The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network (original) (raw)
. Author manuscript; available in PMC: 2012 Jul 17.
Published in final edited form as: Nature. 2008 Aug 20;455(7214):813–817. doi: 10.1038/nature07290
Abstract
Nuclear exclusion of the PTEN tumour suppressor has been associated with cancer progression1-6. However, the mechanisms leading to this aberrant PTEN localization in human cancers are currently unknown. We have previously reported that ubiquitinylation of PTEN at specific lysine residues regulates its nuclear-cytoplasmic partitioning7. Here we show that functional PML-nuclear bodies co-ordinate PTEN localization by opposing the action of a novel PTEN-deubiquitinylating enzyme, HAUSP, and that the integrity of this molecular framework is required for PTEN to be able to enter the nucleus. We find that PTEN is aberrantly localized in acute promyelocytic leukaemia (APL), where PML function is disrupted by the PML-RARα fusion oncoprotein. Remarkably, treatment with drugs that trigger PML-RARα degradation such as all-trans retinoic acid or arsenic trioxide, restore nuclear PTEN. We demonstrate that PML opposes the activity of HAUSP towards PTEN, through a mechanism involving the adaptor protein DAXX. In support of this paradigm, we show that HAUSP is overexpressed in human prostate cancer and is associated with PTEN nuclear exclusion. Thus our results delineate a novel PML-DAXX-HAUSP molecular network controlling PTEN deubiquitinylation and trafficking, which is perturbed by oncogenic cues in human cancer, in turn defining a new deubiquitinylation-dependent model for PTEN subcellular compartmentalization.
PTEN (Phosphatase and Tensin homologue deleted on chromosome 10) is a non-redundant lipid phosphatase, essential to oppose the highly oncogenic pro-survival PI3K/AKT pathway8, 9. PTEN is frequently mutated in multiple sporadic cancers and its mutation in the germline increases cancer susceptibility, as observed in several disorders referred to as the PTEN hamartoma tumour syndrome10. Although originally thought to be functional solely in the cytoplasm, there is now compelling evidence that nuclear PTEN is also essential for tumour suppression7,11-14. In fact, a nuclear-exclusion phenotype of PTEN expression is associated with more aggressive disease in patients with cancers of various histology1-6. Likewise, we have recently reported that mutation of Lysine 289 (K289; one of two major sites for PTEN ubiquitinylation) impairs nuclear localization and increases cancer susceptibility as demonstrated in intestinal polyps from a patient with Cowden syndrome7.
To our surprise, we observed that human APL biopsies harbouring the t(15;17) chromosomal translocation, encoding for PML-RARα fusion protein, exhibited a predominantly cytoplasmic PTEN distribution by immunofluorescence (IF; Fig. 1a and Supplementary Fig. 1), as identified by using an antibody specific for human and mouse PTEN (to ensure specificity we have performed a systematic assessment of all the available PTEN antibodies, see Supplemental Methods Summary and Supplementary Fig. 2). Clinical cases of APL are commonly treated with all-trans retinoic acid (ATRA), an agent which induces degradation of PML-RARα and restores PML nuclear-bodies (PML-NBs; Fig. 1b (insert), Supplementary Fig. 3c, 4a and 5a,b and Ref. 15). Examination of biopsies from APL patients before and after ATRA treatment demonstrated that ATRA effectively restored the nuclear localization of PTEN (Fig 1b and Supplementary Fig 3a,b). Similarly, NB4 cells treated with ATRA showed a redistribution of PTEN such that a greater fraction was localized to the nucleoplasm (Fig. 1c and Supplementary Fig. 4a) with no effect on total PTEN levels (Supplementary Fig. 5b,c). Like ATRA, arsenic trioxide (ATO) is also a powerful therapy for APL and treatment with this compound leads to a dual effect on PML-NBs; short-term treatment of ATO promotes the selective proteasome-dependent degradation of PML-RARα fusion protein and recovery of NBs, whereas long exposure induces the degradation of PML and the consequent disassembly of the NBs16-18. Indeed, this was observed upon treatment of NB4 cells with this compound; specifically, 4 hours of treatment with ATO reconstituted NBs and restored PTEN localization to the nucleus, whereas 24 hours of treatment abolished the NBs with a concomitant cytoplasmic PTEN localization (Fig 1c,d and Supplementary Figs. 4 and 5), with no significant change of PTEN protein levels (Supplementary Fig. 5b). Furthermore, ATRA was not able to relocalize PTEN to the nucleus in ATRA-resistant NB4 (NB4R) cells, in contrast ATO could influence PTEN localization in NB4R as well as in ATRA-sensitive cells (Fig. 1d and Supplementary Fig. 4b).
Figure 1. Aberrant localization of PTEN in APL and _Pml_-null MEFs.
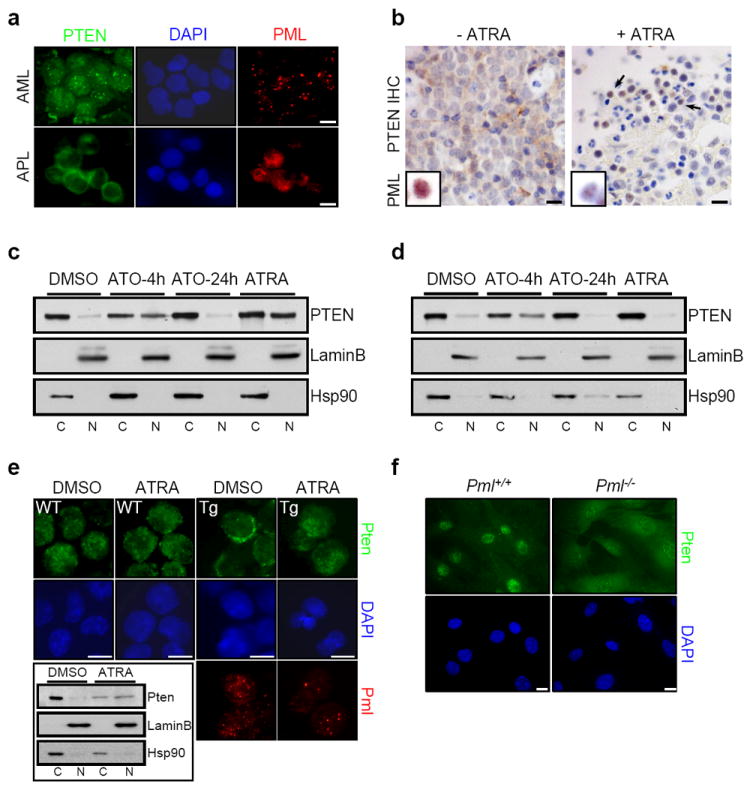
a, Immunofluorescence (IF) analysis of PTEN (green) and PML (red) in AML and APL patient derived bone marrow smears demonstrates cytoplasmic localization of PTEN in APL, but not AML patient samples. Scale bars, 10 μm. Percentage of cases and other representative images of AML and APL are shown in Supplementary Figure 1. b, Immunohistochemical (IHC) analysis of PTEN and PML in APL patient derived biopsies before and after treatment with ATRA shows the restoration of nuclear PTEN localization in vivo by ATRA. Percentage of cases of APL biopsies before and after treatment with ATRA is shown in Supplementary Figure 3a. Inset indicates PML localization (full-size of images are shown in Supplementary Figure 3c). Negative control for PTEN staining is shown in Supplementary Figure 3b. Scale bars, 20 μm. c, Nuclear (N) and cytoplasmic (C) fractionation of PTEN in APL cell line NB4 after treatment with ATO (4 or 24 hours) or ATRA (72 hours). Lamin B and Hsp90 serve as nuclear and cytoplasmic markers, respectively. d, Nuclear-cytoplasmic fractionation of PTEN in ATRA-resistant NB4 (NB4R) cells after treatment with ATO or ATRA. e, IF analysis of Pten and Pml in promyelocytes of wild-type (WT) and MRP8-PML-RARα transgenic (Tg) mice after treatment with ATRA and nuclear-cytoplasmic fractionation analysis of Pten in Tg mice (inset). Scale bars, 10 μm. f, IF analysis of Pten in Pml+/+ and _Pml_-/- MEFs. Scale bars, 10 μm.
To further validate our results, we utilized a transgenic MRP8-PML-RARα (Tg) mouse model of APL, in which we also observed a predominant cytoplasmic localization of Pten in pre-neoplastic promyelocytes expressing the transgenic fusion protein compared to wild type promyelocytes (Fig. 1e). Interestingly, ATRA treatment restored nuclear localization of Pten in Tg promyelocytes but did not alter Pten distribution in the wild type counterparts (Fig. 1e).
To demonstrate the specificity of PML-RARα on Pten, Pml heterozygous mouse embryonic fibroblasts (MEFs) were transduced with a retrovirus conferring stable expression of the fusion protein. Indeed PML-RARα was effective at driving the nuclear exclusion of Pten (Supplementary Fig. 6a,b), and as expected ATRA treatment of PML-RARα–expressing MEFs restored the nuclear localization of Pten (Supplementary Fig. 6c,d). This demonstrates that disassembly of the PML-NBs by PML-RARα can alter Pten localization. Since Pml is essential for proper NB formation19 we analyzed the localization of Pten in wild type and _Pml_-null MEFs. In _Pml_-null MEFs, the fraction of Pten localized to the cytoplasm was significantly increased, as measured by IF and nuclear-cytoplasmic fractionation (Fig. 1f and Supplementary Fig. 6e,f) and accompanied by a slight reduction of PTEN expression levels (Supplementary Fig. 6f right panel). Notably, ATRA treatment of NB4 cells induced an enrichment of PTEN in well-defined nuclear structures, which upon further examination showed colocalization with PML (Supplementary Fig. 7a). Moreover, PTEN co-localized to NBs in wild type, but not _Pml_-null MEF (Supplementary Fig. 7b).
These results are the first to highlight a role for PML and PML-NBs in the regulation of PTEN localization and imply that disrupted PTEN localization may have relevance in malignancies where PML and PML-NBs are compromised as in APL.
Similar to other tumour suppressor proteins like FOXO4 and p53, PTEN localization can be modulated by ubiquitinylation (reviewed in 20). Therefore, we investigated the ubiquitinylation status of PTEN protein in APL and _Pml_-null MEFs. Strikingly, ATRA treatment increased Pten monoubiquitinylation in NB4 cells (Fig. 2a). Moreover, cells lacking Pml displayed reduced levels of Pten monoubiquitinylation (Supplementary Fig. 7c), which correlated with reduced nuclear localization of Pten (Fig. 1f and Supplementary. Fig. 6e) in agreement with our previous report7.
Figure 2. HAUSP interacts with and deubiquitinylates PTEN.
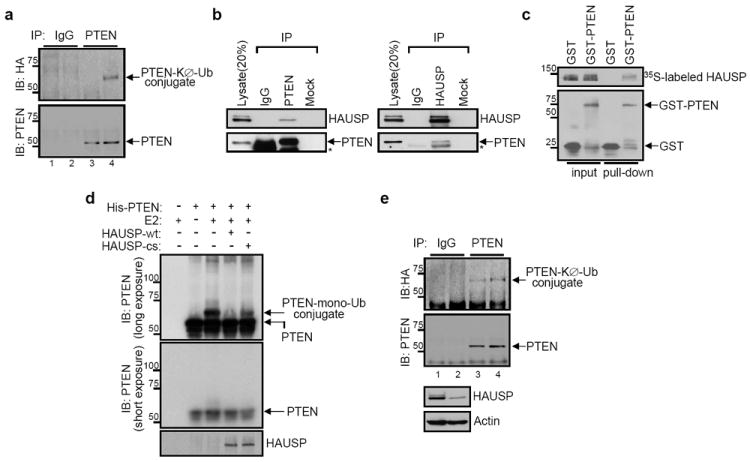
a, ATRA-induced PTEN monoubiquitinylation. Lysates from HA-tagged KØ-Ub-transfected NB4 cells with either DMSO (lanes 1 and 3) or ATRA (lanes 2 and 4) were immunoprecipitated (IP) with control IgG (lanes 1 and 2) or anti-PTEN (lanes 3 and 4), and then analyzed for monoubiquitinylation by immunoblotting with anti-HA. Molecular weights are in kDa. b, Interaction between PTEN and HAUSP in vivo. Immunobloting of U2OS cell lysates after IP with anti-PTEN or anti-HAUSP antibodies. Asterisks indicate heavy chain of IgG c, Interaction between PTEN and HAUSP in vitro. GST pull-down assay of the purified GST-PTEN protein with 35S-labeled _in vitro_-translated HAUSP protein. Molecular weights are in kDa. d, Deubiquitinylation of PTEN in vitro by HAUSP. The purified PTEN protein was incubated with the purified recombinant proteins of either wild-type (wt) or mutant form (cs) of HAUSP. Molecular weights are in kDa. e, Increase of PTEN monoubiquitinylation by HAUSP depletion. Lysates from HA-KØ-Ub and either luciferase (control, lanes 1 and 3) or HAUSP siRNA (lanes 2 and 4)-cotransfected U2OS cells were immunoprecipitated (IP) with control IgG (lanes 1 and 2) or anti-PTEN (lanes 3 and 4), and then analysed for monoubiquitinylation by immunoblotting with anti-HA. Molecular weights are in kDa.
To identify the means by which PML-NBs regulate PTEN ubiquitinylation, we focused our attention on NB-associated proteins that may modulate ubiquitinylation. Herpesvirus-associated ubiquitin protease (HAUSP, also known as USP7) has been reported to co-localize to the NBs, and has been shown to critically regulate protein localization through ubiquitinylation21. We confirmed that a fraction of HAUSP localizes to PML-NBs (Supplementary Fig. 7d) in wild type, but not in _Pml_-null MEF, and that Hausp co-immunoprecipitates with Pml (Supplementary Fig. 7e). Moreover, like PTEN ATRA treatment relocalized HAUSP to PML nuclear bodies in NB4 cells (Supplementary Fig. 7f).
Next, we examined the physical and functional interactions between PTEN and HAUSP. We found that both endogenous and exogenously expressed PTEN and HAUSP co-immunoprecipitated (Fig. 2b and Supplementary Fig. 8a) and this interaction was shown to be direct by means of an in vitro binding assay (Fig. 2c). Importantly, HAUSP reduced monoubiquitinylation of PTEN as measured by an in vitro (Fig. 2d) and in vivo (Supplementary Fig. 8b) ubiquitinylation assay. This event was demonstrated to be dependent on enzymatic activity, since the catalytically inactive (CS) HAUSP-mutant, which can still bind to PTEN (Supplementary Fig. 8c), did not reduce the monoubiquitinylation of PTEN (Fig. 2d and Supplementary Fig. 8b). Additionally, knock-down of HAUSP by RNA interference (RNAi) increased PTEN monoubiquitinylation (Fig. 2e and Supplementary Fig. 8d). Therefore, these findings uncover HAUSP as the first bona-fide PTEN deubiquitinylase (DUB).
Monoubiquitinylation has been suggested to be a ‘key’ for proteins to gain access into other cellular compartments: for example, FOXO4 monoubiquitinylation allows nuclear import, whereas p53 is localized to the cytoplasm and mitochondria upon ubiquitin conjugation22-24. Accordingly, we examined the effect of HAUSP-mediated deubiquitinylation on the cellular localization of PTEN. We found that HAUSP overexpression resulted in the nuclear-exclusion of GFP-PTEN (Fig. 3a), as well as endogenous Pten protein in MEF (Supplementary Fig. 9a). Fractionation of the nucleus and cytoplasm demonstrated a clear enrichment of cytoplasmic PTEN in HAUSP overexpressing cells compared to controls (Fig. 3b and Supplementary Fig. 9b). Strikingly, forced monoubiquitinylation of PTEN with KØ-Ub, which displays mostly nuclear localization, was rendered predominantly cytoplasmic upon HAUSP overexpression (Fig. 3c,d). Conversely, knock-down of HAUSP by RNAi rendered PTEN mainly nuclear (Fig. 3e,f), in agreement with the notion that monoubiquitinylated PTEN is localized to the nucleus (Fig. 2d,e, Supplementary Fig. 8b.d and Ref. 7). Furthermore, we analyzed PTEN localization in HCT116 colon carcinoma cells in which HAUSP was deleted by somatic gene-targeting25. In parental HCT116 cells, PTEN was localized throughout the cell, however _HAUSP_-null cells had enriched proportion of nuclear PTEN (Fig. 3g,h).
Figure 3. HAUSP regulates PTEN localization.

a, Regulation of PTEN localization by HAUSP. IF analysis of GFP-PTEN in Myc-HAUSP-transfected PC3 cells (*P<0.01; #P<0.05). Arrowhead: HAUSPhigh. Asterisk: HAUSPlow. Scale bars, 10 μm. b, Nuclear-cytoplasmic fractionation of GFP-PTEN in empty vector or Myc-HAUSP-transfected PC3 cells. c, Regulation of PTEN localization by HAUSP-mediated deubiquitinylation. IF analysis of GFP-PTEN in HA-KØ-Ub or/and Myc-HAUSP-transfected PC3 cells. Scale bars, 10 μm. d, Nuclear-cytoplasmic fractionation of GFP-PTEN in HA-KØ-Ub or/and Myc-HAUSP-transfected PC3 cells. e, Effects of HAUSP depletion on PTEN localization. IF analysis of GFP-PTEN in control or HAUSP siRNA-transfected PC3 cells (*P<0.01; #P<0.05). Scale bars, 10 μm. f, Nuclear-cytoplasmic fractionation of GFP-PTEN in control or HAUSP siRNA-transfected PC3 cells. g, Regulation of PTEN localization by HAUSP loss. IF analysis of PTEN in HAUSP+/+ and _HAUSP_-/- HCT116 cells. Scale bars, 10 μm. h, Nuclear-cytoplasmic fractionation of PTEN in HAUSP+/+ and _HAUSP_-/- HCT116 cells. i, IHC analysis of prostate tumor tissue microarray (TMA) for PTEN and HAUSP (left). Arrows point out nuclear PTEN with low HAUSP and arrowheads indicate cytoplasmic PTEN with high HAUSP. PTEN localization in TMA with specimens from 81 prostate cancer patients reveals positive correlation between cytoplasmic PTEN and HAUSP overexpression (right). Statistical significance is indicated.
We have previously identified K289 and K13 as two major conserved sites for PTEN ubiquitinylation, and demonstrated that ubiquitin conjugation to these sites are important for the nuclear-cytoplasmic shuttling of PTEN7. To examine the role of HAUSP for the deubiquitinylation of K289 we utilized a mutant GFP-PTENK289E expression construct, which displays a nuclear-exclusion phenotype (Supplementary Fig. 9c,d). Surprisingly, HAUSP silencing or HAUSP deletion rendered PTENK289E predominantly nuclear (Supplementary Fig 9c,d). As demonstrated previously by Trotman et al. (2007), forced monoubiquitinylation of PTENK289E with KØ-Ub promoted its nuclear localization (Supplementary Fig. 9e,f). Unexpectedly, HAUSP overexpression was able to effectively relocalize PTENK289E-KØ-Ub to the cytoplasm, thereby suggesting the presence of other monoubiquitinylated residues on PTEN (Supplementary Fig. 9e,f). In agreement with this hypothesis, PTENK13E was similarly modulated by HAUSP levels (Supplementary Fig. 10a-c). Importantly, the double mutant PTEN_K13,289E_ was resistant to HAUSP-mediated nuclear localization (Supplementary Fig. 10d-f).
Nuclear exclusion of PTEN has been associated with more aggressive or late-stage cancers suggesting that cytoplasmic PTEN lacks full tumor suppressive function1-6. Importantly, nuclear exclusion of a PTEN germ-line mutant, which is catalytically active but unable to accumulate in the nucleus, leads to Cowden syndrome and tumor susceptibility7. To address the role of nuclear PTEN in tumor suppression, we expressed GFP-tagged wild type or K13,289E PTEN in (PTEN_-null) PC3 prostate cancer cells and analyzed apoptosis by Terminal Transferase dUTP Nick End Labeling (TUNEL) (Supplementary Fig. 10g) and confirmed by Annexin V flow cytometric analysis (data not shown). As expected PTEN expression increased apoptosis 12.5-fold over vector control cells (2.5 ± 0.5% and 0.2 ± 0.02%, respectively). However, the expression of a nuclear excluded mutant PTEN (PTEN_K13,289E) resulted in a lower apoptotic index (6.4 fold over vector control; 1.28 ± 0.7%). Importantly, forced monoubiquitinylation and subsequent nuclear localization of PTEN by co-expression of KØ-Ub further increased the apoptotic potential (22.7 fold over KØ-Ub control cells; 6.35 ± 2.1% vs 0.28 ± 0.1%, respectively and 2.5 fold over PTEN transfection). By contrast, co-expression of KØ-Ub with PTEN_K13,289E_ (which remains cytoplasmic; Supplementary Fig. 10e,f) did not increase the apoptotic potential of PTEN_K13,289E_ expression alone (0.85 fold; 1.09 ± 0.8%). These results further support the notion that nuclear excluded PTEN has a decreased apoptotic potential, and therefore less tumor suppressive ability. These findings are of great relevance since the HAUSP/PTEN regulatory network is frequently aberrant in human prostate cancer (see below).
These results in fact imply that deubiquitinylation of PTEN protein by HAUSP, presumably on the previously identified lysine residues 13 and 289 (Ref. 7), plays a crucial role in PTEN localization and function. This in turn implies that alterations in the levels of PTEN DUBs may account for the nuclear exclusion of PTEN observed in many cancer types. To test this hypothesis we analysed gene expression databases as well as human prostate tumour tissue microarrays (TMA) for PTEN and HAUSP status26. We found that HAUSP is overexpressed in prostate cancer and more importantly, that high levels of HAUSP are directly correlated with tumour aggressiveness (Supplementary Fig. 11 and Ref. 22). Moreover, the association between HAUSP levels and PTEN localization indicated the existence of a direct relationship (p < 0.000008; χ2-test) between HAUSP levels and nuclear exclusion of PTEN (Fig. 3i). These results are the first to report that HAUSP is overexpressed in cancer and directly associated with tumour aggressiveness. Furthermore, the correlation with PTEN localization lends support to the notion that overexpression of this DUB impacts on the correct localization of the tumor suppressor PTEN, and therefore on tumourigenesis.
Collectively our findings led us to hypothesize that PML and HAUSP exert opposite functions with respect to PTEN monoubiquitinylation and localization. To further investigate this possibility we tested the ability of PML to oppose HAUSP-mediated PTEN deubiquitinylation. Strikingly, the ability of HAUSP overexpression to localize PTEN to the cytoplasm was drastically inhibited by co-overexpression of PML, such that PTEN was predominantly localized to the nucleus (Fig. 4a) and partially co-localized with PML and HAUSP (Supplementary Fig. 12a). Furthermore, PML was able to inhibit the deubiquitinylation of PTEN (Fig 4b) observed upon HAUSP overexpression alone (Fig. 4b). Unlike PML, PML-RARα overexpression was not able to prevent HAUSP-mediated deubiquitinylation of PTEN (Fig. 4b).
Figure 4. PML opposes HAUSP-mediated PTEN deubiquitinylation.
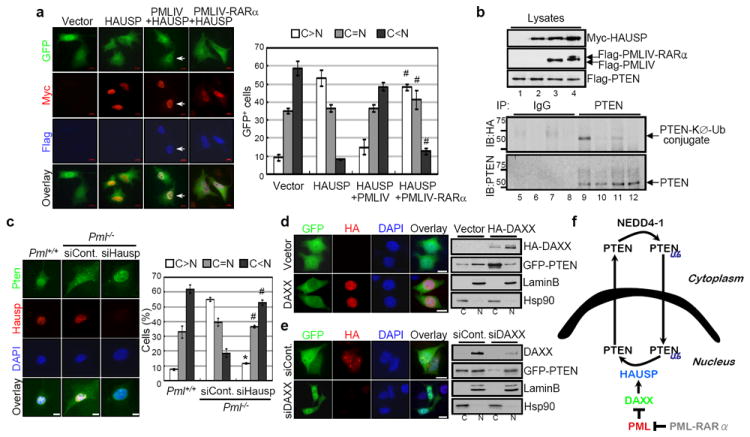
a, Inhibition of HAUSP-mediated nuclear exclusion of PTEN by PML. IF analysis of GFP-PTEN in Myc-HAUSP, Flag-PMLIV or PMLIV-RARα-transfected PC3 cells (#P<0.05). Magnified images (arrows) are shown in Supplementary Figure 12a. b, Inhibition of HAUSP-mediated deubiquitinylation of PTEN by PML. Lysates from immortalized _Pml_-/- MEFs transfected with Flag-PTEN alone (lanes 1, 5 and 9), Flag-PTEN and Myc-HAUSP (lanes 2, 6 and 10), Flag-PTEN, Myc-HAUSP and Flag-PMLIV (lanes 3, 7 and 11) or Flag-PTEN, Myc-HAUSP and Flag-PMLIV-RARα (lanes 4, 8 and 12) in the presence of HA-KØ-Ub were immunoprecipitated (IP) with control IgG (lanes 5 to 8) or anti-PTEN (lanes 9 to 12), and then analyzed for monoubiquitinylation by immunoblotting with anti-HA. Molecular weights are in kDa. c, IF analysis of Pten in control or Hausp siRNA-transfected primary _Pml_-/- MEFs (*P<0.01; #P<0.05). Scale bars, 10 μm. d, Regulation of PTEN localization by DAXX. IF analysis of GFP-PTEN in HA-DAXX-transfected PC3 cells (left). Scale bars, 10 μm. Nuclear-cytoplasmic fractionation of GFP-PTEN in empty vector or HA-DAXX-transfected PC3 cells (right). e, Effects of DAXX depletion on PTEN localization. IF analysis of GFP-PTEN in control or DAXX siRNA-transfected PC3 cells (left). Scale bars, 10 μm. Nuclear-cytoplasmic fractionation of GFP-PTEN in control or DAXX siRNA-transfected PC3 cells (right). f, A model for PTEN monoubiquitinylation and localization by NEDD4-1, HAUSP, DAXX and PML. Cytoplasmic PTEN is monoubiquitinylated (Ub) by NEDD4-1 and subsequently translocated into the nucleus, whereas HAUSP induces deubiquitinylation and nuclear export of PTEN. PML inhibits HAUSP-mediated deubiquitinylation of PTEN and can rescue nuclear PTEN localization through DAXX.
Based on these data, we reasoned that PML-deficiency would render HAUSP overactive, thereby leading to nuclear exclusion of PTEN. Accordingly, Hausp silencing in _Pml_-null MEF resulted in a redistribution of GFP-PTEN as well as endogenous Pten from being predominantly cytoplasmic to one that was highly reminiscent of wild type control cells (Fig. 4c and Supplementary Fig. 12b). Overall, we have identified a novel pathway by which PML regulates PTEN compartmentalization through the inhibition of HAUSP-mediated deubiquitinylation.
To elucidate the mechanism by which PML opposes HAUSP function towards PTEN we first examined the consequences of expression of 3 different PML-mutant proteins on PTEN localization (Supplementary Fig. 13a). Surprisingly, sumoylation deficient PML species, which have been shown to form non-functional NBs27, were unable to localize PTEN to the nucleus as compared to the wild type control and a cs mutant (Supplementary Fig 13b). This suggests that the presence of functional NBs is requisite for HAUSP function. On this basis, we focused on DAXX, a NB-residing protein which has been demonstrated to modulate HAUSP function, and fails to be repressed by the sumoylation defective PML mutants28. Indeed, DAXX overexpression led to cytoplasmic PTEN localization (Fig 4d) whereas DAXX silencing increased nuclear fraction of PTEN (Fig 4e).
Taken together, our findings allowed us to reach a number of relevant conclusions:
- We identify HAUSP as a critical and essential enzyme for PTEN deubiquitinylation and subcellular localization. Nuclear exclusion of PTEN is associated with a more aggressive disease in various tumour types1-7 Also, PTEN has been recently proposed to regulate genomic stability from the nucleus12, and its forced nuclear expression can oppose anchorage independent growth in transformation assays29. In line with this idea, our data show that monoubiquitinylated PTEN, which is mainly nuclear, posses greater apoptotic potential. This suggests that the tumour suppressive activity of PTEN is at least in part due to its ability to reside in the nucleus. A recent study examining a cancer-associated germline PTEN mutation shed light on a ubiquitinylation-dependent mechanism of nuclear-cytoplasmic transport and reported that inhibition of ubiquitin-conjugation by cancer associated mutations of critical lysine residues on PTEN has direct consequences for its localization and tumour suppressor function7. To date, only the E3-ligase NEDD4-1 has been reported to ubiquitinylate PTEN30, while additional E3s may also serve this purpose. Consequently, the notion of ubiquitin-dependency for localization implies that other enzymes capable of deubiquitinylating PTEN may also play an important role in PTEN localization. Therefore, the identification of HAUSP as a critical DUB for PTEN subcellular localization lends strong support to the notion that cellular localization of PTEN is controlled by its ubiquitinylation status.
- We propose a new model for PTEN trafficking (Fig. 4f) whereby in physiological conditions PTEN ubiquitinylation by E3 ligases (such as NEDD4-1) would allow PTEN to accumulate in the nucleus, while PTEN deubiquitinylation by HAUSP and other yet unidentified DUBs would favour PTEN accumulation in the cytoplasm. According to this model, PML would play a critical regulatory role by inhibiting HAUSP activity through DAXX, which in turn favours PTEN nuclear localization.
- These findings have important implications for human cancer as the novel regulatory network we have identified can be perturbed at multiple levels in oncogenic conditions (Fig. 4f). First, HAUSP (a DUB previously known to modify several important genes including p53 and FOXO4) overexpression leads to a predominantly nuclear-excluded PTEN. That this phenotype is associated with more aggressive prostate cancer implies that HAUSP may behave as an oncogene when aberrantly expressed through its ability to disrupt PTEN function through delocalization. Indeed, our finding that HAUSP is overexpressed in human cancer, and that overexpression of HAUSP is associated with nuclear exclusion of PTEN and more aggressive disease supports this hypothesis. Secondly, the fact that this network includes the tumour suppressor PML suggests that PTEN delocalization may be an important consequence in _PML_-loss associated cancers, which include both APL, through the oncogenic activity of PML-RARα, and multiple solid tumours19. Lastly, the fact that in APL blasts, PTEN is excluded from the nucleus implicates loss of PTEN nuclear function in the pathogenesis of this and possibly other forms of leukaemia. The identification of such network and its deregulated hubs in human cancers will ultimately allow the development of drugs that restore normal PTEN localization towards effective therapy as epitomized by ATRA and ATO treatment in APL cells.
Methods Summary
Cell culture
Primary Pml+/+, Pml+/− and _Pml_−/− MEFs were prepared from embryos at day 13.5 of development (E13.5). Early passage (P2-P5) MEFs or SV40 immortalized MEFs were used in all experiments, except as indicated. NB4 cells, a human APL cell line bearing the t(15;17) and ATRA-resistant NB4 (NB4R) cells, were a gift of M. Lanotte. HAUSP+/+ and _HAUSP_−/− HCT116 cells were kindly provided by B. Vogelstein. All-trans retinoic acid (ATRA; Sigma) and arsenic trioxide (ATO; Sigma) was prepared at a concentration of 1 mmol l-1 in PBS and 1 mol l-2 in NaOH, respectively.
In vitro promyelocytes culture
Bone marrow (BM) cells from wild type (n=5) and MRP8-PML-RARα transgenic mice (n=5) were obtained by flushing the femur and tibia BM in RPMI containing 15% FBS. Promyelocytes were collected using the MACS separation column (Miltenyi Biotec) after incubation with anti-CD11b (M1170) and anti-Gr-1 (Rb6-8c5, eBioscience), cultured in RPMI and then treated with DMSO or ATRA for 72 hours.
Immunohistochemistry on patient samples and tumor tissue microarrays
BM smear slides from 9 patients with AML and 9 patients with APL were analyzed by immunofluorescence with anti-PTEN (6H2.1, Cascade) and PML (Ab1370, Chemicon) as previously described7. APL patient sample slides before (n=40) and after (n=9) ATRA treatment were also stained for PTEN and PML. Prostate tumor tissue microarrays were constructed using a fully automated Beecher Instrument, ATA-27. The study cohort comprised APL and prostate tumors consecutively ascertained at the Memorial Sloan-Kettering Cancer Center (MSKCC). All biopsies were evaluated at MSKCC, and the histological diagnosis was based on established criteria.
Plasmids and recombinant proteins
pEGFP-PTEN, HA-KØ-Ub, pCMV-Tag2B-PTEN, -PMLIV, -PMLIV-RARα, and HA-DAXX were described previously7, 27. Recombinant GST-PTEN and His-PTEN protein were kindly provided by X. Jiang30. Myc-HAUSP wild type (wt) and the mutant form (cs) were a generous gift from K.H. Baek. For purification of recombinant HAUSP, Flag-HAUSP-wt and -cs were expressed in 293T cells and purified on M2 column.
siRNA transfection
SMARTpool HAUSP (Dharmacon) represents four pooled SMART-selected siRNA duplexes that target HAUSP (sense sequences are: CUAAGGACCCUGCAAAUUAUU; GUGGUUACGUUAUCAAAUAUU; UGACGUGUCUCUUGAUAAAUU; GAAGGUACUUUAAGAGAUCUU). Cells were transfected with 200 nM of either luciferase or scrambled siRNA (controls) or HAUSP siRNA using the DharmaFECT transfection reagent (Thermo Scientific). Twenty-four hours after transfection, cells were plated for additional transfection for immunofluorescence or immunoblotting.
Immunoblotting, immunoprecipitation and in vitro binding assay
Immunoblotting, (Co-)immunoprecipitation and in vitro binding assay were performed as described7. The following antibodies were used for immunoblotting: PTEN (1:1,000, Cell Signaling; 1:1,000, Cascade Bioscience), Lamin B (1:2,000, Abcam), Hsp90 (1:2,000, BD Transduction Laboratories), Actin (1:5,000, Sigma), Myc (1:2,000, Cell Signaling), Flag (1:2,000, Sigma), GST (1:5,000, Santa Cruz), HA (1:2,000, Covance), HAUSP (1:2,000, Bethyl Laboratories), and DAXX (1:5,000, Epitomics).
Immunofluorescence
Immunofluorescence was performed as described7. The following antibodies were used: PTEN (1:200, 6H2.1, Cascade Bioscience), PML (1:500, Ab1370, Chemicon), HAUSP (1:500), Myc (1:1,000), Flag (1:1,000), HA (1:1,000) and DAXX (1:500, M-112, Santa Cruz). Secondary antibodies used were: Alexa Fluor-488 or -594 (Molecular Probes) or AMCA-conjugated IgG (Jackson ImmunoResearch Laboratories). Coverslips were analyzed by an inverted microscope (Zeiss Axiovert 200) or a confocal laser-scanning microscope (Leica TSC STED). Data were processed with Adobe Photoshop 7.0 software.
Nuclear/cytoplasmic fractionation
Nuclear/cytoplasmic fractionation was performed as described7. Nuclear to cytoplasmic ratios were calculated using the following densities [PTEN-nuc/LaminB]/[PTEN-cyto/Hsp90].
Ubiquitinylation assays
In vivo ubiquitinylation was performed as described7. Immunoprecipitates with anti-PTEN were washed twice with 0.5 M LiCl in TBS and twice with TBS, and then resolved by SDS-PAGE. In vitro ubiquitinylation was performed as described7.
Statystical Analysis
Statistical significance was evaluated by using the student’s t-test and Chi square analysis when required.
Supplementary Material
supp fig and legends
supp files
Acknowledgments
We thank B. Vogelstein, K. H. Baek, M. Lanotte and X. Jiang for sharing valuable reagents and W. Gu for critical discussions. We are grateful to all members of the Pandolfi lab, in particular we thank K. Ito, S. Majid and L. Poliseno for technical support, advice and discussion. This work was supported by NIH grants to P.P.P. L.S. is supported by a Long-Term Fellowship from the International Human Frontier Science Program Organization, and A.C. is supported by a Long-Term Fellowship Award from the European Molecular Biology Organization.
Footnotes
Contributions
The experiments were conceived and designed by M.S.S., L.S., A.C. and P.P.P. Experiments were performed by M.S.S., L.S., A.E. and A.C. F.L.C. provided APL and AML samples. J.T.F. provided and scored IHC of prostate cancer tissue microarray and APL samples. Data were analyzed by M.S.S., L.S., A.C., J.T.F. and P.P.P. The paper was written by M.S.S., L.S., A.C. and P.P.P.
References
- 1.Tachibana M, et al. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94:1955–60. doi: 10.1002/cncr.0678. [DOI] [PubMed] [Google Scholar]
- 2.Whiteman DC, et al. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–7. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- 3.Zhou XP, et al. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–8. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou XP, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol. 2002;161:439–47. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perren A, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097–103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridberg M, et al. Protein expression and cellular localization in two prognostic subgroups of diffuse large B-cell lymphoma: Higher expression of ZAP70 and PKC-beta II in the non-germinal center group and poor survival in patients deficient in nuclear PTEN. Leuk Lymphoma. 2007:1–12. doi: 10.1080/10428190701636443. [DOI] [PubMed] [Google Scholar]
- 7.Trotman LC, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 9.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 10.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 11.Baker SJ. PTEN enters the nuclear age. Cell. 2007;128:25–8. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 12.Shen WH, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 13.Carracedo A, Salmena L, Pandolfi PP. SnapShot: PTEN signaling pathways. Cell. 2008;133:550 e1. doi: 10.1016/j.cell.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Tallman MS. Acute promyelocytic leukemia as a paradigm for targeted therapy. Semin Hematol. 2004;41:27–32. doi: 10.1053/j.seminhematol.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Lallemand-Breitenbach V, et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med. 2001;193:1361–71. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lallemand-Breitenbach V, et al. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol. 2008;10:547–55. doi: 10.1038/ncb1717. [DOI] [PubMed] [Google Scholar]
- 18.Tatham MH, et al. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol. 2008;10:538–46. doi: 10.1038/ncb1716. [DOI] [PubMed] [Google Scholar]
- 19.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 20.Salmena L, Pandolfi PP. Changing venues for tumour suppression: balancing destruction and localization by monoubiquitylation. Nat Rev Cancer. 2007;7:409–13. doi: 10.1038/nrc2145. [DOI] [PubMed] [Google Scholar]
- 21.Everett RD, et al. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. Embo J. 1997;16:1519–30. doi: 10.1093/emboj/16.7.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, et al. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–5. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 23.van der Horst A, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–73. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 24.Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. Embo J. 2007;26:923–34. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummins JM, et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1 p following 486. doi: 10.1038/nature02501. [DOI] [PubMed] [Google Scholar]
- 26.Rhodes DR, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006;24:331–9. doi: 10.1016/j.molcel.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin DY, et al. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell. 2006;24:341–54. doi: 10.1016/j.molcel.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 29.Liu JL, et al. Nuclear PTEN-mediated growth suppression is independent of Akt down-regulation. Mol Cell Biol. 2005;25:6211–24. doi: 10.1128/MCB.25.14.6211-6224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–39. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supp fig and legends
supp files