristie Schmidt and Brooke Burns for assistance with the collection of mouse tissue, Christine Bax and Karen Strat for assistance with Clockdel constructs, and patients, parents, and families for continued support of our research. We also thank Satchin Panda, Christopher Vollmers, and Luchiano DiTacchio for assistance with real-time bioluminescence measurement of circadian rhythm. This study was supported, in part, by funds from Virginia Commonwealth University and by PRISMS (Parents and Researchers Interested in Smith-Magenis Syndrome).
Supplemental Data
Document S1. Table S1 and Supplemental Methods
Web Resources
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
- Student's t test, http://www.physics.csbsju.edu/stats/t-test_bulk_form.html
- UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Greenberg F., Lewis R.A., Potocki L., Glaze D., Parke J., Killian J., Murphy M.A., Williamson D., Brown F., Dutton R. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2) Am. J. Med. Genet. 1996;62:247–254. doi: 10.1002/(SICI)1096-8628(19960329)62:3<247::AID-AJMG9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 2.Smith A.C., Dykens E., Greenberg F. Sleep disturbance in Smith-Magenis syndrome (del 17 p11.2) Am. J. Med. Genet. 1998;81:186–191. [PubMed] [Google Scholar]
- 3.Gropman A.L., Elsea S., Duncan W.C., Jr., Smith A.C. New developments in Smith-Magenis syndrome (del 17p11.2) Curr. Opin. Neurol. 2007;20:125–134. doi: 10.1097/WCO.0b013e3280895dba. [DOI] [PubMed] [Google Scholar]
- 4.Boudreau E.A., Johnson K.P., Jackman A.R., Blancato J., Huizing M., Bendavid C., Jones M., Chandrasekharappa S.C., Lewy A.J., Smith A.C., Magenis R.E. Review of disrupted sleep patterns in Smith-Magenis syndrome and normal melatonin secretion in a patient with an atypical interstitial 17p11.2 deletion. Am. J. Med. Genet. A. 2009;149A:1382–1391. doi: 10.1002/ajmg.a.32846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Leersnyder H. Inverted rhythm of melatonin secretion in Smith-Magenis syndrome: From symptoms to treatment. Trends Endocrinol. Metab. 2006;17:291–298. doi: 10.1016/j.tem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.De Leersnyder H., de Blois M.C., Bresson J.L., Sidi D., Claustrat B., Munnich A. [Inversion of the circadian melatonin rhythm in Smith-Magenis syndrome] Rev Neurol (Paris). 2003;159(11 Suppl) 6S21–26. [PubMed] [Google Scholar]
- 7.De Leersnyder H., De Blois M.C., Claustrat B., Romana S., Albrecht U., Von Kleist-Retzow J.C., Delobel B., Viot G., Lyonnet S., Vekemans M., Munnich A. Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome. J. Pediatr. 2001;139:111–116. doi: 10.1067/mpd.2001.115018. [DOI] [PubMed] [Google Scholar]
- 8.Potocki L., Glaze D., Tan D.X., Park S.S., Kashork C.D., Shaffer L.G., Reiter R.J., Lupski J.R. Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J. Med. Genet. 2000;37:428–433. doi: 10.1136/jmg.37.6.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boone P.M., Reiter R.J., Glaze D.G., Tan D.X., Lupski J.R., Potocki L. Abnormal circadian rhythm of melatonin in Smith-Magenis syndrome patients with RAI1 point mutations. Am. J. Med. Genet. A. 2011;155A:2024–2027. doi: 10.1002/ajmg.a.34098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walz K., Spencer C., Kaasik K., Lee C.C., Lupski J.R., Paylor R. Behavioral characterization of mouse models for Smith-Magenis syndrome and dup(17)(p11.2p11.2) Hum. Mol. Genet. 2004;13:367–378. doi: 10.1093/hmg/ddh044. [DOI] [PubMed] [Google Scholar]
- 11.Elsea S.H., Williams S.R. Smith-Magenis syndrome: Haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathways. Expert Rev. Mol. Med. 2011;13:e14. doi: 10.1017/S1462399411001827. [DOI] [PubMed] [Google Scholar]
- 12.Oshima R.G., Pellett O.L., Robb J.A., Schneider J.A. Transformation of human cystinotic fibroblasts by SV40: Characteristics of transformed cells with limited and unlimited growth potential. J. Cell. Physiol. 1977;93:129–136. doi: 10.1002/jcp.1040930116. [DOI] [PubMed] [Google Scholar]
- 13.Juyal R.C., Kuwano A., Kondo I., Zara F., Baldini A., Patel P.I. Mosaicism for del(17)(p11.2p11.2) underlying the Smith-Magenis syndrome. Am. J. Med. Genet. 1996;66:193–196. doi: 10.1002/(SICI)1096-8628(19961211)66:2<193::AID-AJMG13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 14.Vlangos C.N., Yim D.K., Elsea S.H. Refinement of the Smith-Magenis syndrome critical region to approximately 950kb and assessment of 17p11.2 deletions. Are all deletions created equally? Mol. Genet. Metab. 2003;79:134–141. doi: 10.1016/s1096-7192(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 15.Girirajan S., Elsas L.J., 2nd, Devriendt K., Elsea S.H. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J. Med. Genet. 2005;42:820–828. doi: 10.1136/jmg.2005.031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girirajan S., Truong H.T., Blanchard C.L., Elsea S.H. A functional network module for Smith-Magenis syndrome. Clin. Genet. 2009;75:364–374. doi: 10.1111/j.1399-0004.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 17.Bi W., Ohyama T., Nakamura H., Yan J., Visvanathan J., Justice M.J., Lupski J.R. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith-Magenis syndrome. Hum. Mol. Genet. 2005;14:983–995. doi: 10.1093/hmg/ddi085. [DOI] [PubMed] [Google Scholar]
- 18.Bhandari P., Hill J.S., Farris S.P., Costin B., Martin I., Chan C.L., Alaimo J.T., Bettinger J.C., Davies A.G., Miles M.F. Chloride intracellular channels modulate acute ethanol behaviors in Drosophila, Caenorhabditis elegans and mice. Genes Brain Behav. 2012 doi: 10.1111/j.1601-183X.2012.00765.x. Published online January 13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vollmers C., Panda S., DiTacchio L. A high-throughput assay for siRNA-based circadian screens in human U2OS cells. PLoS ONE. 2008;3:e3457. doi: 10.1371/journal.pone.0003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Straume M. DNA microarray time series analysis: Automated statistical assessment of circadian rhythms in gene expression patterning. Methods Enzymol. 2004;383:149–166. doi: 10.1016/S0076-6879(04)83007-6. [DOI] [PubMed] [Google Scholar]
- 21.Burns B., Schmidt K., Williams S.R., Kim S., Girirajan S., Elsea S.H. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum. Mol. Genet. 2010;19:4026–4042. doi: 10.1093/hmg/ddq317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amr S., Heisey C., Zhang M., Xia X.J., Shows K.H., Ajlouni K., Pandya A., Satin L.S., El-Shanti H., Shiang R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007;81:673–683. doi: 10.1086/520961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang E.E., Liu A.C., Hirota T., Miraglia L.J., Welch G., Pongsawakul P.Y., Liu X., Atwood A., Huss J.W., 3rd, Janes J. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier B., Wendt S., Vanselow J.T., Wallach T., Reischl S., Oehmke S., Schlosser A., Kramer A. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev. 2009;23:708–718. doi: 10.1101/gad.512209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novakova M., Nevsimalova S., Prihodova I., Sladek M., Sumova A. Alteration of the Circadian Clock in Children with Smith-Magenis Syndrome. J. Clin. Endocrinol. Metab. 2012;97:E312–E318. doi: 10.1210/jc.2011-2750. Published online December 7, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Bi W., Yan J., Shi X., Yuva-Paylor L.A., Antalffy B.A., Goldman A., Yoo J.W., Noebels J.L., Armstrong D.L., Paylor R., Lupski J.R. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum. Mol. Genet. 2007;16:1802–1813. doi: 10.1093/hmg/ddm128. [DOI] [PubMed] [Google Scholar]
- 27.Girirajan S., Elsea S.H. Distorted Mendelian transmission as a function of genetic background in Rai1-haploinsufficient mice. Eur. J. Med. Genet. 2009;52:224–228. doi: 10.1016/j.ejmg.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Zheng B., Albrecht U., Kaasik K., Sage M., Lu W., Vaishnav S., Li Q., Sun Z.S., Eichele G., Bradley A., Lee C.C. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 29.Crumbley C., Burris T.P. Direct regulation of CLOCK expression by REV-ERB. PLoS ONE. 2011;6:e17290. doi: 10.1371/journal.pone.0017290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi J.S., Hong H.K., Ko C.H., McDearmon E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang W., Ramsey K.M., Marcheva B., Bass J. Circadian rhythms, sleep, and metabolism. J. Clin. Invest. 2011;121:2133–2141. doi: 10.1172/JCI46043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamont E.W., Coutu D.L., Cermakian N., Boivin D.B. Circadian rhythms and clock genes in psychotic disorders. Isr. J. Psychiatry Relat. Sci. 2010;47:27–35. [PubMed] [Google Scholar]
- 33.Turek F.W., Joshu C., Kohsaka A., Lin E., Ivanova G., McDearmon E., Laposky A., Losee-Olson S., Easton A., Jensen D.R. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salvatore P., Ghidini S., Zita G., De Panfilis C., Lambertino S., Maggini C., Baldessarini R.J. Circadian activity rhythm abnormalities in ill and recovered bipolar I disorder patients. Bipolar Disord. 2008;10:256–265. doi: 10.1111/j.1399-5618.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 35.Mendlewicz J. Disruption of the circadian timing systems: Molecular mechanisms in mood disorders. CNS Drugs. 2009;23(Suppl 2):15–26. doi: 10.2165/11318630-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 36.DiTacchio L., Le H.D., Vollmers C., Hatori M., Witcher M., Secombe J., Panda S. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333:1881–1885. doi: 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elsea S.H., Girirajan S. Smith-Magenis syndrome. Eur. J. Hum. Genet. 2008;16:412–421. doi: 10.1038/sj.ejhg.5202009. [DOI] [PubMed] [Google Scholar]
- 38.De Leersnyder H., Bresson J.L., de Blois M.C., Souberbielle J.C., Mogenet A., Delhotal-Landes B., Salefranque F., Munnich A. Beta 1-adrenergic antagonists and melatonin reset the clock and restore sleep in a circadian disorder, Smith-Magenis syndrome. J. Med. Genet. 2003;40:74–78. doi: 10.1136/jmg.40.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Leersnyder H., Claustrat B., Munnich A., Verloes A. Circadian rhythm disorder in a rare disease: Smith-Magenis syndrome. Mol. Cell. Endocrinol. 2006;252:88–91. doi: 10.1016/j.mce.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 40.Goto M., Oshima I., Tomita T., Ebihara S. Melatonin content of the pineal gland in different mouse strains. J. Pineal Res. 1989;7:195–204. doi: 10.1111/j.1600-079x.1989.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 41.Darvekar S., Johnsen S.S., Eriksen A.B., Johansen T., Sjottem E. Identification of Two Independent Nucleosome Binding Domains in the Transcriptional Co-activator SPBP. Biochem. J. 2012;442:65–75. doi: 10.1042/BJ20111230. [DOI] [PubMed] [Google Scholar]
- 42.Sekiya T., Muthurajan U.M., Luger K., Tulin A.V., Zaret K.S. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Table S1 and Supplemental Methods