Staphylococcus aureus Activation of Caspase 1/Calpain Signaling Mediates Invasion Through Human Keratinocytes (original) (raw)
Abstract
The USA300 strains of Staphylococcus aureus are the major cause of skin and soft tissue infection in the United States. Invasive USA300 infection has been attributed to several virulence factors, including protein A and the α-hemolysin (Hla), which cause pathology by activating host signaling cascades. Here we show that S. aureus exploits the proinflammatory bias of human keratinocytes to activate pyroptosis, a caspase 1–dependent form of inflammatory cell death, which was required for staphylococci to penetrate across a keratinocyte barrier. Keratinocyte necrosis was mediated by calpains, Ca2+-dependent intracellular proteases whose endogenous inhibitor, calpastatin, is targeted by Hla-induced caspase 1. Neither Panton-Valentine leukocidin nor protein A expression was essential, but inhibition of either calpain or caspase 1 activity was sufficient to prevent staphylococcal invasion across the keratinocytes. These studies suggest that pharmacological interruption of specific keratinocyte signaling cascades as well as targeting the Hla might prevent invasive skin infection by staphylococci.
(See the editorial commentary by Otto, on pages 1483–5.)
The morbidity and mortality associated with the epidemic USA300 strains of methicillin-resistant Staphylococcus aureus (MRSA) in the United States has been well documented [1]. In addition to the invasive infections that are associated with significant morbidity and mortality, there is also substantial economic cost associated with the skin and soft tissue infections due to these strains [2]. Exactly which of the many USA300 virulence factors cause this excessive morbidity is widely debated but of great interest in vaccine development [3]. Participation of the α-hemolysin (Hla) in the pathogenesis of skin infection is well documented [4–6], but it remains unclear exactly how these nonmotile bacteria invade through the barrier posed by the multiple layers of proliferating and cornified keratinocytes that comprise normal human skin. Staphylococcus aureus has been shown to invade into human keratinocytes and cause both necrotic and apoptotic forms of cell death, a process attributed to several adhesins and virulence factors [7]. Murine models of skin infection are problematic because staphylococci have difficulty obtaining iron from murine hemoglobin [8] and mouse models require large intradermal inoculations of bacteria, obviating the relevance of the physical and immunological barrier properties of normal human skin [4].
Human keratinocytes are dynamic cells involved in a highly ordered set of developmental activities; initially proliferation, then maturation, cornification, and shedding [9]. As active participants in innate immune signaling, they express Toll-like receptors (TLRs) either constitutively or by induction [10], NODs, and caspases, components of the NLRP3 inflammasome [11, 12]. In response to pathogens, keratinocytes rapidly produce antimicrobial peptides as well as chemokines and cytokines to recruit and activate phagocytes. Given these defenses, normal human skin is remarkably resistant to bacterial infection, even that associated with USA300 MRSA strains. Staphylococcus aureus strains have evolved with their human hosts and are especially adept at exploiting the immune responses that they evoke. Much of the pathology they induce is not necessarily due to the direct toxicity of their virulence factors but instead is due to the immune response elicited. Recent studies have demonstrated that S. aureus protein A, by activating epithelial RhoA/MLC and calpain signaling, mediates staphylococcal invasion through the paracellular junctions of the airway epithelium [13]. We postulated that staphylococcal exploitation of keratinocyte signaling could be responsible for penetration of these organisms through the barrier posed by human keratinocytes and characterized the signaling pathways that are involved.
METHODS
Bacterial Strains and Cell Line
MRSA USA300 (LAC) wild-type (WT) and protein A (SpA−) mutant were grown as described elsewhere [14]. USA300 WT and Hla− mutant were provided by Juliane Bubeck-Wardenburg (University of Chicago), and USA300 WT and Panton-Valentine leukocidin (PVL−) mutant were provided by Frank DeLeo (National Institute of Allergy and Infectious Diseases). The human keratinocyte HaCaT cell line was obtained from Angela Christiano (Columbia University) and grown in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% fetal bovine serum.
Organotypic Cultures and Microscopy
Organotypic cultures of human keratinocytes in primary culture were obtained from the Cell and Tissue Kinetics Core of the Columbia University Department of Dermatology Skin Disease Research Center. Human keratinocytes were grown at an air-liquid interface supported by a dermal substitute matrix as a 3-dimensional model system composed of the dermal and the epidermal compartments. Following 24 hours of stimulation with USA300 or phosphate-buffered saline (PBS), human organotypic skin equivalents were stained with hematoxylin-eosin.
Bacterial Transmigration, Confocal Microscopy, and Dextran Permeability
All analyses were performed as described elsewhere [13] with the following inhibitors: calpeptin (20 μM), cytochalasin D (20 μM), TAPI (50 μM), GM6001 (20 μM), or caspase 3 inhibitor I (10 μM) from Calbiochem, EMD; Z-VAD-FMK (10 μM) or Z-WEHD-FMK (10 μM) from G-Biosciences; and Z-YVAD-FMK (10 μM) from Enzo.
Ca2+ Imaging
HaCaT cells were grown to 80% confluence in coverglass chamberslides and loaded for 1 hour at room temperature with 2 μM Fluo-3/AM in the presence of 0.02% pluronic acid in Minimum Essential Medium Eagle. Cells were washed with PBS and incubated at 37°C for 1 hour in RPMI 1640 medium. Immediately after adding 108 colony-forming units (CFUs) of USA300, Fluo-3/AM fluorescence imaging was obtained and collected at 6-second intervals using a Zeiss LSM 510 META scanning confocal microscope and analyzed using the ImageJ program.
Apoptosis-Pyroptosis Assays
HaCaT cells were grown to confluence in chamber slides, and 108 CFU/mL bacteria at 1:4 dilution was added to chambers. For inhibitor studies, 20 μM calpeptin (calpain inhibitor) was incubated with cells for 30 minutes prior to bacterial stimulation. Pan-caspase inhibitor, Z-VAD-FMK, and the specific caspase 1 inhibitor Z-WEHD-FMK were added at the same time as bacteria. After 2 hours of incubation at 37°C, cells were washed 3 times with cold PBS, then incubated for 30 minutes on ice with Molecular Probes YO-PRO-1 + PI assay according to the manufacturer's instructions. For fluorescence microscopy, after 3 cold PBS washes, 1% paraformaldehyde in PBS was added to wells, which were then imaged by confocal microscopy using a Zeiss LSM 510 Meta Inverted microscope. All detector gain and amplification settings were identical for each image. For flow cytometry, after incubation with YO-PRO-1 + PI and PBS washes, cells were incubated with 0.02% ethylene glycol tetraacetic acid in PBS to induce detachment. Cells were scraped and transferred to tubes containing paraformaldehyde to a final concentration of 1% for flow cytometry analysis by means of a BD FACSCalibur using Cell Quest software (Becton Dickinson).
Statistics
Samples with normal distribution were analyzed by Student t test. Differences between groups were considered significant at P < .05. Multiple comparisons were analyzed using 1-way analysis of variance with an appropriate posttest. Statistical analysis was performed using GraphPad Instat version 3.0.
RESULTS
USA300 Accumulates Within Keratinocytes
We examined the effects of S. aureus USA300, the epidemic MRSA clone responsible for the majority of skin as well as invasive infection in the United States [1], on human keratinocytes in primary organotypic culture (Figure 1A). Following a 24-hour incubation with USA300 S. aureus, human keratinocytes in primary culture remained intact; the basal layers of keratinocytes showed vacuolization and the proliferating layers were obviously thickened, an effect that has been attributed to Hla-EGFR interactions [15]. Confocal images demonstrated USA300 on the surface of polarized HaCaT cells, a human keratinocyte cell line, at 4 hours but no evidence of internalization or paracellular invasion at that time (Figure 1). At 24 hours staphylococci appeared to have accumulated within keratinocytes, in a focal distribution pattern, primarily at the apical surface of the cell cultures (Figure 1B and 1_C_). Staphylococci were not observed within the paracellular spaces.
Figure 1.

Interactions of Staphylococcus aureus strain USA300 and human keratinocytes. A, Light microscopic images of organotypic cultures of human keratinocytes grown at an air-liquid interface supported by a dermal substitute matrix as a 3-dimensional model system composed of the dermal and epidermal compartments under control conditions (phosphate-buffered saline [PBS]) and following a 24-hour incubation with wild-type USA300. B, z_-section confocal images of HaCaT cells grown in a polarized fashion on transwells, following 4-hour and 24-hour incubation with USA300, imaged after staining with phalloidin (red) and anti–_S. aureus (green). C, x-y sections of infected HaCaTs demonstrating focal localization of organisms primarily restricted to the apical surfaces of the cells. Staining is phalloidin (red) and anti–S. aureus (green). Left, enlarged first apical section. Right, series of sections from apical to basal; sequence, left to right, top to bottom.
Participation of S. aureus Virulence Factors in the Activation of Inflammasome Signaling
These images of keratinocytes whose cytoplasm appeared to be entirely replaced by S. aureus suggested the induction of apoptosis/necrosis or pyroptosis, a caspase 1–dependent form of necrotic, proinflammatory cell death [16]. Staphylococcal activation of the NLRP3 inflammasome has been well documented in immune cells [17] and mouse models of skin infection [11], although there are data to suggest the participation of additional pathways in the induction of apoptosis and necrosis as well [7, 12]. Staphylococcus aureus activation of the NLRP3 inflammasome is thought to involve 2 stimuli, TLR2- or NOD2-associated induction of pro–interleukin 1β (IL-1β) production and Hla pore-dependent K+ efflux inducing a sufficient cell stress to activate caspase 1 [18]. Caspase 1 activity results in production of IL-1β, a potent proinflammatory cytokine that is critical for polymorphonuclear leukocyte (PMN) recruitment and eventual clearance of staphylococcal skin infection [6]. Whether virulence factors such as protein A, which activates TNFR1 signaling and thus is potentially linked to apoptosis [19], or the PVL toxin might also contribute to inflammasome signaling is not established.
We postulated that constitutive expression of pro–IL-1β by keratinocytes [20] as shown (Figure 2A) would prime them for activation of the inflammasome. Because caspase 1 is required for pro–IL-1β processing, we similarly documented the production of caspases 1, 3, and 7, which were induced by WT USA300, PVL− mutant, or SpA− mutant (which lack the conserved surface protein that interacts with TNFR1 [19]) with the exception of the Hla− strain that induced significantly less caspase 1 (Figure 2B), which was quantified by a colorimetric assay (G-Biosciences) (Figure 2C). In response to these S. aureus strains, there was significant production of IL-1β that was significantly decreased in cells treated with a pan-caspase inhibitor (Figure 2D), indicating that the majority of IL-1β production in this setting is from inflammasome activation.
Figure 2.

Activation of keratinocyte inflammasome signaling by Staphylococcus aureus USA300 strains. A, Constitutive production of pro–interleukin 1β (IL-1β) shown by immunoblot of cell lysates following exposure to wild-type (WT) USA300 and mutant strains. B, Immunodetection of caspase 1, 3, and 7 production in keratinocytes 2 hours after exposure to USA300 strains. C, Quantification of caspase 1 activity in culture supernatants by colorimetric assay (G-Biosciences) following incubation with USA300 strains for 20 hours. *P < .01 compared with WT (1-way analysis of variance, Dunnett posttest). Data are mean (SD). D, Secretion of IL-1β into culture supernatant following 2-hour exposure to USA300 strains and effect of the pan-caspase inhibitor Z-VAD-FMK (Z-VAD) quantified by enzyme-linked immunosorbent assay. *P < .001 compared with WT; **P < .001 compared with untreated (1-way analysis of variance, Tukey posttest); n= 6 for each. Data are mean (SD). Abbreviations: Hla, α-hemolysin; PVL, Panton-Valentine leukocidin; SpA, protein A.
USA300 Activation of Keratinocyte Calpain Activity
Caspase 1 also targets calpastatin, the endogenous inhibitor of the calpains, intracellular proteases that participate in apoptosis and degradation of intracellular components [21]. To determine whether caspase expression in human keratinocytes was associated with the activation of calpains, we first confirmed the expression of calpains 1 and 2 and calpastatin in the HaCaT cells (Figure 3A) as well as the generation of Ca2+ flux (Figure 3B) required for calpain activity in response to USA300 [22]. We predicted that WT USA300, but not the Hla− mutant, would activate the degradation of calpastatin, through caspase 1 activation. This was confirmed in immunoblots showing calpastatin breakdown in keratinocytes stimulated with WT S. aureus that are capable of activating caspase 1, but not in response to the Hla− mutant or in cells pretreated with calpeptin, a calpain inhibitor expected to interfere with caspase binding (Figure 3C).
Figure 3.

Calpain expression in keratinocytes. A, Calpain expression in HaCaT cells was demonstrated by reverse-transcription polymerase chain reaction using primers for calpain 1 (C1), calpain 2 (C2), calpastatin (Cst), and an actin control (A). B, Activation of Ca2+ fluxes in Fluo-3 loaded HaCaTs following the addition of Staphylococcus aureus strain USA300 or thapsigargin (Thaps) as a positive control. C, Immunodetection of calpastatin in HaCaT lysates following 6-hour incubation with wild-type (WT) alone or pretreated with calpeptin (Calp) or α-hemolysin (Hla−) mutant as compared with a medium control (M).
USA300 Activates Apoptosis and Pyroptosis in Human Keratinocytes
Having demonstrated caspase 1 and inflammasome activation in HaCaT cells exposed to WT but not Hla− S. aureus, we next examined the biological consequences of this cellular response to infection. We differentiated the induction of pyroptosis, or necrosis associated with caspase 1 activity, which typically involves the rapid loss of the integrity of the plasma membrane [16, 23] from apoptosis associated with caspases 3 and 7, using differential fluorescent labeling (Vybrant apoptosis assay kit, YO-PRO-1/propidium iodide, Molecular Probes). After 2 hours of exposure to WT USA300 but not the Hla− mutant, both apoptotic and necrotic keratinocytes were observed (Figure 4A). PVL− and SpA− mutants activated apoptosis and necrosis at rates similar to the WT strain (data not shown). In the presence of a pan-caspase or a caspase-1–specific inhibitor, the numbers of both apoptotic and necrotic/pyroptotic cells decreased significantly (Figure 4B).
Figure 4.

Staphylococcus aureus strain USA300 induces pyroptosis in keratinocytes. A, Fluorescence imaging was used to detect apoptotic keratinocytes permeable to YO-PRO-1 (green) that binds DNA of cells with semipermeable cytoplasmic membranes and pyroptotic, fully permeable necrotic cells (PI) (red) following a 2-hour exposure to USA300: i, YO-PRO-1(+)—apoptotic (green); ii, PI(+)—pyroptotic (red); iii, transmission brightfield image; iv, merged. B, Flow cytometric quantification of apoptotic (YO-PRO-1+) (green) vs necrotic/pyroptotic (PI+) (red) cells as identified in (A), from cultures treated with wild-type (WT) USA300 under control conditions or with pan-caspase inhibitor Z-VAD-FMK (ZV), a caspase 1–specific inhibitor, Z-WEHD-FMK (ZW), or calpeptin (Cpep), as compared with the effects of the α-hemolysin (Hla−) mutant strain. *P < .01 compared with WT (1-way analysis of variance, Dunnett posttest). Data are mean (SD), representative of 2 individual experiments. C, Immunodetection of HMGB1 in keratinocyte supernatants following 6-hour incubation with WT or Hla− USA300. Abbreviation: MFI, mean fluorescence activity.
An additional marker of cell necrosis is HMGB1, an alarmin that is released from the cell nucleus and a potent activator of inflammation [24]. Release of HMGB1 from the cells exposed to WT USA300 but not the Hla− mutant further indicated the induction of necrosis and activation of another proinflammatory signal (Figure 4C).
Calpain or Caspase 1 Inhibitors Rescue Keratinocytes From Hla-Mediated Pyroptosis
We postulated that targeting the effectors of cell necrosis, blocking either caspase 1 or the calpains, would prevent keratinocyte cell death. Treatment of the keratinocytes with calpeptin significantly decreased the numbers of both apoptotic and necrotic/pyroptotic cells, consistent with the predicted role of caspase 1 in activating calpains that contribute to cell autolysis. By fluorescence-activated cell sorting analysis, a population of smaller, condensed keratinocytes was readily apparent in the HaCaTs exposed to WT but not Hla− staphylococci (Figure 5A). Pretreatment of the keratinocytes with either calpeptin or a caspase 1 inhibitor, but not a caspase 3 inhibitor, significantly decreased this fraction of necrotic, and in this case specifically pyroptotic, cells (Figure 5B). Despite the generation of pyroptotic cells, the tissue culture wells appear to remain fully populated with intact monolayers of keratinocytes as viewed with transmission microscopy; the cells exposed to WT staphylococci for 6 hours had condensed nuclei, rounded cytoplasm, and scattered permeability to trypan blue and were distinct from the larger cells with well-defined borders observed in the Hla− or medium-exposed controls (Figure 5C).
Figure 5.

Calpain and caspase 1 inhibitors prevent keratinocyte pyroptosis. A, Fluorescence-activated cell sorting analysis of HaCaTs incubated for 2 hours with medium alone, wild-type (WT) Staphylococcus aureus strain USA300, or α-hemolysin (Hla−) mutant USA300 demonstrating the generation of a population of smaller, condensed cells (circled). B, Quantification of the numbers of condensed cells following exposure to WT USA300 in the presence of calpeptin (Cpep), a caspase 1 inhibitor (C1inh), or a caspase 3 inhibitor (C3inh) as compared with cells exposed to the Hla− mutant strain, expressed as a percentage of the total cells. *P < .01 compared with WT (1-way analysis of variance, Dunnett posttest). Data are mean (SD), representative of 2 individual experiments. C, Transmission light microscopy of representative trypan blue–stained wells of HaCaTs after 6 hours of exposure to WT or Hla− staphylococci and medium alone as a control. Blue staining due to membrane permeability indicated apoptosis/necrosis in WT, less in Hla−, and not at all in medium-treated cells.
Pyroptotic Keratinocytes Facilitate S. aureus Penetration Through Keratinocytes
Pyroptotic keratinocytes could provide a focus for S. aureus to accumulate within the dead and dying cells, eventually reaching sufficient numbers to breach the keratinocyte barrier. This hypothesis was tested by comparing the numbers of WT versus Hla− mutants able to penetrate through HaCaTs grown on Transwells (Corning-Costar) (Figure 6A). There was significantly greater transmigration of the WT USA300 strain than the Hla− mutant (P < .0001), whereas the SpA− strain invaded as well as the WT control. The PVL− strain was also less capable of transmigration but not as deficient as the Hla− strain. To assess the integrity of the keratinocyte barrier, we monitored translocation of 3000 MW fluorescent dextran from the apical to the basal chamber of the transwells (Figure 6B). Only the Hla− strain was associated with significantly less dextran permeability, suggesting that Hla but not PVL was critical in the disruption of the integrity of the keratinocyte barrier.
Figure 6.
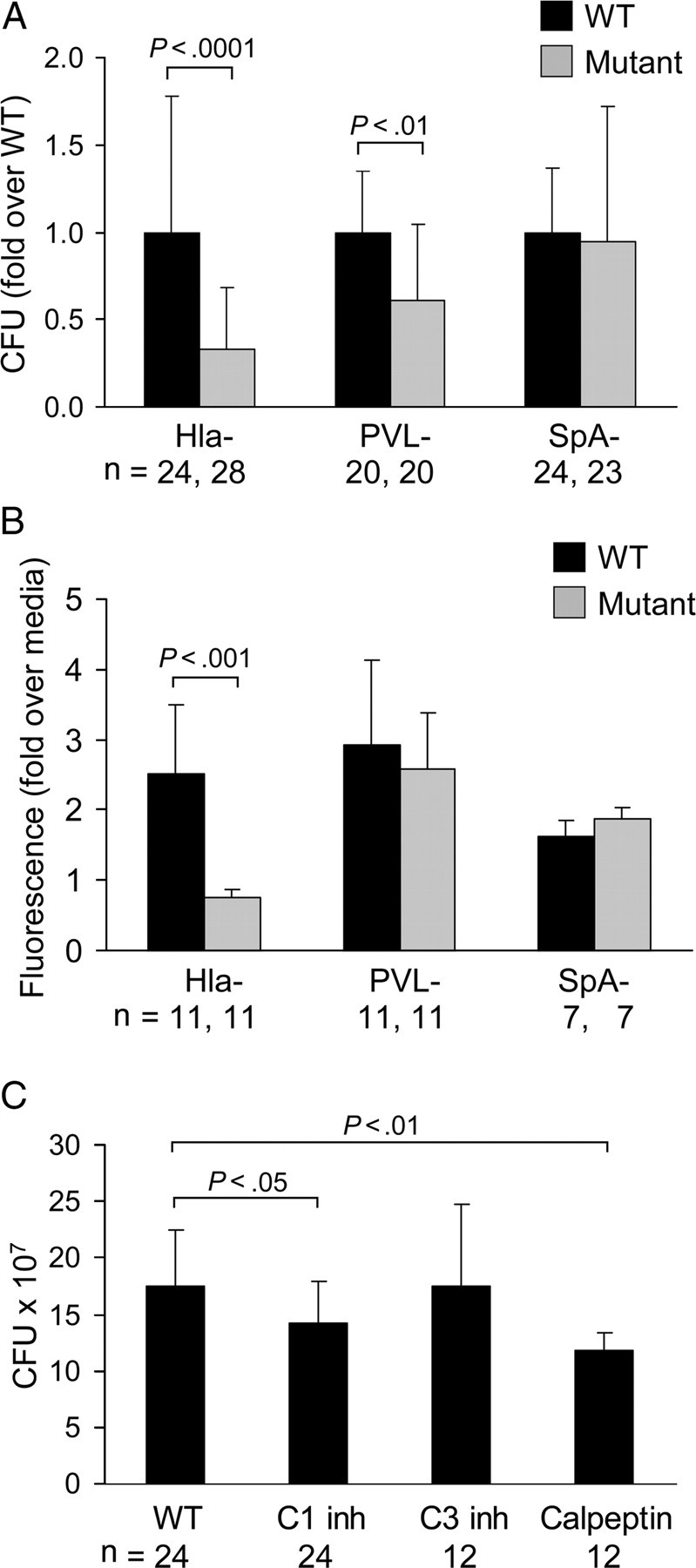
Staphylococcus aureus invasion through keratinocytes is decreased by calpain and caspase 1 inhibition. A, Quantification (in colony-forming units [CFUs]) of organisms recovered from the basal compartment of transwells 24 hours after application of a 108-CFU inoculum in the upper chamber. Approximately 90% of the added inoculum of wild-type (WT) strain was retrieved in the basal compartment following 24-hour incubation. Mutant strains were compared with their matching WT strain (P values are shown; Student t test). Data are mean (SD), representative of at least 3 individual experiments. B, Recovery of fluorescent 3000 MW dextran from the basal compartment of transwells incubated with the staphylococcal mutant and WT paired strains indicated. All were compared with medium alone–treated HaCaT cells (P value shown is for mutant compared with WT; Student t test). Data are mean (SD), representative of at least 2 individual experiments. C, Effects of caspase 1 inhibitor (C1 inh), caspase 3 inhibitor (C3 inh), and calpeptin treatment on S. aureus transmigration, showing quantification of CFUs of WT USA300 recovered from the basal compartment of transwells treated with the inhibitors. *P values shown are compared with WT (1-way analysis of variance, Dunnett posttest). Data are mean (SD), representative of at least 2 individual experiments. Abbreviations: Hla, α-hemolysin; PVL, Panton-Valentine leukocidin; SpA, protein A.
Caspase 1 or Calpain Inhibitors Prevent USA300 Penetration Through Keratinocytes
The predicted mechanism of staphylococcal penetration through the keratinocytes, via caspase 1–mediated cleavage of calpastatin, activation of calpains, and pyroptosis, was further documented. USA300 transmigration across HaCaTs treated with calpeptin, a calpain inhibitor, or a caspase 1 inhibitor was significantly decreased whereas treatment of the keratinocytes with a caspase 3 inhibitor did not alter rates of transmigration (Figure 6C). Cytochalasin D treatment of the HaCaTs, which prevents the endocytosis of S. aureus or its components [25], the metalloproteinase inhibitor TAPI, and a general protease inhibitor, GM6001, did not inhibit transmigration (data not shown). Thus, inhibition of caspase 1 or calpain activity protected the keratinocytes from pyroptosis and staphylococcal penetration.
DISCUSSION
These experiments further characterize the signaling cascades exploited by S. aureus to cause skin infection. Our data suggest that it is the proinflammatory bias of human keratinocytes and the expression of specific staphylococcal toxins, particularly Hla, that result in focal skin infection. Human keratinocytes, even in the absence of recruited phagocytes, are remarkably resistant to S. aureus penetration. Keratinocytes have several unique features that contribute to their ability to withstand invasion by S. aureus and other common pathogens. Despite the high prevalence of S. aureus nasal colonization, even with the potentially virulent USA300 strains, remarkably few individuals actually become infected. Production of several types of antimicrobial peptides is likely to substantially inhibit superficial colonization by these organisms [26]. However, once staphylococci are sensed by the keratinocytes, they are poised to undergo pyroptosis to eradicate infected cells. In contrast to macrophages, which require the internalization of particulate peptidoglycan to activate inflammasome signaling [17], keratinocytes constitutively express IL-1β and thus are primed for activation by surface contact with staphylococci. While the keratinocytes are undergoing degradation, the organisms appear to proliferate intracellularly, having activated the destruction of the keratinocyte and evaded the toxicity of the keratinocyte products.
The ability of keratinocytes to proliferate in response to S. aureus is also likely to prevent invasive infection. As we observed, by 24 hours organotypic cultures of human keratinocytes appeared to be substantially thicker when exposed to these bacteria. As S. aureus Hla stimulates epithelial proliferation via EGFR [15], keratinocyte infection does not result in widespread loss of barrier function but instead results in focal areas of infection and induction of more keratinocytes to replace the necrotic cells. This was evident in the transmission microscopy of the HaCaT cultures, which despite the induction of pyroptosis remained remarkably intact for at least 24 hours following infection. This is in contrast to what occurs at more vulnerable mucosal sites, such as the airway, that do not have such proliferative capacity and would be rapidly destroyed by a similar exposure [13].
It is interesting that different components of staphylococci appear to mediate invasion through specific tissues. Hla production is clearly critical for skin infection, whereas protein A, which was entirely dispensable in the activation of pyroptosis of keratinocytes, was essential to enable staphylococci to penetrate across an airway epithelial barrier [13]. Hla also contributes to barrier dysfunction in A549 cells through its interaction with ADAM-10 and cleavage of E-cadherin [27]. This interaction may also contribute to penetration through the skin, accounting for the transmigration that is not inhibited in the presence of calpain or caspase 1 inhibitors. It is also noteworthy that neither SpA nor Hla is unique to the USA300 strains of staphylococci.
The PVL− strain was less effective in transmigration across the keratinocytes, but the same mutant was as effective as the parental strain in stimulating pyroptosis. It is possible that PVL pore formation acts in concert with Hla to enhance keratinocyte pyroptosis but is not a critical factor.
The induction of keratinocyte signaling and specifically calpain activity was critical for staphylococcal invasion. The participation of calpains in mediating inflammation in response to bacterial infection has become increasingly recognized. We have previously found that calpains participate in PMN recruitment across airway epithelial barriers [22]. In those studies, the stimulus for calpain activity was the Ca2+ flux induced through TLR2 signaling, a factor that is relevant to staphylococcal skin infection. Calpains are also central to the pathology that is caused by staphylococcal infection of the skin. Although our in vitro system was restricted to the keratinocytes themselves, in vivo, PMNs would be recruited and activated through inflammasome-mediated signaling: by IL-1β and by calpain-mediated cleavage of paracellular junctional proteins to facilitate their recruitment [22]. As a consequence of inflammasome activation in the skin, focal accumulation of PMNs results in the pustule or abscess formation characteristic of S. aureus skin infection. Recognition that the caspase 1/calpain cascade in keratinocytes is a major cause of the pathology induced by S. aureus could provide new targets for topical antistaphylococcal therapy, as well as provide new impetus for drugs that inhibit S. aureus Hla activity.
Notes
Acknowledgments. We thank Taylor Cohen for help with statistical analyses.
Financial support. This work was supported by the National Institutes of Health (grant number RO1 HL79395 to A. P.) and the Columbia University Department of Dermatology Pilot and Feasibility Grant Program (grant number P30AR44535).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–71. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Talan DA, Krishnadasan A, Gorwitz RJ, et al. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin Infect Dis. 2011;53:144–9. doi: 10.1093/cid/cir308. [DOI] [PubMed] [Google Scholar]
- 3.Otto M. A MRSA-terious enemy among us: end of the PVL controversy? Nat Med. 2011;17:169–70. doi: 10.1038/nm0211-169. [DOI] [PubMed] [Google Scholar]
- 4.Hruz P, Zinkernagel AS, Jenikova G, et al. NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc Natl Acad Sci U S A. 2009;106:12873–8. doi: 10.1073/pnas.0904958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy AD, Bubeck Wardenburg J, Gardner DJ, et al. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis. 2010;202:1050–8. doi: 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller LS, Pietras EM, Uricchio LH, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–42. doi: 10.4049/jimmunol.179.10.6933. [DOI] [PubMed] [Google Scholar]
- 7.Mempel M, Schnopp C, Hojka M, et al. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br J Dermatol. 2002;146:943–51. doi: 10.1046/j.1365-2133.2002.04752.x. [DOI] [PubMed] [Google Scholar]
- 8.Pishchany G, McCoy AL, Torres VJ, et al. Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe. 2010;8:544–50. doi: 10.1016/j.chom.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houben E, De Paepe K, Rogiers V. A keratinocyte's course of life. Skin Pharmacol Physiol. 2007;20:122–32. doi: 10.1159/000098163. [DOI] [PubMed] [Google Scholar]
- 10.Kollisch G, Kalali BN, Voelcker V, et al. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology. 2005;114:531–41. doi: 10.1111/j.1365-2567.2005.02122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–8. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craven RR, Gao X, Allen IC, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soong G, Martin FJ, Chun J, Cohen TS, Ahn DS, Prince A. Staphylococcus aureus protein A mediates invasion across airway epithelial cells through activation of RhoA GTPase signaling and proteolytic activity. J Biol Chem. 2011 doi: 10.1074/jbc.M111.295386. 10.1074/jbc.M1111.295386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin FJ, Gomez MI, Wetzel DM, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest. 2009;119:1931–9. doi: 10.1172/JCI35879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haugwitz U, Bobkiewicz W, Han SR, et al. Pore-forming Staphylococcus aureus alpha-toxin triggers epidermal growth factor receptor-dependent proliferation. Cell Microbiol. 2006;8:1591–600. doi: 10.1111/j.1462-5822.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 16.Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimada T, Park BG, Wolf AJ, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walev I, Martin E, Jonas D, et al. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–9. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez MI, Lee A, Reddy B, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10:842–8. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 20.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 21.Kashio Y, Nakamura K, Abedin MJ, et al. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol. 2003;170:3631–6. doi: 10.4049/jimmunol.170.7.3631. [DOI] [PubMed] [Google Scholar]
- 22.Chun J, Prince A. TLR2-induced calpain cleavage of epithelial junctional proteins facilitates leukocyte transmigration. Cell Host Microbe. 2009;5:47–58. doi: 10.1016/j.chom.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pisetsky DS, Erlandsson-Harris H, Andersson U. High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. 2008;10:209. doi: 10.1186/ar2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards AM, Potter U, Meenan NA, Potts JR, Massey RC. Staphylococcus aureus keratinocyte invasion is dependent upon multiple high-affinity fibronectin-binding repeats within FnBPA. PLoS One. 2011;6:e18899. doi: 10.1371/journal.pone.0018899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev Dermatol. 2010;5:183–95. doi: 10.1586/edm.10.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoshima I, Inoshima N, Wilke GA, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17:1310–4. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]