The neurovascular unit and combination treatment strategies for stroke (original) (raw)
. Author manuscript; available in PMC: 2013 Aug 1.
Published in final edited form as: Trends Pharmacol Sci. 2012 May 16;33(8):415–422. doi: 10.1016/j.tips.2012.04.006
Abstract
Tissue plasminogen activator (tPA) administered within 4.5 hours of symptom onset restores cerebral blood flow and promotes neurological recovery of stroke patients. However, the narrow therapeutic time window and the risk of intracerebral hemorrhage after tPA treatment pose major hurdles to its clinical usage. In light of the failures of neuroprotective therapies in clinical trials, emerging concepts suggest that neuroprotection alone without restoration of tissue perfusion and vascular integrity may not be adequate for treatment of acute stroke. Here we review evidence of the use of adjuvant pharmacological agents to extend the therapeutic window for tPA via targeting the neurovascular unit and the underlying mechanisms of the combination therapy in experimental stroke.
Stroke
Stroke is a serious health problem worldwide and shows increasing prevalence as the population ages. Despite a wealth of insight into the pathogenesis of stroke, current therapies for this devastating disease are far from optimal. The only treatment approved for stroke by the USA Food and Drug Administration (FDA) is intravenous tissue plasminogen activator (tPA), which dissolves the obstructive clot to restore blood flow 1. However, only a small percentage of stroke patients are eligible for tPA, largely due to the narrow therapeutic window 2–4. In addition, thrombolysis with tPA carries a higher risk of symptomatic hemorrhagic complications, which hampers the clinical use of tPA 5, 6. Thus, alternative or complementary therapeutic approaches aimed at expanding the therapeutic window for thrombolysis are of great clinical importance.
Stroke is a highly complex process which involves cerebrovasculature and parenchymal tissues through the interaction of multiple mechanisms 7–9. There have been substantial advances in understanding ischemic neuronal injury. Although neuroprotection has been validated in experimental stroke, clinical trials show that many neuroprotective drugs failed to achieve desirable clinical benefit for treatment of acute stroke 10–14. In addition to multifactor effects, such as clinical trial design inconsistent with preclinical studies and lack of rigorous preclinical investigation, that may have lead to failure of the clinical trials, evidence from these trials suggests that neuroprotection alone without restoration of tissue perfusion and vascular integrity may have compromised the therapeutic benefit of promising neuroprotective agents for treatment of acute stroke13–16. The neurovascular unit is a functional and structurally interdependent multi-cellular complex, comprised of endothelial cells, the basal lamina, pericytes, astrocytes, and neurons 17–19. A successful therapy for acute stroke must restore the normal function of the neurovascular unit by rapidly reestablishing cerebral blood flow in the microvascular bed, preserving vascular integrity, and minimizing neuronal death. Indeed, in experimental stroke, thrombolytic agents in combination with compounds that target the neurovascular unit show synergistic effects on reduction of ischemic brain damage 20–24. In this review, we discuss the neurovascular protective effects of these multi-target approaches in experimental stroke and their potential as adjuvant agents in extending the therapeutic window for stroke.
Neurovascular dysfunction: a role for ischemic penumbra?
Abrupt interruption of blood supply in the brain leads to irreversible tissue damage in the ischemic core, whereas the immediate peripheral regions with less severe blood perfusion deficits contain potentially salvageable ischemic tissue - termed ischemic penumbra which gradually evolves into infarction within days in the absence of reperfusion 25, 26. Thus, the ischemic penumbra represents the most clinically relevant therapeutic target, and becomes the focal point of stroke research for treatment of acute stroke. It is well accepted that the evolution of ischemic brain damage is a heterogeneous and dynamic process, which is manifested by the interplay of multiple cell types through a cascade of molecular events. Although some of these events coincide spatially and temporally with the gradual recruitment of potentially salvageable tissue to the ischemic core, the others are associated with endogenous protective and repair mechanisms, and thus, generate multiple molecular penumbras of ischemic brain 16, 27. Emerging evidence suggests that the neurovascular unit is not only the target of these molecular events, but also actively participates in the pathogenesis of stroke. Situated at the interface between the circulating blood and brain parenchyma, endothelial cells structurally and functionally interact with basal lamina, astrocytic end-foot processes, and pericytes to form the blood-brain barrier (BBB), all of which are essential for maintaining circulatory homeostasis and neural function via facilitating neurovascular coupling, modulating vessel tone, and regulating the barrier functions of the BBB 9, 28–31.
Shortly after onset of focal cerebral ischemia, neurovascular dysfunction is manifested by the disruption of BBB integrity and function 32, 33. On the cellular level, endothelial cells rapidly convert into a pro-inflammatory/pro-thrombotic state via upregulating protease-activated receptor 1 (PAR-1) and tissue factor (TF), and matrix metalloproteinase (MMP) gene expression in the ischemic core and boundary, which facilitates inflammation and BBB disruption 34–36. In addition, the pericyte contraction following ischemic insult results in persistent microvascular constriction, despite successful reopening of the occluded vessel, indicating that pericytes are involved in the microvascular hemodynamic dysfunction in stroke37. Furthermore, stroke induces a rapid loss of β1-integrin expression in the endothelial cells and astrocytes, which coincide with increased cerebrovascular permeability 38–40.
More recently, activation of the immune system has been implicated in neurovascular dysfunction following stroke 41–48. Toll-like receptors (TLRs) are the key receptors of innate immunity 49, 50. Following ischemic insult, the release of damage-associated molecular patterns (DAMPs) from damaged tissue, including heat shock proteins, high mobility group box 1 (HMGB1), and fibrinogen, trigger the activation of TLRs. The TLRs in turn signal through transcription factors NF-κB and activator protein-1 (AP-1) to induce the expression of genes encoding for inflammation and apoptosis 51, 52(Figure 1). In the cerebral endothelial cells, the activation of TLRs following oxidative stress causes downregulation of tight junction proteins 53. In addition, mice with a defective TLR4 gene have reduced MMP-9 expression and are less susceptible to ischemic brain injury, suggesting TLR4 signaling plays a crucial role for MMP-9 induction and neurovascular injury 54. Importantly, in a rat model of embolic stroke, the upregulation of interleukin-1 receptor-associated kinase-1 (IRAK-1), an important component of the TLR/NF-kB signaling pathway, is associated with BBB disruption and increased brain hemorrhage 55. Thus, experimental evidence indicates that stroke induces a rapid BBB disruption through multiple cellular and molecular events. In parallel, the importance of neurovascular dysfunction in the pathogenesis of stroke has also been demonstrated in stroke patients. Recent studies in human brain tissues show elevated levels of MMP-9 in microvascular endothelium isolated from the ischemic area, and increased numbers of MMP-9-positive neutrophils accumulated at the perivascular areas, which co-localized with microvessel disruption and hemorrhagic transformation in the ischemic brain33, 56, 57. In patients with acute middle cerebral artery (MCA) stroke, high levels of interleukin-10 (IL-10) and tumor necrosis factor-α (TNF-α), as well as low levels of IL-6 and active MMP-9 are associated with clinical-diffusion mismatch-a surrogate indication of ischemic penumbra based on the assumption that ischemic penumbra is likely present in patients with severe clinical deficits and relatively small lesion volumes measured by diffusion-weighted imaging 58. Furthermore, patients with high levels of TLRs at admission are associated with poor stroke outcome and increased inflammatory response in acute stroke 59. Thus, these data provide clinical support for the hypothesis that activation of vascular disruptive mechanisms occurs acutely at the neurovascular unit, which leads to progression of brain damage and unfavorable outcomes. Owing to the intimate relationship among cellular components, neurovascular dysfunction appears to be a potential target for the prevention of secondary damage following stroke.
Figure 1. Endothelial cell dysfunction and TLR signaling.

In response to ischemic insult, endothelial cells express protease-activated receptor 1 (PAR-1), tissue factor (TF), and matrix metalloproteinases (MMPs). This process facilitates the accumulation of fibrin, platelet, and neutrophil, and results in microvascular obstruction. On the abluminal front, MMPs degrade neurovascular matrix, leading to acute BBB disruption. Release of endogenous ligands from damaged cells lead to the activation of TLRs, which signal through a number of adoptors and kinases, such as MyD88, IRAK, TRAF-6. These signaling cascades lead to the production of proinflammatory cytokines through the activation of transcription factors, such as NF-κB and AP-1, which contribute to neurovascular damage.
Perturbation of neurovascular function following stroke limits the thrombolytic effects of tPA
Successful and timely reperfusion of ischemic brain is essential to salvage the vulnerable tissue 1, 60. To date, intravenous (IV) thrombolysis with tPA is the only proven intervention for acute management of stroke; tPA dissolves the fibrin contained in a clot to reestablish blood flow. However, the thrombolytic effects of tPA are far from optimal. Fewer than 40% of stroke patients establish early reperfusion after IV tPA administration61, 62. In addition, the risk of symptomatic hemorrhage is high in patients receiving tPA, and hemorrhage is associated with a higher mortality and worse outcome in surviving patients 63, 64. Due to the heterogeneous mechanisms of stroke, the thrombolytic efficacy of tPA is largely dependent on the composition of the occlusive clots and vascular conditions. Platelet-rich clots are more resistant to thrombolysis than fibrin-rich clots 65, 66. In addition, stroke patients with increased plasminogen activator inhibitor-1 (PAI-1) levels - the principal inhibitor of tPA - are less susceptible to thrombolytic therapy 67, 68. Alternative reperfusion strategies, including combined intravenous and intra-arterial thrombolysis with tPA with or without thrombectomy, ultrasound-enhanced thrombolysis, alternative thrombolytic agents (desmoteplase and tenecteplase), and combination of thrombolytic and antithrombotic agents may enhance the efficacy and safety of thrombolytic therapy. Their possible merits over IV tPA have been thoroughly reviewed elsewhere 69, and will not be discussed further.
Thus, although proper reperfusion is essential to halt stroke progression, thrombolysis with tPA after stroke has been shown to potentiate neurovascular disruption. Indeed, in experimental stroke, acute upregulation of endogenous tPA at the astrocyte and endothelial interface interacts with low-density lipoprotein-related protein (LRP), which potentiates BBB permeability by directly altering the architecture and function of the neurovascular unit 70, 71. In addition, a number of experimental and clinical studies have indicated that tPA facilitates the degradation of critical components of the BBB via the activation of MMPs 56, 72–75. Furthermore, tPA increases oxidative stress and inflammatory responses at the neurovascular unit, which are important mechanisms in mediating reperfusion injury and damage after stroke 76. Moreover, exogenous tPA may cross the BBB without compromising the integrity of the BBB, and then in the parenchyma, tPA may stimulate excitotoxic neuronal injury 77–81. Thus, as a pleiotropic protein, exogenous tPA elicits neurovascular disruption and parenchymal damage, which may constrain the beneficial effects of thrombolytic therapy in stroke. By contrast, as noted above, neurovascular dysfunction after stroke activates coagulation cascade, which facilitates the accumulation of blood elements including, platelets, fibrin, leukocytes, and erythrocytes at the site of occlusion and in downstream microvessels, and therefore, may counteract the fibrinolytic effects through releasing serpins such as PAI-1 82, 83. Thus, these data suggest that tPA potentiates neurovascular dysfunction, and the thrombolytic effects of tPA are hampered by the activation of anti-fibrinolytic pathway at the site of vascular dysfunction following stroke.
Neurovascular aging
Advanced aging is associated with increased morbidity and mortality in stroke patients, and represents a major risk factor for stroke 84, 85. It is well recognized that during aging, cerebral microvasculature undergoes profound structure and functional changes, which may contribute to increased vulnerability to ischemic insults 86, 87. In the normal aged brain, cerebral blood flow (CBF) reduction, and BBB leakage is a widespread phenomenon 88–90. In addition, tight junction proteins such as claudin 5 and occludin are disassembled 90, 91. Several molecular mechanisms such as oxidative stress and mitochondrial dysfunction are activated in endothelial cells, astrocytes, and microglia 92. A recent genomic profile in the aged brain reveals that TLRs, NF-kB, and expression of pro-inflammatory genes are upregulated, which implicates the disruption of innate immunity during aging 93, 94. In experimental stroke, a recent study shows that aging potentiates BBB permeability detected by quantitative MRI, which is associated with increased claudin 5 and occludin disassembly. tPA treatment results in an early BBB disruption and lesion expansion, indicating tPA exacerbates BBB disruption and tissue demise in the aged rat after stroke 90. Thus, advanced aging has a great impact on the neurovascular function. As the thrombolytic agent may render further detrimental effects on the already compromised neurovascular function, the use of the aged animal is mandatory for preclinical evaluation of potential neuroprotective agents.
Combination approaches targeting neurovascular units
Neurovascular dysfunction following stroke poses a major hurdle for thrombolytic therapy. Given the complexity of the cellular-biological mechanisms in the pathogenesis of neurovascular dysfunction following stroke, adjuvant pharmacological agents with multi-complementary mechanisms on neurovascular protection may enhance the efficacy of thrombolytic therapy without adversely compromising the integrity of the BBB. In experimental stroke, several pharmacological agents with multi-targeted neurovascular protective effects promote the efficiency and safety of thrombolysis with tPA.
Statins
Statins, also known as 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase inhibitors, are a class of compounds that have pleiotropic effects well beyond their lipid lowering actions 95. In cardiovascular disease and cerebral ischemia, statins have been shown to exert potent vascular protective effects including: improvement of endothelial dysfunction, inhibition of inflammatory and coagulation cascades, increased nitric oxide synthesis, antioxidant properties, and immunomodulatory actions 96–98. Given their direct mechanisms of action on neurovascular units, statins may represent the ideal adjuvant agents for adjuvant thrombolytic therapies. In a rat model of embolic stroke, combination treatment with atorvastatin and tPA extended the therapeutic window for tPA to at least 4 h after stroke onset 22. The neuroprotective effects of the combination treatment are associated with the enhancement of cerebral vascular patency and integrity, whereas tPA monotherapy exacerbates cerebral vascular disruption, indicating that statins provide protection from ischemic brain damage, largely attributed to the cerebrovascular protective mechanisms 20, 22. Indeed, atorvastatin blocked tPA-upregulated PAR-1 and TF in cerebral endothelium, which are key modulators of thrombus formation. In addition, atorvastatin abolished tPA-induced aggravation of BBB disruption via downregulating endothelial MMP-2 and MMP-9 expression. Moreover, although tPA has been shown to potentiate NMDA receptor mediated excitotoxic neuronal death – the principal mechanisms of neuronal death in ischemic brain 99 – statins protect against NMDA-induced neuronal damage 100. In primary rat cortical astrocytes, simvastatin suppresses tPA-induced upregulation of MMP-9 by inhibiting the Rho kinase pathway 101. Treatment with rosuvastatin completely abolished tPA-induced aggravation of ischemic brain damage in mice after stroke 102. Thus, statins protect against the tPA-mediated neurovascular unit dysfunction, and thereby extend the therapeutic window for stroke. The efficacy of statins in lowering the risk of cerebrovascular events is now well established 103, 104. However, only a few clinical studies addressed the question of whether statins may improve stroke outcome following thrombolytic therapy, and the results are controversial 105. In an observational study of 145 patients, Alvarez-Sabin et al. indicated that prior statin treatment provides long-term functional benefits in stroke patients receiving intravenous tPA without increasing the incidence of hemorrhagic transformation 106. However, Meier et al. analyzed 311 patients treated with intra-arterial urokinase, in which prior statin use (55 patients) is associated with a higher frequency of hemorrhagic transformation without influencing the clinical outcome 107. Moreover, others have reported that statin use in stroke patients treated with tPA is not associated with favorable clinical outcome, and did not increase the risk of hemorrhagic transformation 108, 109. A recent retrospective study on 178 patients showed that statin treatment started within 24h after intravenous thrombolysis improves stroke outcome, whereas prophylactic statin treatment is associated with increased the risk of hemorrhagic transformation 110. Thus, the clinical feasibility of using statins as an adjuvant agent for thrombolytic therapy requires further rigorous investigation.
Proteasome inhibitors
The ubiquitin-proteasome pathway is an important pathway for intracellular protein degradation 111. This pathway regulates central mediators of coagulation and inflammation that are fundamental mechanisms in the development of neurovascular dysfunction after stroke 112. Proteasome inhibitors target the ubiquitin-proteasome pathway, and they have been shown to provide significant therapeutic benefit in experimental stroke. Two different classes of proteasome inhibitor have been employed in the treatment of experimental stroke. Velcade (bortezomib, PS-341) is a dipeptidyl boronic acid, which has been clinically approved and well characterized for the treatment of patients with relapsed multiple myeloma 113, 114. PS-519 (MLN519) is a lactacystin derivative, which demonstrates anti-inflammatory actions in a variety of inflammatory-related pathological conditions 115–117. Both agents are known to effectively suppress immuno-inflammatory cascade by a mechanism involving the inhibition of NF-κB activation in animal models of stroke. In addition, proteasome inhibitors upregulate eNOS, an important regulatory enzyme that maintains endothelium-dependent vasodilatation and also mediates antithrombotic actions 118, 119. Moreover, proteasome inhibitors induce antioxidative enzyme expression, which protects neurons astrocytes, and endothelial cells from oxidative stress 118, 120–123. Thus, proteasome inhibitors may have multiple mechanisms of action on neurovascular units that make it attractive adjuvant agents for thrombolytic therapy. In a rat model of embolic stroke, thrombolysis with tPA at 4 h after stroke increased hemorrhage transformation without reduction of cerebral infarction, but combination treatment with PS-519 and tPA at 2, 4, or 6 h after embolization reduced cerebral infarction without increasing the incidence of hemorrhage transformation124. Similarly, the combination treatment with bortezomib and tPA enhanced vascular patency and vascular integrity, and concomitantly extended the therapeutic window to at least 6 hours after stroke without increasing hemorrhagic transformation 125. The neurovascular protective effect of the combination of bortezomib and tPA were further demonstrated in aged rats after stroke 21. Bortezomib neutralized tPA aggravated BBB disruption via blocking the upregulation of MMP-9. In addition, bortezomib blocked the microvascular secondary thrombus formation, and thereby amplified the thrombolytic effects of tPA (Figure 2) 21. Collectively, these studies indicate that the proteasome inhibitor acts on multiple neurovascular protective mechanisms, which not only counteract tPA potentiated BBB disruption, but also enhanced the thrombolytic effects of tPA. As an important risk factor for stroke, advanced age is associated with lower thrombolytic efficacy and an increased risk of hemorrhagic transformation, which hamper the clinical use of thrombolytic therapy 35, 126, 127, 128. Thus, the beneficial effects of the combination therapy in aged rats may have important clinical implications for stroke treatment.
Figure 2. Effects of combination treatment with BORTEZOMIB and tPA on thrombolysis and vascular patency.
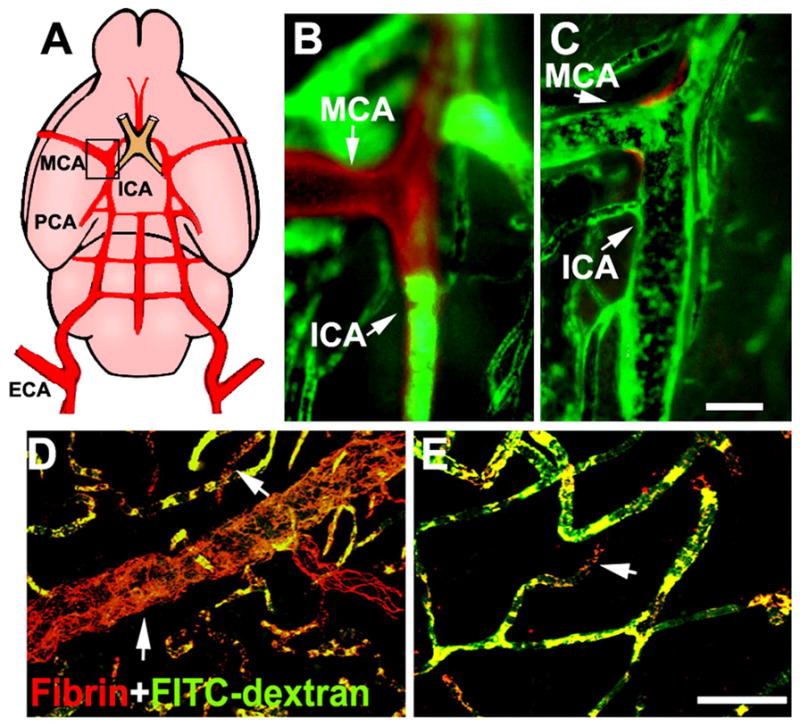
Panel A shows a schematic representation of the ICA and the MCA. A large fragment of Evans blue labeled residual embolus (red) was detected within the origin of the MCA and intracranial segment of the ICA, which blocked plasma perfusion (green) from representative saline-treated rats (B). The combination treatment with bortezomib and tPA resulted in only a small fragment of residual embolus (C, red) at the origin of the MCA where was well perfused by FITC-dextran (C, green). Panels D and E are two-dimensional projections of fibrin/fibrinogen immunoreactivity (red) and vascular plasma perfusion (green) acquired from representative rats treated with saline (D), and combination of BORTEZOMIB and tPA (E). Intravascular fibrin/fibrinogen immunoreactivity was associated with the absence of plasma perfusion in saline treated rats (Arrows in D), suggesting intravascular fibrin deposition blocks plasma perfusion. The combination treatment with bortezomib and tPA reduced number of fibrin/fibrinogen immunoreactive vessels and preserved plasma perfusion compared with saline treated rats. Bars = 400μm for panels B and C, and 100μm for panels D and E.
Minocycline
Minocycline is a broad-spectrum tetracycline antibiotic, which has been widely used for the treatment of various types of bacterial infections. In addition to its well documented anti-inflammatory, anti-apoptotic, and antioxidant properties in the experimental stroke, minocycline exerts potent vascular-protective effects via suppressing stroke induced MMP activation 129–131. Adjuvant administration of minocycline suppresses tPA-potentiated upregulation of plasma levels of MMP-9 and hemorrhagic complications, and thereby extends the thrombolytic time window to 6 hours after embolic stroke in rats 132. In a transient ischemic model of stroke, treatment with minocycline attenuates tPA induced MMPs activation and basal lamina protein degradation without compromising the proteolytic activity of t-PA. In light of these promising preclinical findings, the clinical efficacy of minocycline is currently under investigation. Lampl et al indicated that treatment with minocycline within 24 after stroke onset is associated with better clinical outcome 133. More intriguingly, a recent completed early phase, open-label trial-minocycline to improve neurologic outcome in stroke (MINOS) suggests that minocycline is safe to use alone and in combination with tPA, and can effectively lower plasma MMP-9 levels in patients with or without tPA treatment 134, 135. Thus, the beneficial effects of minocycline in the acute management of stroke and its desirable safety profile make it an attractive candidate for use as an adjunctive therapy to thrombolysis. However, these encouraging early findings should be confirmed by further investigations.
Free radical scavengers
The excessive generation of free radicals following stroke is a major contributor of neurovascular dysfunction following stroke 136, 137. In experimental stroke, free radical scavengers are proven to be effective in reducing BBB disruption as well as ischemic brain damage 138, 139. In addition, treatment with various free radical scavengers markedly reduced t-PA-induced hemorrhage and exerted potent neuroprotective effects 140–142. Thus, free radical scavengers represent potential adjuvant agents for thrombolytic therapy. So far, several free radical scavengers are under various stages of clinical testing (recently reviewed in 143). Despite the promising result from SAINT-I in which acute administration of disodium 2,4-disulphophenyl-_N_-tert-butylnitrone (NXY-059) decreased incidence of tPA-related hemorrhagic complications, pooled data from SAINT-I and SAINT-II trials failed to show meaningful clinical benefit. Recently, a multicenter, randomized, open-labeled study reported that co-administration of edaravone with low dose tPA (0.6mg/kg) is associated with early recanalization and good clinical outcome as compared with patients treated with tPA alone, indicating that the use of free radical scavengers may potentially enhanced the thrombolytic efficacy of tPA 144. Clinical trials free radical scavengers are ongoing and further studies are needed to determine whether free radical scavengers will benefit stroke patients with thrombolytic therapy.
Erythropoietin (EPO)
The cytokine (EPO) is a naturally occurring hormone which was originally described as a primary regulator of erythropoiesis. In the ischemic brain, EPO has been reported to induce neurovascular protection through a broad range of cellular and biological properties. EPO treatment protects against ischemia-induced BBB disruption through down-regulating Flk-1 expression and the response to the VEGF pathway 145. In addition, EPO attenuates inflammatory response, and exerts antiapoptotic and anti-oxidant effect in the animal model of stroke 146–148. However, despite the robust beneficial effects of EPO observed in experimental stroke, the clinical translation of EPO as a neuroprotective agent failed. A double-blind, placebo-controlled, randomized German Multicenter EPO Stroke Trial 149 reported that EPO treatment failed to improve clinical outcomes and increased mortality rate when compared with patients treated with placebo. Subgroup analysis of patients reveal that although patients receiving EPO without tPA treatment showed a better clinical outcome, a significantly increased mortality rate was found in the tPA population, suggesting a adverse EPO and tPA interaction. The detrimental effects of EPO and tPA combintion were further demonstrated in experimental stroke55, 150. In mice subjected to a 90 min transient MCA occlusion, EPO monotherapy reduced brain edema, whereas combination treatment with EPO and tPA exacerbated the BBB permeability and brain edema. Although the combination treatment with EPO and tPA initiated 2h after embolic stroke reduced ischemic brain damage without compromise the BBB integrity, the delayed combination treatment at 6h carries a higher risk of intracerebral hemorrhage without reduction of ischemic brain damage. Upregulation of IRAK1 and activation of NF-κB in cerebral endothelial cells likely contribute to exacerbate neurovascular damage after the delayed combination treatment 55. These findings are consistent with the clinical finding in which a negative influence of tPA protocol violation including treatment beyond the 3h time window (16%) may have contributed to the high mortality rate in patients treated with EPO 149. Taken together, these studies suggest that the clinical translation of adjuvant agents with thrombolytic therapy requires preclinical assessment for their feasibility, safety, and efficacy.
Concluding remarks
In conclusion, neurovascular dysfunction after stroke is multifaceted, which may contribute to a board pathological impairment in the ischemic brain. Here, we described the effects of multi-target neurovascular protective agents in experimental stroke and their potential to enhance the beneficial effects of thrombolytic therapy while minimize the hemorrhagic complication. Although the combination of thrombolytic therapy and pharmacological agents with pleiotropic actions on neurovascular unit appears to be a rational strategy for the treatment of stroke, successful translation of therapies into the clinical will need rigorous investigation of possible detrimental interaction.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hacke W, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 2.Adeoye O, et al. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: a doubling of treatment rates over the course of 5 years. Stroke. 2011;42:1952–1955. doi: 10.1161/STROKEAHA.110.612358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleindorfer D, et al. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke. 2008;39:924–928. doi: 10.1161/STROKEAHA.107.490375. [DOI] [PubMed] [Google Scholar]
- 4.Fonarow GC, et al. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123:750–758. doi: 10.1161/CIRCULATIONAHA.110.974675. [DOI] [PubMed] [Google Scholar]
- 5.Lee M, et al. Blood-brain barrier permeability derangements in posterior circulation ischemic stroke: Frequency and relation to hemorrhagic transformation. J Neurol Sci. 2012;313:142–146. doi: 10.1016/j.jns.2011.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 7.Moskowitz MA, et al. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavez JC, et al. Pharmacologic interventions for stroke: looking beyond the thrombolysis time window into the penumbra with biomarkers, not a stopwatch. Stroke. 2009;40:e558–563. doi: 10.1161/STROKEAHA.109.559914. [DOI] [PubMed] [Google Scholar]
- 9.Lo EH, et al. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 10.Lapchak PA, et al. Effects of the spin trap agent disodium- [tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (generic NXY-059) on intracerebral hemorrhage in a rabbit Large clot embolic stroke model: combination studies with tissue plasminogen activator. Stroke. 2002;33:1665–1670. doi: 10.1161/01.str.0000017145.22806.aa. [DOI] [PubMed] [Google Scholar]
- 11.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2009;40:S4–7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ginsberg MD. Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke. 2009;40:S111–114. doi: 10.1161/STROKEAHA.108.528877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Collins VE, et al. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 15.Feuerstein GZ, et al. Missing steps in the STAIR case: a Translational Medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J Cereb Blood Flow Metab. 2008;28:217–219. doi: 10.1038/sj.jcbfm.9600516. [DOI] [PubMed] [Google Scholar]
- 16.Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- 17.Zacchigna S, et al. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- 18.del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 20.Zhu H, et al. Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. J Cereb Blood Flow Metab. 2010;30:1137–1146. doi: 10.1038/jcbfm.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, et al. Combination treatment with VELCADE and low-dose tissue plasminogen activator provides potent neuroprotection in aged rats after embolic focal ischemia. Stroke. 2010;41:1001–1007. doi: 10.1161/STROKEAHA.109.577288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, et al. Multitargeted effects of statin-enhanced thrombolytic therapy for stroke with recombinant human tissue-type plasminogen activator in the rat. Circulation. 2005;112:3486–3494. doi: 10.1161/CIRCULATIONAHA.104.516757. [DOI] [PubMed] [Google Scholar]
- 23.Fujiwara N, et al. Combination therapy with normobaric oxygen (NBO) plus thrombolysis in experimental ischemic stroke. BMC Neurosci. 2009;10:79. doi: 10.1186/1471-2202-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34:198–209. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos-Cabrer P, et al. Targeting the ischemic penumbra. Stroke. 2011;42:S7–11. doi: 10.1161/STROKEAHA.110.596684. [DOI] [PubMed] [Google Scholar]
- 26.Fisher M. The ischemic penumbra: a new opportunity for neuroprotection. Cerebrovasc Dis. 2006;21(Suppl 2):64–70. doi: 10.1159/000091705. [DOI] [PubMed] [Google Scholar]
- 27.Sharp FR, et al. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 29.Mae M, et al. Getting to know the cast - cellular interactions and signaling at the neurovascular unit. Curr Pharm Des. 2011;17:2750–2754. doi: 10.2174/138161211797440113. [DOI] [PubMed] [Google Scholar]
- 30.Ballabh P, et al. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Lok J, et al. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32:2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- 32.del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98:73–81. doi: 10.1016/s0049-3848(00)00218-8. [DOI] [PubMed] [Google Scholar]
- 33.Rosell A, et al. Mechanisms and markers for hemorrhagic transformation after stroke. Acta Neurochir Suppl. 2008;105:173–178. doi: 10.1007/978-3-211-09469-3_34. [DOI] [PubMed] [Google Scholar]
- 34.Liu XS, et al. Atorvastatin downregulates tissue plasminogen activator-aggravated genes mediating coagulation and vascular permeability in single cerebral endothelial cells captured by laser microdissection. J Cereb Blood Flow Metab. 2006;26:787–796. doi: 10.1038/sj.jcbfm.9600227. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ZG, et al. Cerebral microvascular obstruction by fibrin is associated with upregulation of PAI-1 acutely after onset of focal embolic ischemia in rats. J Neurosci. 1999;19:10898–10907. doi: 10.1523/JNEUROSCI.19-24-10898.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 38.del Zoppo GJ, Milner R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. 2006;26:1966–1975. doi: 10.1161/01.ATV.0000232525.65682.a2. [DOI] [PubMed] [Google Scholar]
- 39.Tagaya M, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21:835–846. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Wagner S, et al. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28:858–865. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- 41.Constantin D, et al. Neisseria meningitidis-induced death of cerebrovascular endothelium: mechanisms triggering transcriptional activation of inducible nitric oxide synthase. J Neurochem. 2004;89:1166–1174. doi: 10.1111/j.1471-4159.2004.02393.x. [DOI] [PubMed] [Google Scholar]
- 42.Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology. 2004;201:197–207. doi: 10.1016/j.tox.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 44.Zhou ML, et al. Expression of Toll-like receptor 4 in the brain in a rabbit experimental subarachnoid haemorrhage model. Inflamm Res. 2007;56:93–97. doi: 10.1007/s00011-006-6035-9. [DOI] [PubMed] [Google Scholar]
- 45.Ziegler G, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 46.Tang SC, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abe T, et al. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41:898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsh BJ, et al. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–1020. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medzhitov R, et al. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 50.Akira S, et al. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 51.Nagyoszi P, et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 57:556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 52.Caso JR, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 53.Nagyoszi P, et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 54.Qiu J, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 41:2077–2082. doi: 10.1161/STROKEAHA.110.590463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia L, et al. Erythropoietin in combination of tissue plasminogen activator exacerbates brain hemorrhage when treatment is initiated 6 hours after stroke. Stroke. 2010;41:2071–2076. doi: 10.1161/STROKEAHA.110.586198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, et al. Targeting extracellular matrix proteolysis for hemorrhagic complications of tPA stroke therapy. CNS Neurol Disord Drug Targets. 2008;7:235–242. doi: 10.2174/187152708784936635. [DOI] [PubMed] [Google Scholar]
- 57.Rosell A, et al. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez-Yanez M, et al. Early biomarkers of clinical-diffusion mismatch in acute ischemic stroke. Stroke. 2011;42:2813–2818. doi: 10.1161/STROKEAHA.111.614503. [DOI] [PubMed] [Google Scholar]
- 59.Brea D, et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011;31:1424–1431. doi: 10.1038/jcbfm.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis SM, et al. Effects of alteplase beyond 3 h after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET): a placebo-controlled randomised trial. Lancet Neurol. 2008;7:299–309. doi: 10.1016/S1474-4422(08)70044-9. [DOI] [PubMed] [Google Scholar]
- 61.del Zoppo GJ, et al. Recombinant tissue plasminogen activator in acute thrombotic and embolic stroke. Ann Neurol. 1992;32:78–86. doi: 10.1002/ana.410320113. [DOI] [PubMed] [Google Scholar]
- 62.von Kummer R, Hacke W. Safety and efficacy of intravenous tissue plasminogen activator and heparin in acute middle cerebral artery stroke. Stroke. 1992;23:646–652. doi: 10.1161/01.str.23.5.646. [DOI] [PubMed] [Google Scholar]
- 63.Berger C, et al. Hemorrhagic transformation of ischemic brain tissue: asymptomatic or symptomatic? Stroke. 2001;32:1330–1335. doi: 10.1161/01.str.32.6.1330. [DOI] [PubMed] [Google Scholar]
- 64.Larrue V, et al. Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European-Australasian Acute Stroke Study (ECASS II) Stroke. 2001;32:438–441. doi: 10.1161/01.str.32.2.438. [DOI] [PubMed] [Google Scholar]
- 65.Pancioli AM. Combination pharmacotherapy for achievement and maintenance of vascular patency. Stroke. 2009;40:S99–102. doi: 10.1161/STROKEAHA.108.529800. [DOI] [PubMed] [Google Scholar]
- 66.Molina CA, et al. Differential pattern of tissue plasminogen activator-induced proximal middle cerebral artery recanalization among stroke subtypes. Stroke. 2004;35:486–490. doi: 10.1161/01.STR.0000110219.67054.BF. [DOI] [PubMed] [Google Scholar]
- 67.Ribo M, et al. Acute hyperglycemia state is associated with lower tPA-induced recanalization rates in stroke patients. Stroke. 2005;36:1705–1709. doi: 10.1161/01.STR.0000173161.05453.90.9f. [DOI] [PubMed] [Google Scholar]
- 68.Kim SH, et al. Plasma fibrinolysis inhibitor levels in acute stroke patients with thrombolysis failure. J Clin Neurol. 2005;1:142–147. doi: 10.3988/jcn.2005.1.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vivien D, et al. Impact of tissue plasminogen activator on the neurovascular unit: from clinical data to experimental evidence. J Cereb Blood Flow Metab. 2011;31:2119–2134. doi: 10.1038/jcbfm.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Polavarapu R, et al. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yepes M, et al. Tissue-type plasminogen activator in the ischemic brain: more than a thrombolytic. Trends Neurosci. 2009;32:48–55. doi: 10.1016/j.tins.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 72.Lapchak PA, et al. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- 73.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- 74.Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34:2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. [DOI] [PubMed] [Google Scholar]
- 75.Montaner J, et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- 76.Warach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption. Stroke. 2004;35:2659–2661. doi: 10.1161/01.STR.0000144051.32131.09. [DOI] [PubMed] [Google Scholar]
- 77.Nicole O, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 78.Benchenane K, et al. Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation. 2005;111:2241–2249. doi: 10.1161/01.CIR.0000163542.48611.A2. [DOI] [PubMed] [Google Scholar]
- 79.Wang YF, et al. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 80.Tsirka SE, et al. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- 81.Lo EH, et al. tPA and proteolysis in the neurovascular unit. Stroke. 2004;35:354–356. doi: 10.1161/01.STR.0000115164.80010.8A. [DOI] [PubMed] [Google Scholar]
- 82.Fay WP, et al. High concentrations of active plasminogen activator inhibitor-1 in porcine coronary artery thrombi. Arterioscler Thromb Vasc Biol. 1996;16:1277–1284. doi: 10.1161/01.atv.16.10.1277. [DOI] [PubMed] [Google Scholar]
- 83.Robbie LA, et al. Proteins of the fibrinolytic system in human thrombi. Thromb Haemost. 1996;75:127–133. [PubMed] [Google Scholar]
- 84.Kelly-Hayes M, et al. The influence of gender and age on disability following ischemic stroke: the Framingham study. J Stroke Cerebrovasc Dis. 2003;12:119–126. doi: 10.1016/S1052-3057(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 85.Rothwell PM, et al. Population-based study of event-rate, incidence, case fatality, and mortality for all acute vascular events in all arterial territories (Oxford Vascular Study) Lancet. 2005;366:1773–1783. doi: 10.1016/S0140-6736(05)67702-1. [DOI] [PubMed] [Google Scholar]
- 86.Park L, et al. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 87.Mayhan WG, et al. Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation. 2008;15:225–236. doi: 10.1080/10739680701641421. [DOI] [PubMed] [Google Scholar]
- 88.Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557–565. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- 89.Simpson JE, et al. Alterations of the blood-brain barrier in cerebral white matter lesions in the ageing brain. Neurosci Lett. 2010;486:246–251. doi: 10.1016/j.neulet.2010.09.063. [DOI] [PubMed] [Google Scholar]
- 90.Kaur J, et al. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood-brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab. 2011;31:1874–1885. doi: 10.1038/jcbfm.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mooradian AD, et al. Age-related changes in rat cerebral occludin and zonula occludens-1 (ZO-1) Mech Ageing Dev. 2003;124:143–146. doi: 10.1016/s0047-6374(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 92.Dei R, et al. Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer’s disease. Acta Neuropathol. 2002;104:113–122. doi: 10.1007/s00401-002-0523-y. [DOI] [PubMed] [Google Scholar]
- 93.Letiembre M, et al. Innate immune receptor expression in normal brain aging. Neuroscience. 2007;146:248–254. doi: 10.1016/j.neuroscience.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 94.Berchtold NC, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maron DJ, et al. Current perspectives on statins. Circulation. 2000;101:207–213. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 96.Mooradian AD, et al. Statins ameliorate endothelial barrier permeability changes in the cerebral tissue of streptozotocin-induced diabetic rats. Diabetes. 2005;54:2977–2982. doi: 10.2337/diabetes.54.10.2977. [DOI] [PubMed] [Google Scholar]
- 97.Sacco RL, Liao JK. Drug Insight: statins and stroke. Nat Clin Pract Cardiovasc Med. 2005;2:576–584. doi: 10.1038/ncpcardio0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 99.Liot G, et al. 2,7-Bis-(4-amidinobenzylidene)-cycloheptan-1-one dihydrochloride, tPA stop, prevents tPA-enhanced excitotoxicity both in vitro and in vivo. J Cereb Blood Flow Metab. 2004;24:1153–1159. doi: 10.1097/01.WCB.0000134476.93809.75. [DOI] [PubMed] [Google Scholar]
- 100.Zacco A, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci. 2003;23:11104–11111. doi: 10.1523/JNEUROSCI.23-35-11104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang S, et al. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke. 2006;37:1910–1912. doi: 10.1161/01.STR.0000226923.48905.39. [DOI] [PubMed] [Google Scholar]
- 102.Kilic E, et al. Aggravation of focal cerebral ischemia by tissue plasminogen activator is reversed by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor but does not depend on endothelial NO synthase. Stroke. 2005;36:332–336. doi: 10.1161/01.STR.0000152273.24063.f7. [DOI] [PubMed] [Google Scholar]
- 103.Amarenco P, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–559. doi: 10.1056/NEJMoa061894. [DOI] [PubMed] [Google Scholar]
- 104.Palnum KH, et al. Use of Secondary Medical Prophylaxis and Clinical Outcome Among Patients With Ischemic Stroke: A Nationwide Follow-Up Study. Stroke. 2011 doi: 10.1161/STROKEAHA.111.635342. [DOI] [PubMed] [Google Scholar]
- 105.Review: Insufficient evidence exists to determine the benefits and risks of statins for acute stroke or TIA. Ann Intern Med. 2011;155:JC606. doi: 10.7326/0003-4819-155-12-201112200-02006. [DOI] [PubMed] [Google Scholar]
- 106.Alvarez-Sabin J, et al. Prior statin use may be associated with improved stroke outcome after tissue plasminogen activator. Stroke. 2007;38:1076–1078. doi: 10.1161/01.STR.0000258075.58283.8f. [DOI] [PubMed] [Google Scholar]
- 107.Meier N, et al. Prior statin use, intracranial hemorrhage, and outcome after intra-arterial thrombolysis for acute ischemic stroke. Stroke. 2009;40:1729–1737. doi: 10.1161/STROKEAHA.108.532473. [DOI] [PubMed] [Google Scholar]
- 108.Miedema I, et al. Statin use and functional outcome after tissue plasminogen activator treatment in acute ischaemic stroke. Cerebrovasc Dis. 29:263–267. doi: 10.1159/000275500. [DOI] [PubMed] [Google Scholar]
- 109.Makihara N, et al. Effect of Serum Lipid Levels on Stroke Outcome after rt-PA Therapy: SAMURAI rt-PA Registry. Cerebrovasc Dis. 33:240–247. doi: 10.1159/000334664. [DOI] [PubMed] [Google Scholar]
- 110.Cappellari M, et al. Does statin in the acute phase of ischemic stroke improve outcome after intravenous thrombolysis? A retrospective study. J Neurol Sci. 2011;308:128–134. doi: 10.1016/j.jns.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 111.Coux O, et al. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 112.Wojcik C, Di Napoli M. Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke. 2004;35:1506–1518. doi: 10.1161/01.STR.0000126891.93919.4e. [DOI] [PubMed] [Google Scholar]
- 113.Adams J, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 114.Kane RC, et al. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 115.Campbell B, et al. Cardioprotective effects of a novel proteasome inhibitor following ischemia and reperfusion in the isolated perfused rat heart. J Mol Cell Cardiol. 1999;31:467–476. doi: 10.1006/jmcc.1998.0880. [DOI] [PubMed] [Google Scholar]
- 116.Conner EM, et al. Proteasome inhibition attenuates nitric oxide synthase expression, VCAM-1 transcription and the development of chronic colitis. J Pharmacol Exp Ther. 1997;282:1615–1622. [PubMed] [Google Scholar]
- 117.Vanderlugt CL, et al. Treatment of established relapsing experimental autoimmune encephalomyelitis with the proteasome inhibitor PS-519. J Autoimmun. 2000;14:205–211. doi: 10.1006/jaut.2000.0370. [DOI] [PubMed] [Google Scholar]
- 118.Stangl K, Stangl V. The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc Res. 2010;85:281–290. doi: 10.1093/cvr/cvp315. [DOI] [PubMed] [Google Scholar]
- 119.Huang PL, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 120.Yamamoto N, et al. Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: relevance to Parkinson disease. J Biol Chem. 2007;282:4364–4372. doi: 10.1074/jbc.M603712200. [DOI] [PubMed] [Google Scholar]
- 121.Chen J, Regan RF. Increasing expression of heme oxygenase-1 by proteasome inhibition protects astrocytes from heme-mediated oxidative injury. Curr Neurovasc Res. 2005;2:189–196. doi: 10.2174/1567202054368344. [DOI] [PubMed] [Google Scholar]
- 122.Lorenz M, et al. Proteasome inhibition prevents experimentally-induced endothelial dysfunction. Life Sci. 2009;84:929–934. doi: 10.1016/j.lfs.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 123.Dreger H, et al. Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc Res. 2010;85:395–403. doi: 10.1093/cvr/cvp279. [DOI] [PubMed] [Google Scholar]
- 124.Zhang L, et al. Postischemic (6-Hour) treatment with recombinant human tissue plasminogen activator and proteasome inhibitor PS-519 reduces infarction in a rat model of embolic focal cerebral ischemia. Stroke. 2001;32:2926–2931. doi: 10.1161/hs1201.100207. [DOI] [PubMed] [Google Scholar]
- 125.Zhang L, et al. Treatment of embolic stroke in rats with bortezomib and recombinant human tissue plasminogen activator. Thromb Haemost. 2006;95:166–173. [PubMed] [Google Scholar]
- 126.Minnema MC, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101:10–14. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huynh T, et al. Predictors of intracranial hemorrhage with fibrinolytic therapy in unselected community patients: a report from the FASTRAK II project. Am Heart J. 2004;148:86–91. doi: 10.1016/j.ahj.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 128.Abbate R, et al. Age-related changes in the hemostatic system. Int J Clin Lab Res. 1993;23:1–3. doi: 10.1007/BF02592271. [DOI] [PubMed] [Google Scholar]
- 129.Yenari MA, et al. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 130.Machado LS, et al. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koistinaho M, et al. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab. 2005;25:460–467. doi: 10.1038/sj.jcbfm.9600040. [DOI] [PubMed] [Google Scholar]
- 132.Murata Y, et al. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke. 2008;39:3372–3377. doi: 10.1161/STROKEAHA.108.514026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lampl Y, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 134.Fagan SC, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 41:2283–2287. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Switzer JA, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke. 2012;42:2633–2635. doi: 10.1161/STROKEAHA.111.618215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chan PH. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994;4:59–65. doi: 10.1111/j.1750-3639.1994.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 137.Heo JH, et al. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 138.Kim GW, et al. The cytosolic antioxidant, copper/zinc superoxide dismutase, attenuates blood-brain barrier disruption and oxidative cellular injury after photothrombotic cortical ischemia in mice. Neuroscience. 2001;105:1007–1018. doi: 10.1016/s0306-4522(01)00237-8. [DOI] [PubMed] [Google Scholar]
- 139.Schmid-Elsaesser R, et al. Superior neuroprotective efficacy of a novel antioxidant (U-101033E) with improved blood-brain barrier permeability in focal cerebral ischemia. Stroke. 1997;28:2018–2024. doi: 10.1161/01.str.28.10.2018. [DOI] [PubMed] [Google Scholar]
- 140.Lapchak PA, et al. Pharmacological effects of the spin trap agents N-t-butyl-phenylnitrone (PBN) and 2,2,6, 6-tetramethylpiperidine-N-oxyl (TEMPO) in a rabbit thromboembolic stroke model: combination studies with the thrombolytic tissue plasminogen activator. Stroke. 2001;32:147–153. doi: 10.1161/01.str.32.1.147. [DOI] [PubMed] [Google Scholar]
- 141.Yagi K, et al. Edaravone, a free radical scavenger, inhibits MMP-9-related brain hemorrhage in rats treated with tissue plasminogen activator. Stroke. 2009;40:626–631. doi: 10.1161/STROKEAHA.108.520262. [DOI] [PubMed] [Google Scholar]
- 142.Yamashita T, et al. Dissociation and protection of the neurovascular unit after thrombolysis and reperfusion in ischemic rat brain. J Cereb Blood Flow Metab. 2009;29:715–725. doi: 10.1038/jcbfm.2008.164. [DOI] [PubMed] [Google Scholar]
- 143.Amaro S, Chamorro A. Translational stroke research of the combination of thrombolysis and antioxidant therapy. Stroke. 42:1495–1499. doi: 10.1161/STROKEAHA.111.615039. [DOI] [PubMed] [Google Scholar]
- 144.Kimura K, et al. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients - a preliminary study. J Neurol Sci. 313:132–136. doi: 10.1016/j.jns.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 145.Li Y, et al. Erythropoietin prevents blood brain barrier damage induced by focal cerebral ischemia in mice. Neurochem Res. 2007;32:2132–2141. doi: 10.1007/s11064-007-9387-9. [DOI] [PubMed] [Google Scholar]
- 146.Wang Y, et al. Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br J Pharmacol. 2007;151:1377–1384. doi: 10.1038/sj.bjp.0707285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Siren AL, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–4049. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Byts N, Siren AL. Erythropoietin: a multimodal neuroprotective agent. Exp Transl Stroke Med. 2009;1:4. doi: 10.1186/2040-7378-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ehrenreich H, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- 150.Zechariah A, et al. Combination of tissue-plasminogen activator with erythropoietin induces blood-brain barrier permeability, extracellular matrix disaggregation, and DNA fragmentation after focal cerebral ischemia in mice. Stroke. 2010;41:1008–1012. doi: 10.1161/STROKEAHA.109.574418. [DOI] [PubMed] [Google Scholar]