Depletion of p31comet Protein Promotes Sensitivity to Antimitotic Drugs (original) (raw)
Background: Slippage promotes premature mitotic exit during mitotic block.
Results: Expression of p31comet regulates mitotic slippage and sensitivity to spindle poisons.
Conclusion: Inactivation of p31comet delays mitotic slippage and increases cytotoxicity of spindle poisons.
Significance: p31comet can be a target for antimitotic therapies.
Keywords: Cell Cycle, Cell Division, Checkpoint Control, Chemoresistance, Mitosis, Spindle Poisons, Spindle Assembly Checkpoint
Abstract
Antimitotic spindle poisons are among the most important chemotherapeutic agents available. However, precocious mitotic exit by mitotic slippage limits the cytotoxicity of spindle poisons. The MAD2-binding protein p31comet is implicated in silencing the spindle assembly checkpoint after all kinetochores are attached to spindles. In this study, we report that the levels of p31comet and MAD2 in different cell lines are closely linked with susceptibility to mitotic slippage. Down-regulation of p31comet increased the sensitivity of multiple cancer cell lines to spindle poisons, including nocodazole, vincristine, and Taxol. In the absence of p31comet, lower concentrations of spindle poisons were required to induce mitotic block. The delay in checkpoint silencing was induced by an accumulation of mitotic checkpoint complexes. The increase in the duration of mitotic block after p31comet depletion resulted in a dramatic increase in mitotic cell death upon challenge with spindle poisons. Significantly, cells that are normally prone to mitotic slippage and resistant to spindle disruption-mediated mitotic death were also sensitized after p31comet depletion. These results highlight the importance of p31comet in checkpoint silencing and its potential as a target for antimitotic therapies.
Introduction
Mitotic exit is driven by the anaphase-promoting complex/cyclosome (APC/C),4 which mediates the destruction of substrates, including cyclin B and securin (1). Although phosphorylation by CDK1 and binding to CDC20 are involved in APC/C activation, complete activation is initiated only when all of the chromosomes have achieved proper bipolar spindle attachment. Unattached kinetochores or the absence of tension between the paired kinetochores activates the spindle assembly checkpoint (2). Essential components of the checkpoint include a diffusible complex containing MAD2, BUBR1, BUB3, and CDC20 (called the mitotic checkpoint complex (MCC)), which promotes the inhibition of APC/C-CDC20 by MAD2 (3). Binding to CDC20 requires a conformational change in MAD2 from an open (O-MAD2) to a closed (C-MAD2) conformation (4).
Several mechanisms have been implicated in switching off the checkpoint after it is satisfied. In mammalian cells, there is compelling evidence that MAD2 is neutralized by binding to a protein called p31comet (also called MAD2L1BP) (5). Although overexpression of p31comet disrupts the spindle assembly checkpoint, knockdown of p31comet leads to a delay in mitotic exit (5–7). Recently, p31comet was shown to be important for the destabilization of CDC20 as part of the checkpoint inactivation mechanism (8). Nonetheless, the precise mechanism of how p31comet inactivates the checkpoint remains incompletely understood. Complexes containing p31comet, MAD2, and APC/C can be detected, indicating that p31comet does not interfere with MAD2-APC/C interaction (6). It is believed that p31comet preferably binds MAD2 in the C-MAD2 conformation, thereby blocking checkpoint activation by competing with O-MAD2 for C-MAD2 binding (9, 10). It has been demonstrated that p31comet is involved in the removal of MAD2 from MCC (11). It has also been suggested that p31comet is involved in the disassembly of MCC by an ATP-dependent process (12). The crystal structure of p31comet reveals a folding with striking similarity to C-MAD2, suggesting that structural mimicry may be involved in checkpoint silencing (9).
Agents that suppress microtubule dynamics can artificially activate the spindle assembly checkpoint by leaving the kinetochores unoccupied. These include spindle poisons that inhibit microtubule depolymerization (e.g. Taxol) or polymerization (e.g. vincristine and nocodazole). Spindle poisons are among the most important chemotherapeutic agents available for a variety of cancers. Persistent activation of the spindle assembly checkpoint results in a special form of CDK1-dependent apoptosis, often termed mitotic catastrophe (13). Although a direct link between CDK1 activity and mitotic catastrophe has yet to be fully established, it is clear that cell death can be promoted by a continue trap in mitosis. The responses to prolonged exposure to spindle poisons vary greatly between cancer cells (14). After a prolonged block in mitosis, some cells can inactivate cyclin B1-CDK1 precociously and exit mitosis without chromosome segregation through a process termed mitotic slippage (also called adaptation) (15, 16). Hence, strategies that delay mitotic slippage should be able to shift the fate of spindle poison-treated cells to mitotic cell death (14).
In this study, we present evidence of a correlation between the expression of p31comet and MAD2 with mitotic slippage and spindle poison-mediated cytotoxicity in different cancer cell lines. By targeting p31comet, the balance between mitotic slippage and mitotic cell death was shifted, resulting in an increase in sensitivity to spindle poisons.
EXPERIMENTAL PROCEDURES
Materials
All reagents were obtained from Sigma-Aldrich unless stated otherwise. GST-3C protease was purified as described previously (17).
Cell Culture
H1299 (non-small cell lung carcinoma), HeLa (cervical carcinoma), Hep3B (hepatocellular carcinoma), HepG2 (hepatoblastoma), and IMR90 (normal fibroblasts) cells were obtained from American Type Culture Collection (Manassas, VA). The HeLa cell line used in this study was a clone expressing the tetracycline transactivator repressor chimera (18). The U2OS (osteosarcoma) Tet-On cell line was obtained from Clontech (Palo Alto, CA). HCT116 (colorectal carcinoma) and p53−/− HCT116 cells were gifts from Dr. Bert Vogelstein (The Johns Hopkins University). HONE1 (nasopharyngeal carcinoma) and NP460 (immortalized normal nasopharyngeal epithelial) cells were gifts from Dr. George Tsao (University of Hong Kong). To generate p31comet-expressing cells, HeLa cells were transfected with FLAG-p31comet in pUHD-P1/PUR in the presence of doxycycline. The cells were selected with puromycin. Individual colonies were isolated and propagated in the absence of puromycin. HeLa cells (7) or H1299, HCT116, or U2OS cells (19) stably expressing histone H2B-GFP were used for live cell imaging. The same approach was used to generate Hep3B cells expressing histone H2B-GFP. Synchronization with a double thymidine block was performed as described (20). For synchronization at prometaphase with nocodazole, cells were first synchronized with a double thymidine block and released, nocodazole was added after 2 h, and mitotic cells were isolated by mechanical shake-off after another 10 h. Unless stated otherwise, cells were treated with the following reagents at the indicated final concentrations: doxycycline (2 μg/ml), nocodazole (0.1 μg/ml), pan-caspase inhibitor benzyloxycarbonyl-VAD(OMe)-fluoromethyl ketone (Enzo Life Sciences, Farmingdale, NY), and Taxol (0.5 μg/ml). Cells were transfected with plasmids and siRNAs using a calcium phosphate precipitation method (21) and Lipofectamine RNAiMAX (Invitrogen), respectively. For combination of siRNA transfection and double thymidine synchronization, cells were transfected immediately after the release of the first thymidine block; the cells were then treated with a second thymidine block at 9 h for 13 h. Cell-free extracts were prepared as described (22).
DNA Constructs and siRNAs
FLAG-p31comet in pUHD-P1/PUR was constructed as described previously (19). FLAG-p31comet in pUHD-P1 (19) was cut with NcoI-BamHI and ligated into pUHD-P3T/PUR (23) to generate FLAG-3C-p31comet in pUHD-P3T/PUR. The p31comet variant 1 cDNA (I.M.A.G.E. clone ID 3533182) was amplified by PCR with 5′-CCCATGGCCCGCGTGCCGCTGGGGCGGAGT-3′ and 5′-CGGATCCTCACTCGCGGAAGCCTTT-3′; the PCR product was cut with NcoI-BamHI and ligated into pUHD-P3T (23) to generate FLAG-3C-p31comet variant 1 in pUHD-P3T. FLAG-p31comet in pUHD-P1 was cut with NcoI-HindIII and ligated into pGEX-KG to generate GST-p31comet in pGEX-KG. MAD2 was amplified by PCR with 5′-GGGATCCATGGCGCTGCAGCTCT-3′ and 5′-TGAATTCCCCAGTCATTGACAGGA-3′; the PCR product was cut with BamHI-EcoRI and ligated into pcDNA3.1/Myc-HisC. It was then cut with NcoI-NotI and ligated into pET21d for bacterial expression. Stealth siRNAs against MAD2 (GCCACUGUUGGAAGUUUCUUGUUCA) and p31comet (siRNA1, CCAAGGAGUUCUAUGAACUCGACUU; and siRNA2, GGAGUGGUAUGAGAAGUCCGAAGAA) were obtained from Invitrogen. Unless specified otherwise, p31comet siRNA1 was used.
Live Cell Imaging
The setup and conditions of time-lapse microscopy of living cells were as described previously (19).
Flow Cytometry
Flow cytometry analysis after propidium iodide staining was performed as described previously (24).
Antibodies and Immunological Methods
Monoclonal antibodies against β-actin (25) and FLAG (26) and polyclonal antibodies against FLAG (18) and MAD2 (27) were obtained from sources as described. Polyclonal antibodies against BUBR1 (Bethyl Laboratories, Montgomery, TX) and phospho-histone H3 Ser-10 (sc-8656R, Santa Cruz Biotechnology, Santa Cruz, CA) and monoclonal antibodies against CDC20 (sc-5296, Santa Cruz Biotechnology), CDC27 (BD Transduction Laboratories), and MAD2 (BD Transduction Laboratories) were obtained from the indicated suppliers. Chicken polyclonal antibodies against p31comet was obtained from Abcam (Cambridge, United Kingdom). Rabbit polyclonal antibodies against p31comet was raised against bacterially expressed GST-p31comet. To purify the anti-p31comet antibodies, GST-p31comet was coupled to CNBr-activated Sepharose 4B (GE Healthcare) according to the manufacturer's instructions. The antiserum was diluted 1:10 in PBS and applied 20 times at 25 °C to the GST-p31comet-Sepharose column using gravity. After extensive washing with PBS, the antibodies were eluted with 100 mm glycine (pH 2.4), followed by neutralization with 1 m Tris-HCl (pH 8.5). Immunoblotting and immunoprecipitation were performed as described previously (22).
RESULTS
Correlation between p31comet and MAD2 Expression and Mitotic Slippage
To gain insight into the role of p31comet in intrinsic mitotic slippage, we first examined the expression of endogenous p31comet and its target MAD2 in different cell lines. Different cell lines displayed variations in the expression of p31comet (Fig. 1A). In particular, p31comet was highly expressed in cell lines such as Hep3B. As the expression of MAD2 also varied, the ratio between MAD2 and p31comet varied markedly between different cell lines. Using bacterially expressed MAD2 and p31comet standards, we found that MAD2 was present at ∼16-fold molar excess over p31comet in HeLa cell lysates (supplemental Fig. S1). In contrast, the MAD2/p31comet ratio was only ∼4 in Hep3B cells (Fig. 1A).
FIGURE 1.
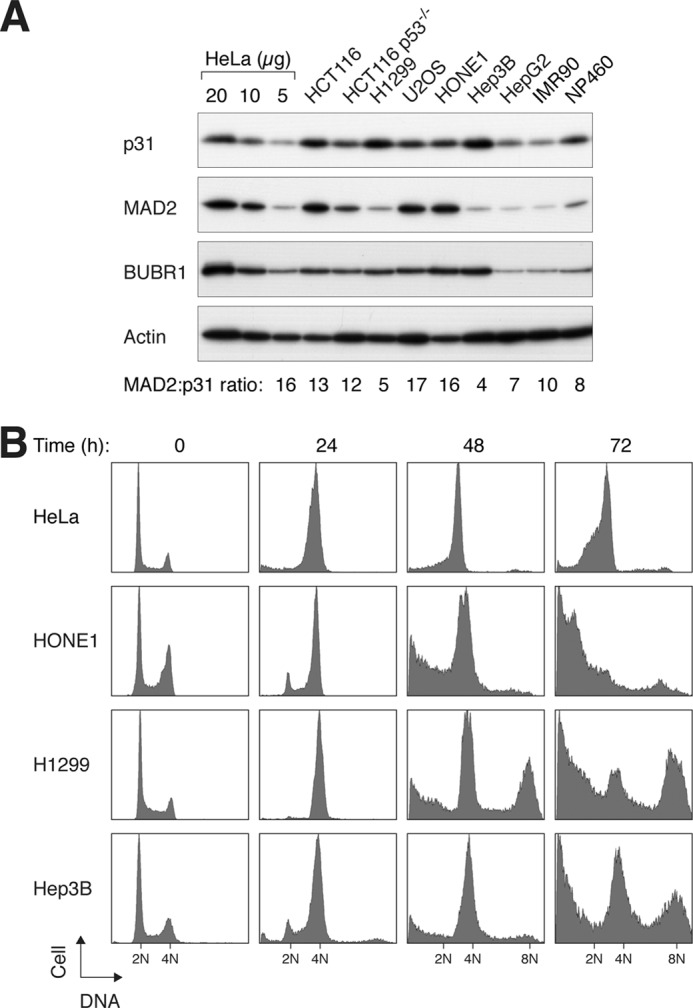
Expression of p31comet and MAD2 correlates with timing of mitotic slippage. A, expression of p31comet and MAD2 in normal and cancer cell lines. Lysates of various cell lines were prepared and analyzed by immunoblotting for p31comet, MAD2, and BUBR1. Normal fibroblasts (IMR90) and immortalized nasopharyngeal epithelial cells (NP460) were included in the analysis. Serial dilutions of HeLa cell lysates were used to generate standard curves to quantify the relative levels of MAD2 and p31comet in different cell lines. The ratios of MAD2 to p31comet in different cell lines (relative to the 16-fold molar excess found in HeLa cells; see supplemental Fig. S1) are indicated. B, cell lines that express relatively high levels of p31comet are prone to mitotic slippage. HeLa, HONE1, H1299, and Hep3B cells were incubated with nocodazole. At the indicated time points, the cells were harvested and analyzed by flow cytometry. Cells that were unable to maintain the spindle assembly checkpoint underwent mitotic slippage and reduplicated their DNA (indicated by the 8N DNA contents).
As p31comet is implicated in the inactivation of the spindle assembly checkpoint, a prediction is that cells containing a high level of p31comet will not be able to maintain a state of checkpoint activation and readily undergo mitotic slippage. Exposure of HeLa cells to the microtubule-disrupting agent nocodazole induced a block in mitosis, as indicated by an increase in mitotic markers and mitotic index (see below), as well as the accumulation of cells with 4N DNA contents (Fig. 1B). Mitotic slippage in HeLa cells was relatively slow, as indicated by the persistence of the 4N DNA contents up to 72 h. Another cell line, HONE1, which expressed a similar p31comet/MAD2 ratio as the HeLa cell line (Fig. 1A), was also resistant to mitotic slippage (Fig. 1B). In the absence of mitotic slippage, a large number of HONE1 cells underwent apoptosis (as indicated by the sub-G1 population).
Compared with HeLa and HONE1 cells, H1299 cells contained a relatively high concentration of p31comet but a low concentration of MAD2 (Fig. 1A). Similar to other cell lines, H1299 could be trapped in mitosis at 24 h after the nocodazole challenge (Fig. 1B), but unlike HeLa cells, DNA reduplication (as indicated by the appearance of >4N DNA contents) was detected at 48 h after nocodazole treatment. These data indicate that H1299 cells were unable to sustain the mitotic block and readily undergo mitotic slippage. Likewise, Hep3B cells, another cell line that expressed a high level of p31comet, were also susceptible to mitotic slippage. The relationship between genome reduplication and p31comet was not a simple one, as other factors, including p53, also play critical roles in regulating genome reduplication (supplemental Fig. S2). Collectively, these data suggest that the level of p31comet may play a role in determining the rate of mitotic slippage.
Rate of Mitotic Slippage Is Determined by Expression of p31comet
Given the potential importance of the abundance of p31comet in mitotic slippage, we next investigated how much excess p31comet is required to promote mitotic slippage. Two isoforms of p31comet are predicted in the human genome. Variant 1 is larger owing to a 32-residue N-terminal extension (supplemental Fig. S3). When ectopically expressed, both variants could bind endogenous MAD2 (Fig. 2A) and uncouple the spindle assembly checkpoint (Fig. 2B). As only a single band of p31comet was detected in different cell lines (Fig. 1A), we next determined which isoform was expressed by using both variants as standards. After removing the FLAG epitope tag, the shorter variant 2 co-migrated with endogenous p31comet (Fig. 2C), indicating that variant 2 is the major p31comet expressed in cell lines. Variant 2 was used for the rest of this study.
FIGURE 2.

Mitotic slippage is promoted by small increase in p31comet. A, both p31comet variants can interact with MAD2. Lysates from HeLa cells transfected with FLAG-3C-tagged p31comet variants 1 and 2 were immunoprecipitated (IP) with either normal rabbit serum (NRS) or FLAG antiserum. Both the total lysates and the immunoprecipitates were immunoblotted with antibodies against FLAG and MAD2. B, the spindle assembly checkpoint can be disrupted by both p31comet variants. HeLa cells were transfected with plasmids expressing FLAG-tagged p31comet variant 1 or 2. The cells were then treated with nocodazole for 48 h before harvesting for flow cytometry analysis. Untreated cells were included as controls. The positions of 2N, 4N, and 8N DNA contents are indicated. Cells with ectopically expressed p31comet were unable to maintain the spindle assembly checkpoint, and they underwent mitotic slippage and reduplicated their DNA. C, variant 2 is the major p31comet isoform expressed in human cell lines. FLAG-3C-tagged p31comet variants 1 and 2 were expressed in HeLa cells. The FLAG-3C tag was cleaved by the addition of GST-3C protease to the lysates. The samples were then analyzed by immunoblotting to compare the size of the recombinant p31comet variants and endogenous p31comet. As the p31comet antiserum was raised against a GST fusion protein, GST-3C protease was also detected. D, ectopic expression of p31comet bypasses the checkpoint. HeLa cells were transiently transfected with either control or FLAG-p31comet-expressing plasmids. The cells were treated with either buffer or nocodazole (NOC) for 16 h before both floating and attached cells were collected. Lysates were prepared and analyzed by immunoblotting. H3(S10-p), phosphorylated histone H3 Ser-10. E, titration of p31comet to disrupt the checkpoint. HeLa cells were transfected with different amounts of FLAG-p31comet (F-p31)-expressing plasmids. (A plasmid expressing histone H2B-GFP and a blasticidin-resistant gene was cotransfected in all of the samples.) Transfected cells were enriched by transient selection with blasticidin. The cells were then treated with either buffer or nocodazole for 16 h. Lysates were prepared and analyzed by immunoblotting.
As expected, overexpression of p31comet by transient transfection abolished nocodazole-mediated phosphorylation of BUBR1 and histone H3 Ser-10 (Fig. 2D). By expressing different amounts of p31comet, we found that the checkpoint could not be maintained when FLAG-p31comet was expressed to a level similar to endogenous p31comet (Fig. 2E, lane 5). These results indicate that a 2-fold increase in p31comet is sufficient to uncouple the spindle assembly checkpoint induced by nocodazole.
Down-regulation of p31comet Promotes Spindle Poison-mediated Checkpoint Activation and Cell Death
We next examined the effects of p31comet down-regulation on mitotic slippage. Fig. 3A shows that p31comet could be reduced with two different siRNAs. Although the overall cell cycle profile was not affected by the siRNAs (Fig. 3B), the duration of mitosis was significantly increased from ∼50 to ∼100 min (Fig. 3C). As a control, depletion of MAD2 accelerated mitosis irrespective of the presence or absence of p31comet.
FIGURE 3.

p31comet modulates sensitivity of spindle poison-induced checkpoint. A, depletion of p31comet with siRNAs. HeLa cells were transfected either with control siRNA or with two different p31comet siRNAs and harvested after 24 h. Lysates were prepared and analyzed by immunoblotting. The asterisk indicates a cross-reactive band. Uniform loading was confirmed by immunoblotting for actin. The expression of MAD2 was not affected by the siRNAs. B, cell cycle progression is not significantly affected by p31comet depletion. Cells were treated as described for A. The DNA contents of the cells were analyzed by flow cytometry. C, depletion of p31comet lengthens mitosis. HeLa cells expressing histone H2B-GFP were transfected with siRNAs against p31comet and MAD2. At 24 h after transfection, the cells were harvested, and protein expression was analyzed by immunoblotting (left panel). The cells were also analyzed by time-lapse microscopy for 24 h (right panel). The duration of mitosis was quantified (mean ± 90% confidence interval; n = 50). D, depletion of p31comet sensitizes cells to the nocodazole-induced checkpoint. HeLa cells were transfected either with control siRNA or with two different p31comet siRNAs. At 24 h after transfection, the cells were treated with different concentrations of nocodazole (NOC). After another 24 h, the cells were harvested and analyzed by flow cytometry. E, depletion of p31comet sensitizes cells to the vincristine-induced checkpoint. HeLa cells were transfected with either control or p31comet siRNAs. At 24 h after transfection, the cells were treated with different concentrations of vincristine. After another 24 h, the cells were harvested and analyzed by flow cytometry.
To test whether p31comet affects the sensitivity to spindle poisons, cells were challenged with different concentrations of nocodazole in the presence or absence of p31comet depletion. For normal cells, at least 100 ng/ml nocodazole was required to induce mitotic arrest (Fig. 3D). In marked contrast, about 10 ng/ml nocodazole was already sufficient to trigger G2/M arrest in p31comet-depleted cells.
Similar to nocodazole, the anticancer therapeutic agent vincristine (Oncovin) also disrupts the assembly of microtubules. Fig. 3E shows that the sensitivity to vincristine was also enhanced in the absence of p31comet, indicating that the effect of p31comet siRNA was not just limited to nocodazole. Taken together, these data indicate that by depleting p31comet, lower concentrations of spindle poisons are needed to induce mitotic block and subsequent mitotic cell death.
The increase in sensitivity of p31comet-depleted cells to the nocodazole-activated checkpoint resulted in more cell death, as indicated by cleaved poly(ADP-ribose) polymerase and trypan blue exclusion analysis (Fig. 4A). Time-lapse fluorescence microscopy of cells stably expressing histone H2B-GFP indicated that whereas control cells were able to exit mitosis after exposure to 10 ng/ml nocodazole, p31comet-depleted cells were trapped in mitosis, resulting in massive mitotic cell death (Fig. 4B). Collectively, these data indicate that by depleting p31comet, lower concentrations of spindle poisons are needed to induce mitotic block and subsequent mitotic cell death.
FIGURE 4.

Down-regulation of p31comet promotes spindle poison-induced checkpoint activation and cell death. A, depletion of p31comet sensitizes cells to nocodazole-mediated cell death. HeLa cells were transfected with either control or p31comet siRNAs. At 24 h after transfection, the cells were treated with 10 ng/ml nocodazole. At the indicated time points, the cells were harvested and analyzed by immunoblotting and trypan blue exclusion assays. PARP, poly(ADP-ribose) polymerase. B, depletion of p31comet promotes mitotic cell death. HeLa cells expressing histone H2B-GFP were transfected with either control or p31comet siRNAs and incubated with 10 ng/ml nocodazole (NOC). Individual cells were tracked by time-lapse microscopy for 24 h (n = 50). Each horizontal line represents one cell. Gray, interphase; black, mitosis (from DNA condensation to anaphase or cell death); truncated bars, cell death. The time of cell death after mitosis is defined by the death of one of the daughter cells. C, depletion of p31comet stimulates MCC formation after low concentration nocodazole treatment. Control or p31comet siRNA-transfected HeLa cells were treated with the indicated concentrations of nocodazole. Mitotic cells were collected by mechanical shake-off. MCC was then analyzed by MAD2 immunoprecipitation (IP). D, overexpression of p31comet reduces MCC after nocodazole treatment. HeLa cells expressing FLAG-p31comet under the control of doxycycline (Dox) were generated. The cells were incubated with 2 μg/ml, 0.1 ng/ml, and 0.05 ng/ml doxycycline to control the expression of FLAG-p31comet. The cells were then synchronized at prometaphase with nocodazole as described under “Experimental Procedures.” Lysates were prepared and subjected to immunoprecipitation with anti-MAD2 and anti-BUBR1 antibodies.
To understand how p31comet affects the sensitivity to spindle poisons, we next analyzed the level of MCC after p31comet was depleted. It is known that MCC components, including CDC20, can be specifically immunoprecipitated using anti-MAD2 antibodies (27). Exposure of normal cells to a low concentration of nocodazole (10 ng/ml) typically does not trigger mitotic arrest (28), and MCC was not detectable.5 In contrast, p31comet-depleted cells were trapped in mitosis by 10 ng/ml nocodazole (Fig. 4B). Moreover, MCC was present at a similar level as in cells treated with 100 ng/ml nocodazole (Fig. 4C). Over 60% of p31comet-depleted mitotic cells were able to form metaphase plates following treatment with 10 ng/ml nocodazole (data not shown), suggesting defects in checkpoint silencing. We next performed the converse experiments by overexpressing p31comet. We found that expression of p31comet reduced the level of MCC during nocodazole challenge (Fig. 4D), further supporting the regulation of MCC abundance by p31comet. Thus, the enhanced sensitivity to spindle poisons induced by p31comet siRNAs is likely to be caused by an accumulation of MCC and a delay in checkpoint silencing.
To exclude the possibility that the increase in sensitivity to spindle poisons after p31comet depletion was only a phenomenon limited to HeLa cells, we demonstrated that HCT116 cells were also more sensitive to 10 ng/ml nocodazole after p31comet was depleted (Fig. 5A). Finally, to ascertain that the effect of p31comet depletion was not limited to agents that inhibit microtubule assembly, such as nocodazole and vincristine, we also treated cells with Taxol, a spindle poison that inhibits microtubule disassembly (Fig. 5B). A relatively low concentration of Taxol, which did not cause a complete mitotic block on its own, was used. In both Taxol- and nocodazole-treated cells, p31comet depletion increased mitotic block and cell death significantly. Collectively, these data indicate that down-regulation of p31comet promotes the spindle assembly checkpoint induced by various spindle poisons and the ensuring mitotic cell death.
FIGURE 5.
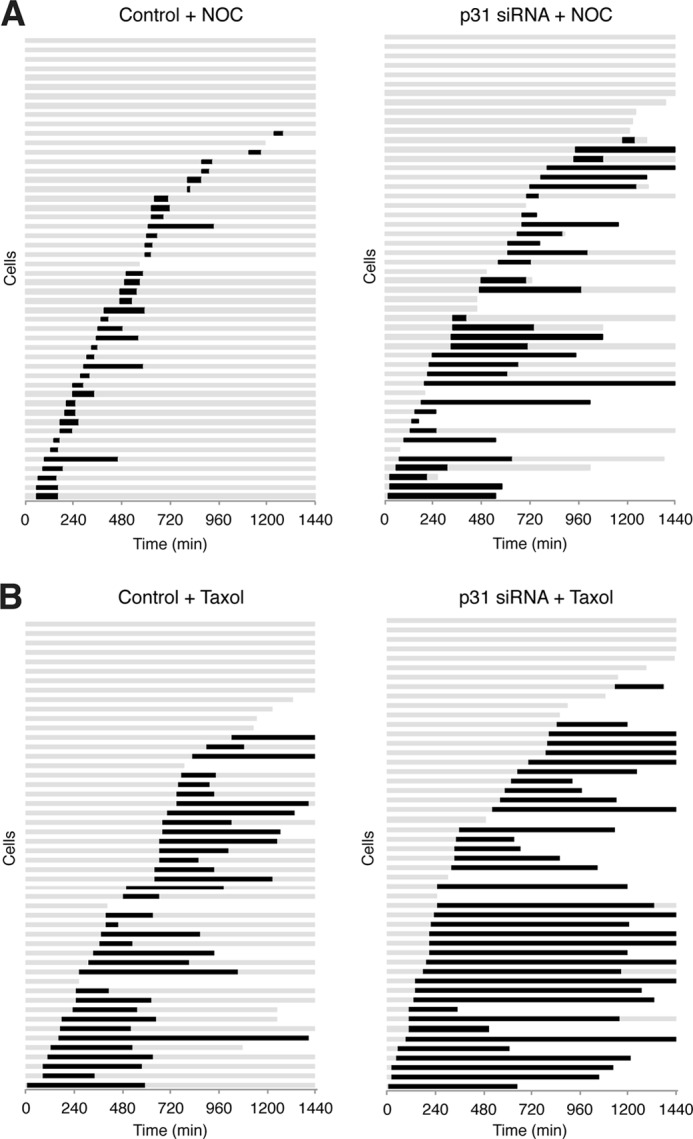
Down-regulation of p31comet sensitizes HCT116 cells to nocodazole and Taxol. A, depletion of p31comet promotes nocodazole-mediated mitotic cell death. HCT116 cells stably expressing histone H2B-GFP were generated. The cells were transfected with either control or p31comet siRNAs and treated with 10 ng/ml nocodazole (NOC). Individual cells were monitored by time-lapse microscopy for 24 h (n = 50). Each horizontal line represents one cell. Gray, interphase; black, mitosis (from DNA condensation to anaphase or cell death); truncated bars, cell death. The time of cell death after mitosis is defined by the death of one of the daughter cells. B, depletion of p31comet promotes Taxol-mediated mitotic cell death. HCT116 cells were transfected as described for A. The cells were incubated with 20 ng/ml Taxol and analyzed by time-lapse microscopy as described for A.
Down-regulation of p31comet Promotes Mitotic Cell Death in Cells That Are Relatively Resistant to Spindle Disruption
As shown above, different cell lines varied widely in their susceptibility to mitotic slippage following spindle disruption (Fig. 1B). Cells that readily undergo mitotic slippage, such as H1299 and Hep3B, are relatively resistant to spindle poison-mediated cell death. Given the correlation of these cell lines with p31comet expression, we next investigated if down-regulation of p31comet could sensitize these cell lines to spindle poisons. Indeed, mitotic block induced by nocodazole was poorly sustained in H1299 cells (Fig. 6A, lanes 1–7). Mitotic slippage occurred at ∼16 h after release from a double thymidine block, as indicated by the degradation of cyclin B1 and dephosphorylation of histone H3 Ser-10 and CDC27. Importantly, mitotic slippage in H1299 cells was delayed after the depletion of p31comet (Fig. 6A). Consistently, the genome reduplication caused by prolonged nocodazole exposure was inhibited after p31comet depletion (Fig. 6B). To examine individual cells, H1299 cells stably expressing histone H2B-GFP were examined with time-lapse fluorescence microscopy. These results validated that p31comet-depleted H1299 cells could be trapped in mitosis by Taxol for a longer duration compared with control cells (Fig. 6C).
FIGURE 6.

Down-regulation of p31comet delays mitotic slippage and promotes cell death in cells that are relatively resistant to spindle poisons. A, mitotic slippage occurs quickly in H1299 cells and is suppressed after p31comet depletion. H1299 cells were transfected with control siRNA or with different p31comet siRNAs. The cells were then synchronized at early S phase with a double thymidine block and released. The cells were treated with nocodazole at 7 h after the release. At 11 h after the release, mitotic cells were isolated by mechanical shake-off, incubated further in the presence of nocodazole, and harvested at the indicated time points. Caspase inhibitors were included to repress the apoptosis induced by the prolonged mitotic arrest. Lysates were prepared and analyzed by immunoblotting. The positions of non-phosphorylated and phosphorylated forms of CDC27 are indicated. H3(S10-p), phosphorylated histone H3 Ser-10. B, depletion of p31comet inhibits genome reduplication. H1299 cells were transfected with control or p31comet siRNAs. The cells were then synchronized at early S phase with a double thymidine block and released in the presence of nocodazole. After 12 and 36 h, the cells were harvested and analyzed by flow cytometry. C, depletion of p31comet inhibits mitotic slippage in H1299 cells. H1299 cells stably expressing histone H2B-GFP were generated. The cells were transfected with either control or p31comet siRNAs before synchronization with a double thymidine block. After release from the block, the cells were treated with Taxol and analyzed by time-lapse microscopy for 36 h. Individual cells were tracked, and the duration of mitosis was quantified (n = 50). D, mitotic slippage in Hep3B cells is abolished after p31comet depletion. Hep3B cells stably expressing histone H2B-GFP were generated. The cells were transfected with either control or p31comet siRNAs and treated with either buffer or nocodazole (NOC). Individual cells were monitored by time-lapse microscopy for 48 h (n = 50). The percentage of cells that underwent mitotic slippage and the accumulative cell death (at the end of imaging) are quantified (upper and middle panels). The full data set for the individual cells is shown in supplemental Fig. S4. The knockdown of p31comet was confirmed by immunoblotting (lower panel).
We next performed a similar analysis using Hep3B cells, another cancer cell line that readily undergoes mitotic slippage after spindle disruption. Histone H2B-GFP was again stably integrated into the genome of Hep3B cells to allow us to monitor individual cells with time-lapse fluorescence microscopy. Mitotic slippage occurred in >70% of the Hep3B cells following nocodazole challenge (Fig. 6D; data for individual cells are shown in supplemental Fig. S4). Transfection of p31comet siRNA abolished the mitotic slippage. More importantly, abolition of mitotic slippage resulted in a dramatic increase in cell death induced by the spindle poison. Taken together, these results indicate that by controlling the expression p31comet, the sensitivity to spindle poisons can be altered markedly.
DISCUSSION
In this study, we have shown that both up- and down-regulation of p31comet exerted profound effects on the spindle assembly checkpoint and modulated the mitotic cell death response to spindle poisons. Overexpression of p31comet overcame the nocodazole-induced checkpoint, as indicated by the loss of phosphorylation of histone H3 Ser-10 (Fig. 2, D and E) and BUBR1 (Fig. 2D) and by genome reduplication (Fig. 2B). These effects have been well established (5). In fact, only a 2-fold increase in p31comet over the endogenous level appears to be sufficient to promote checkpoint bypass (Fig. 2E). Checkpoint disruption by p31comet was not limited only to that induced with nocodazole. The checkpoint activated by depletion of the plus-end-directed microtubule motor Eg5 could also be uncoupled by p31comet.5 Under ectopic conditions, both variants of p31comet could bind MAD2 (Fig. 2A) and uncouple the checkpoint (Fig. 2B). This is consistent with the fact that the MAD2-binding domain was identified to be outside the N-terminal region that distinguishes the two variants (9). Our data indicate that variant 2 is the major p31comet isoform expressed in normal and cancer cell lines (Figs. 1A and 2C).
Consistent with a previous study (8), depletion of p31comet lengthened the unperturbed mitosis (Fig. 3C). In addition, we found that p31comet depletion sensitized cells to G2/M arrest induced by low doses of nocodazole (Fig. 3D). The hypersensitivity to nocodazole after p31comet knockdown was due primarily to an increase in the duration of mitotic block (Fig. 4B). Two p31comet siRNAs against independent regions of the gene were used in this study (Fig. 3A). Although, in general, siRNA1 exerted a marginally stronger effect on mitosis compared with siRNA2 (e.g. Fig. 3D), both siRNAs supported the same conclusions in this study (data from siRNA1 are shown), confirming that the observed effects were not due to off-target effects of the siRNAs.
The stimulation of mitotic block by p31comet siRNAs was not limited to nocodazole. Mitotic block was exacerbated with multiple spindle poisons, including nocodazole (Fig. 3D), vincristine (Fig. 3E), and Taxol (Fig. 5B). One interpretation is that the checkpoint was more susceptible to activation without p31comet, but this would imply that a low amount of spindle stress is normally ignored and that the checkpoint is not activated. A more satisfying argument is that the checkpoint is in fact activated by low concentrations of spindle poisons, but the spindle-kinetochore attachments are quickly corrected, and the checkpoint is inactivated. With insufficient p31comet, however, the checkpoint inactivation step was delayed, and checkpoint-arrested cells were able to accumulate.
Down-regulation of p31comet also prevented mitotic slippage after prolonged mitotic arrest induced by spindle poisons. This effect could be readily observed in cells that are prone to mitotic slippage, including H1299 and Hep3B (Fig. 6, A and D). One possible explanation is that MCC is hyperaccumulated (Fig. 4C) in the absence of p31comet, thereby resulting in prolonged activation of the spindle assembly checkpoint.
Consequent to a more effective mitotic block, p31comet-depleted cells underwent more extensive mitotic cell death compared with control cells. We demonstrated this both in cells that are relatively resistant to mitotic slippage, such as HeLa (Fig. 4) and HCT116 (Fig. 5), and in cells that readily undergo mitotic slippage, such as Hep3B (Fig. 6D). We believe that targeting p31comet, either by RNA interference or with small molecules that break the interaction with MAD2, may be a suitable approach to develop more effective anticancer drugs. As suggested by our cell line models (e.g. Fig. 3, D and E), relatively low concentrations of spindle poisons, themselves not sufficient to induce mitotic block and cell death, were able to kill cells when p31comet was down-regulated. One of the principal side effects of spindle poisons as anticancer drugs is neurotoxicity (29). It is conceivable that by targeting p31comet, lower doses of spindle poisons (hence having fewer side effects) can be used in therapies.
Another implication from this study is that as the levels of p31comet and MAD2 correlated with the susceptibility to mitotic slippage (as endogenous proteins both in different cell lines (Fig. 1) and in experimental models that manipulated their expression), it is possible that the levels of these proteins can help to predict the outcome of antimitotic drug treatments. It is conceivable that this “personalized medicine” approach may assist in making the decision of whether spindle poisons alone or in combination with other agents (for example, p31comet-targeting agents) will be effective in cancer therapies for a particular cancer. In conclusion, the level of p31comet in a given cell line controls the effectiveness of mitotic slippage and mitotic cell death upon spindle poison challenge.
Acknowledgments
We thank Drs. George Tsao and Bert Vogelstein for generous gifts of reagents. We acknowledge the technical assistance provided by Anita Lau, Nelson Lee, and Leanne Leung.
*
This work was supported in part by Research Grants Council Grants 662208, AOE-MG/M-08/06, and HKU7/CRG/09 (to R. Y. C. P.).
5
H. T. Ma, Y. Y. Chan, X. Chen, K. F. On, and R. Y. C. Poon, unpublished data.
4
The abbreviations used are:
APC/C
anaphase-promoting complex/cyclosome
MCC
mitotic checkpoint complex.
REFERENCES
- 1.Pesin J. A., Orr-Weaver T. L. (2008) Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 24, 475–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musacchio A., Salmon E. D. (2007) The spindle assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 3.Vigneron S., Prieto S., Bernis C., Labbé J. C., Castro A., Lorca T. (2004) Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol. Biol. Cell 15, 4584–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo X., Tang Z., Rizo J., Yu H. (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 9, 59–71 [DOI] [PubMed] [Google Scholar]
- 5.Habu T., Kim S. H., Weinstein J., Matsumoto T. (2002) Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 21, 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia G., Luo X., Habu T., Rizo J., Matsumoto T., Yu H. (2004) Conformation-specific binding of p31comet antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 23, 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan Y. W., Ma H. T., Wong W., Ho C. C., On K. F., Poon R. Y. (2008) CDK1 inhibitors antagonize the immediate apoptosis triggered by spindle disruption but promote apoptosis following the subsequent rereplication and abnormal mitosis. Cell Cycle 7, 1449–1461 [DOI] [PubMed] [Google Scholar]
- 8.Varetti G., Guida C., Santaguida S., Chiroli E., Musacchio A. (2011) Homeostatic control of mitotic arrest. Mol. Cell 44, 710–720 [DOI] [PubMed] [Google Scholar]
- 9.Yang M., Li B., Tomchick D. R., Machius M., Rizo J., Yu H., Luo X. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131, 744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mapelli M., Filipp F. V., Rancati G., Massimiliano L., Nezi L., Stier G., Hagan R. S., Confalonieri S., Piatti S., Sattler M., Musacchio A. (2006) Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J. 25, 1273–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westhorpe F. G., Tighe A., Lara-Gonzalez P., Taylor S. S. (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J. Cell Sci. 124, 3905–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teichner A., Eytan E., Sitry-Shevah D., Miniowitz-Shemtov S., Dumin E., Gromis J., Hershko A. (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc. Natl. Acad. Sci. U.S.A. 108, 3187–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chow J. P. H., Poon R. Y. C. (2010) in Cell Cycle Deregulation in Cancer (Enders G., ed) pp. 79–96, Springer, New York [Google Scholar]
- 14.Gascoigne K. E., Taylor S. S. (2008) Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 [DOI] [PubMed] [Google Scholar]
- 15.Rieder C. L., Maiato H. (2004) Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 7, 637–651 [DOI] [PubMed] [Google Scholar]
- 16.Weaver B. A., Cleveland D. W. (2005) Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell 8, 7–12 [DOI] [PubMed] [Google Scholar]
- 17.Fung T. K., Yam C. H., Poon R. Y. (2005) The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle 4, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 18.Yam C. H., Siu W. Y., Lau A., Poon R. Y. (2000) Degradation of cyclin A does not require its phosphorylation by CDC2 and cyclin-dependent kinase 2. J. Biol. Chem. 275, 3158–3167 [DOI] [PubMed] [Google Scholar]
- 19.On K. F., Chen Y., Ma H. T., Chow J. P., Poon R. Y. (2011) Determinants of mitotic catastrophe on abrogation of the G2 DNA damage checkpoint by UCN-01. Mol. Cancer Ther. 10, 784–794 [DOI] [PubMed] [Google Scholar]
- 20.Arooz T., Yam C. H., Siu W. Y., Lau A., Li K. K., Poon R. Y. (2000) On the concentrations of cyclins and cyclin-dependent kinases in extracts of cultured human cells. Biochemistry 39, 9494–9501 [DOI] [PubMed] [Google Scholar]
- 21.Ausubel F., Brent R., Kingston R., Moore D., Seidman J., Smith J., Struhl K. (1991) Current Protocols in Molecular Biology, John Wiley & Sons, New York [Google Scholar]
- 22.Poon R. Y., Toyoshima H., Hunter T. (1995) Redistribution of the CDK inhibitor p27 between different cyclin-CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol. Biol. Cell 6, 1197–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma H. T., Tsang Y. H., Marxer M., Poon R. Y. (2009) Cyclin A2-cyclin-dependent kinase 2 cooperates with the PLK1-SCFβ-TrCP1-EMI1-anaphase-promoting complex/cyclosome axis to promote genome reduplication in the absence of mitosis. Mol. Cell. Biol. 29, 6500–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siu W. Y., Arooz T., Poon R. Y. (1999) Differential responses of proliferating versus quiescent cells to adriamycin. Exp. Cell Res. 250, 131–141 [DOI] [PubMed] [Google Scholar]
- 25.Siu W. Y., Lau A., Arooz T., Chow J. P., Ho H. T., Poon R. Y. (2004) Topoisomerase poisons differentially activate DNA damage checkpoints through ataxia-telangiectasia mutated-dependent and -independent mechanisms. Mol. Cancer Ther. 3, 621–632 [PubMed] [Google Scholar]
- 26.Fung T. K., Siu W. Y., Yam C. H., Lau A., Poon R. Y. (2002) Cyclin F is degraded during G2-M by mechanisms fundamentally different from other cyclins. J. Biol. Chem. 277, 35140–35149 [DOI] [PubMed] [Google Scholar]
- 27.Ma H. T., Poon R. Y. (2011) Orderly inactivation of the key checkpoint protein mitotic arrest-deficient 2 (MAD2) during mitotic progression. J. Biol. Chem. 286, 13052–13059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong J., Fang G. (2006) HURP controls spindle dynamics to promote proper interkinetochore tension and efficient kinetochore capture. J. Cell Biol. 173, 879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan M. A., Wilson L. (2004) Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265 [DOI] [PubMed] [Google Scholar]